Article Text

Abstract
Objective Liver fibrosis and cirrhosis resulting from chronic liver injury represent a major healthcare burden worldwide. Growth differentiation factor (GDF) 11 has been recently investigated for its role in rejuvenation of ageing organs, but its role in chronic liver diseases has remained unknown. Here, we investigated the expression and function of GDF11 in liver fibrosis, a common feature of most chronic liver diseases.
Design We analysed the expression of GDF11 in patients with liver fibrosis, in a mouse model of liver fibrosis and in hepatic stellate cells (HSCs) as well as in other liver cell types. The functional relevance of GDF11 in toxin-induced and cholestasis-induced mouse models of liver fibrosis was examined by in vivo modulation of Gdf11 expression using adeno-associated virus (AAV) vectors. The effect of GDF11 on leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5)+ liver progenitor cells was studied in mouse and human liver organoid culture. Furthermore, in vivo depletion of LGR5+ cells was induced by injecting AAV vectors expressing diptheria toxin A under the transcriptional control of Lgr5 promoter.
Results We showed that the expression of GDF11 is upregulated in patients with liver fibrosis and in experimentally induced murine liver fibrosis models. Furthermore, we found that therapeutic application of GDF11 mounts a protective response against fibrosis by increasing the number of LGR5+ progenitor cells in the liver.
Conclusion Collectively, our findings uncover a protective role of GDF11 during liver fibrosis and suggest a potential application of GDF11 for the treatment of chronic liver disease.
- fibrosis
- myofibroblasts
- chronic liver disease
- stem cells
- hepatic stellate cells
- GDF11 and LGR5
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Liver fibrosis and cirrhosis are common features of most chronic liver diseases and cause millions of deaths worldwide.
Transforming growth factor β family members have been shown to play pivotal roles in fibrogenesis of the liver as well as in other organs.
Till date, the function of growth differentiation factor (GDF) 11, a member of the transforming growth factor β family, during liver fibrosis has not been elucidated.
What are the new findings?
The level of GDF11 is elevated in patients with liver fibrosis.
Identification of activated hepatic stellate cells as the main source of hepatic GDF11 in vivo.
The overexpression of GDF11 by adeno-associated virus vectors attenuates toxin-induced and cholestasis-induced liver fibrosis models, two independent modes of chronic liver injury.
GDF11 attenuates liver fibrosis via augmenting expansion of liver progenitor cells in fibrotic mice.
How might it impact on clinical practice in the foreseeable future?
We demonstrate that GDF11 overexpression reduces liver fibrosis by promoting the number of liver progenitor cells in mice. Our findings provide a foundation for further investigation of the therapeutic utility of GDF11 in the treatment of liver fibrosis.
Introduction
The transforming growth factor β (TGF-β) superfamily of secreted proteins comprises more than 30 structurally related, yet functionally distinct proteins that play critical roles in embryological tissue development and patterning, wound healing and adult tissue maintenance.1 The GDF11 gene, a member of TGF-β superfamily, is located on chromosome 12 in humans and on chromosome 10 in mice and encodes a secreted protein that shares high homology with growth differentiation factor (GDF) 8 (myostatin), a proven negative regulator of muscle mass.2 The knockout of Gdf8 results in muscle hypertrophic animals,2 whereas the Gdf11 knockout mice are perinatal lethal,3 indicating functional differences between the two proteins. The functions of GDF11 in modulation of age-related dysfunction of heart,4 5 skeletal muscle6–8 and brain9 have been recently investigated. The role of GDF11 in acute liver injury has been investigated recently.10 However, till date, the relevance of GDF11 in the pathophysiology of chronic liver disease and its potential therapeutic application therein remain to be understood.
Adult stem/progenitor cells play key roles in organ homeostasis and pathophysiological conditions.11 12 The transplantation of adult stem cells is one of the methods for the treatment of multiple disorders including blood, metabolic, muscle and skin diseases.12 13 Hematopoietic, skeletal muscle and intestinal stem cells represent a class of dedicated stem cells that contribute to maintenance of normal organ function. In contrast, organs such as liver maintain homeostasis by differentiated cells, mainly hepatocytes (HCs) and cholangiocytes. In chronic liver injury, LGR5+ liver progenitor cells (LPCs), which are almost absent in the normal liver, emerge in response to damage.14–16 The factors that are able to increase the number of stem/progenitor cells remain to be identified.
GDF11 is known to regulate progenitor cell growth in different organs such as developing retina,17 pancreas18 and endothelium.19 However, it has remained unexplored whether GDF11 can promote the expansion of LGR5+ LPCs and its impact on progression of chronic liver diseases. Here, we report that hepatic GDF11 is upregulated in patients with fibrotic livers and mouse models of liver fibrosis. We identified hepatic stellate cells (HSCs) as a primary source of hepatic GDF11. The overexpression of GDF11 in the liver exerts a protective response against liver fibrosis in different mouse models. Furthermore, the antifibrotic effect of GDF11 is dependent on the enhanced number of LGR5+ LPCs.
Methods
Ethics statement
Formalin-fixed paraffin-embedded liver tissues from human fibrosis or cirrhosis patients were obtained from Hannover Medical School, Germany. RNA samples of fibrotic human liver were provided by Haikou Hospital, China, and Hannover Medical School, Germany. Human LPC organoids were prepared at Hannover Medical School. Adult male 8- to 12-week-old BALB/c mice were used for all in vivo experiments performed in this study.
In situ hybridisation
Non-radioactive in situ hybridisation analysis of gene expression was performed on 10 µm paraffin sections of the fibrotic and healthy livers of patients and mice using digoxigenin-labelled antisense riboprobes for human GDF11 and mouse Gdf11 as described previously.20 Six liver samples in each group were used for in situ hybridisation. Briefly, after deparaffinisation, liver sections were pretreated with proteinase K, rinsed and re-fixed. Sections were allowed to pre-hybridise and then hybridised in hybridisation mix with digoxigenin-labelled antisense riboprobes. Immunological detection was then performed, followed by dehydration and placing the coverslip. Images were taken using a Nikon camera attached to Olympus microscope.
Isolation of primary cells
Mouse primary HCs were isolated following our previously reported method21 and cultured with hepatocyte maintenance medium (HCM) . HSCs were isolated and either lysed directly in Trizol or cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% Penicillin-Streptomycin and 4 mM L-glutamine.22 Liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs) were isolated following the procedure as described.23 In brief, two-step perfusion of mouse livers was performed. At first, HCs were collected by centrifugation at 300 rpm. HSCs were isolated by Nycodenz gradient centrifugation. LSECs were purified using CD146 microbead-based magnetic activated cell sorting (MACS). KCs were isolated by Percoll density gradient centrifugation. Primary human hepatic myofibroblasts were cultured in DMEM supplemented with 10% heat-inactivated FBS, 1% Penicillin-Streptomycin and 4 mM L-glutamine.
Isolation and transplantation of LGR5+ LPCs
Cell sorting of LGR5+ LPCs was performed as outlined in previous publications.14 24 Liver perfusion was performed by a two-step collagenase (Roche) perfusion method. The digested liver was suspended in 50 mL of DMEM, and the dissociated cells were passed through a 100 µm nylon mesh and centrifuged at 50 g for 5 min at 4°C. The supernatant was collected and centrifuged at 300 g for 5 min at 4°C. Murine or human LGR5+ LPCs were stained with the anti-LGR5 antibody, followed by Alexa-594 conjugated goat anti-rabbit secondary antibody and sorted using MoFlo XDP machine. For in vivo experiments, mice were transplanted with sorted LGR5+ cells via spleen. Briefly, mice were anaesthetised with a continuous supply of isoflurane and oxygen and cut with a lateral abdominal incision. We then injected 100 000 cells per mouse in a volume of 50 µL using 27 g needle into the spleen of mice. A knot was tied to prevent the outward flow of the cell suspension before suturing the mice.
Culture of murine and human LPC organoids
Sorted LGR5+ murine LPCs were mixed with Matrigel (BD Bioscience) and cultured as described.14 Briefly, for the first 3 days after sorting, the medium composition was as follows: AdDMEM/F12 (Invitrogen) supplemented with 1% B27 and 1% N2 (Invitrogen), N-acetylcysteine (1.25 mM, Sigma-Aldrich), gastrin (10 nM, Sigma), EGF (50 ng/mL, Peprotech), RSPO1 (50 ng/mL, R&D), FGF10 (100 ng/mL, Peprotech), nicotinamide (10 mM, Sigma-Aldrich), and HGF (50 ng/mL, Peprotech), 25 ng/mL Noggin (R&D), 25 ng/mL WNT (R&D) and 10 µM Y27632 (Sigma-Aldrich). After 3 days, the medium was changed into the above medium without Noggin, WNT and Y27632 and was changed every other day.
For human organoid culture, we followed a previously published protocol from Dr Clevers’ laboratory.25 Briefly, the medium composition was as follows: AdDMEM/F12 (Invitrogen) supplemented with 1% B27 and 1% N2 (Invitrogen), N-acetylcysteine (1.25 mM, Sigma-Aldrich), gastrin (10 nM, Sigma), EGF (50 ng/mL, Peprotech), RSPO1 (50 ng/mL, R&D), FGF10 (100 ng/mL, Peprotech), nicotinamide (10 mM, Sigma-Aldrich), and HGF (50 ng/mL, Peprotech), 5 µM A83.01 (Tocris) and 10 µM FSK (Tocris). For the establishment of the culture, the first 3 days after isolation, the medium was supplemented with 25 ng/mL Noggin (R&D), 25 ng/mL WNT (R&D) and 10 µM Y27632 (Sigma-Aldrich). Liver organoids were passaged every 5 days in the presence of GDF11, whereas organoids without GDF11 needed 7 days for each passage.
For HC differentiation, liver organoids were cultured 2 days in organoid culture medium supplemented with BMP7 (25 ng/mL). Then, medium was changed to differentiation medium: AdDMEM/F12 medium, 1% N2, 1% B27, EGF (50 ng/mL), gastrin (10 nM, Sigma), HGF (25 ng/mL), FGF19 (100 ng/mL), A8301 (500 nM), DAPT (10 µM), BMP7 (25 ng/mL), dexamethasone (30 µM). Differentiation medium was changed every 3 days. After 12 days, supernatant was collected for ALB secretion analysis.
Histology, immunohistochemistry and immunofluorescence
HCs were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min and stained following a standard protocol for immunofluorescence. For immunohistochemistry, liver tissues were fixed with 4% formalin and embedded in paraffin. Biotinylated goat anti-rabbit and goat anti-rat secondary antibodies (Vectastain, Vector Laboratories) were used. For Sirius Red staining, the sections were deparaffinised and stained with picro-Sirius Red solution for 60 min. Sections were rinsed with water, dehydrated and mounted in xylene. Results of immunofluorescence or immunohistochemistry were quantified by Image-J software in a blinded manner. Lipid droplet accumulation was stained with Oil red O (O0625; Sigma-Aldrich) using frozen liver sections. To calculate the NAS, H&E were analysed blindly according to criteria described by Brunt et al.26 Mouse serum insulin was assessed using ELISA kit (cat 90080; Crystal Chem). Mouse blood glucose concentration was assessed using blood glucose metre (Bayer). A list of antibodies and primers are provided in online supplementary tables 2 and 3, respectively.
Supplemental material
Statistical analysis
Significance was determined with the two-tailed Student’s t-test or two-sided Welch’s t-test. Statistical significance was calculated with error bars representing ±SEM. P<0.05 was considered as significant.
For further methods, please see supplementary information.
Results
The expression of hepatic GDF11 is enhanced during liver fibrosis
To investigate the relevance of GDF11 in liver diseases, we first examined the expression of GDF11 in patients with liver fibrosis, the common feature of most chronic liver diseases.27 In situ hybridisation on formalin-fixed paraffin-embedded sections indicated increased expression compared with respective healthy controls (figure 1A). These results were confirmed by quantitative (q) PCR in human fibrotic samples obtained from two different hospitals: Hannover Medical School, Germany (figure 1B and online supplementary table 1a) and Haikou Hospital, China (figure 1C and online supplementary table 1b).

GDF11 is upregulated during liver fibrosis. (A) In situ hybridisation analysis of human GDF11 expression. Scale bars, 100 µm. (B,C) The qPCR-based analysis of human GDF11 mRNA in patients with fibrosis (n=6) and controls (n=6) from Hannover Medical School, Germany (B) and patients with fibrosis (n=26) and controls (n=6) from Haikou Hospital, China (C). (D) In situ hybridisation for mouse Gdf11 in fibrotic and control livers. (E) The qPCR-based analysis of mouse Gdf11 mRNA in fibrotic (n=7) and control (n=7) livers. (F) Western blot for GDF11 in normal and fibrotic mouse livers and its quantification. (G) Measuring the GDF11 content by ELISA in mouse serum. (H) The qPCR analysis of mouse Gdf11 in various liver cells such as hepatocytes (HC), hepatic stellate cells (HSC), Kupffer cells (KC) and liver sinusoidal endothelial cells (LSEC), isolated from fibrotic livers of different mice. Total RNA from kidney was used as a positive control. (I,J) GDF11 and desmin co-immunofluorescence in fibrotic livers of patients and mouse. Scale bars, 100 µm. (K) qPCR analysis for Gdf11 expression in quiescent and HSCs activated by culturing them in the presence of platelet-derived growth factor (PDGF). Experiments were repeated twice for B, C, H and I, and three times for E and F. The values shown in panels G, J and K are the mean of three independent cell isolations. Data are mean±SEM; two-tailed Student’s t-test (panels E, F, G and K) or two-sided Welch’s t-test (panels B and C). CCl4, carbon tetrachloride; DAPI, 4′,6-diamidino-2-phenylindole; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; GDF11, growth differentiation factor 11; NPC, non-parachamal cells.
Similar to the observed elevated levels of GDF11 in fibrotic human livers, in a mouse model of liver fibrosis, which was induced by injecting mice with carbon tetrachloride (CCl4) for 8 weeks, Gdf11 mRNA and GDF11 protein levels were markedly increased as shown by in situ hybridisation (figure 1D), qPCR (figure 1E) and Western blot analysis (figure 1F and online supplementary figure 1a), respectively. The elevated levels of GDF11 were confirmed in the serum of mice injected with CCl4 or mice fed with the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet for 8 weeks (figure 1G). Since GDF11 exhibits high similarity with GDF8, we also determined the expression of Gdf8 in mouse liver. We found that Gdf8 is expressed in murine embryonic liver (E13.5). However, Gdf8 expression was not detected in the adult liver, neither in purified adult primary HCs nor in non-parenchymal adult liver cells isolated from both control and fibrotic livers (online supplementary figure 1b). In concert with these results, GDF8 expression was unaltered in human fibrotic livers and respective controls (online supplementary figure 1c–d). Thus, a contribution of Gdf8 to fibrosis in livers of humans and mice seems unlikely.
Supplemental material
We next determined the cell type-specific expression of Gdf11 in murine normal and fibrotic livers by qPCR. We isolated HCs, HSCs, KC and LSECs from normal livers (online supplementary figure 1e) and found that HSCs are the major cell of origin that express Gdf11 in normal livers (online supplementary figure 1f). In fibrotic livers also, we observed the highest expression of Gdf11 in activated HSCs followed by KC and LSEC (figure 1H). In contrast, HC, which constitute up to 80% of the liver mass, had minimal expression of Gdf11 (figure 1H). GDF11 expression in activated HSCs (also known as myofibroblasts) was confirmed by co-staining of desmin and GDF11 in fibrotic human livers (figure 1I) and fibrotic mouse livers (figure 1J). Furthermore, expression of Gdf11 was increased in cultured activated primary mouse HSCs in vitro (figure 1K) and in vivo (online supplementary figure 1g–h), indicating a role in the regulation of fibrosis.
In vivo overexpression of hepatic Gdf11 attenuates liver fibrosis
In view of elevated in vivo Gdf11 expression in HSC-derived myofibroblasts, which are key cellular drivers of fibrogenesis in the liver,28 we reasoned that Gdf11 may have regulatory functions in liver fibrosis. To address this, we examined the in vivo function of GDF11 in a CCl4-induced mouse model of liver fibrosis (figure 2A). For this, we administered 1×1011 adeno-associated virus serotype 8 (AAV8) particles encoding Gdf11 (henceforth referred to as AAV.GDF11) to fibrotic BALB/c mice. Control mice were injected with equal numbers of control AAV particles. We first ascertained high transduction efficiency on AAV injection (online supplementary figure 2). Overexpression of Gdf11 mRNA as well as GDF11 protein in AAV.GDF11-injected fibrotic mice was confirmed by qPCR (figure 2B) and ELISA (online supplementary figure 3a–b). We found a significant reduction of fibrosis in AAV.GDF11-injected mice as shown by lower hydroxyproline content (figure 2C), Sirius Red and desmin staining (figure 2D–E) and expression of fibrogenic genes such as Acta2, p75Ntr, Col1a1 and Col2a1 (figure 2F). These results indicate that GDF11 administration mitigates toxin-induced liver fibrosis in mice.
Supplemental material
Supplemental material

Hepatic overexpression of GDF11 attenuates liver fibrosis. (A,G) Schematic overview of experiments analysing the effect of Gdf11 overexpression in CCl4-induced (A) and cholestasis (DDC)-induced (G) mouse models of liver fibrosis (n=8 mice per group of CCl4 and n=8 mice per group of DDC model). (B,H) The qPCR analyses of Gdf11 expression in murine livers after AAV8-GDF11 injection. (C,I) Measurement of the total collagen content by hydroxyproline assay. (D,J) Representative immunohistochemical images of H&E, Sirius Red and desmin staining. Scale bars, 100 µm. (E,K) Quantification of Sirius Red and desmin staining in mice injected with either AAV8-GDF11 or AAV control particles. For each mouse, 6 liver sections were stained in batches and pictures from 12 random fields per section were captured and quantified in a blinded manner using Image-J. (F,I) The qPCR analyses of fibrosis-related genes such as Acta2, p75Ntr, Col1a1 and Col2a1. All experiments shown in this figure were repeated twice. Data are mean±SEM; two-tailed Student’s t-test. AAV8, adeno-associated virus serotype 8; CCl4, carbon tetrachloride; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; GDF11, growth differentiation factor 11.
Cholestasis-induced fibrosis and cirrhosis are other major indications for liver transplantation.29 Hence, we investigated whether GDF11 overexpression is able to mitigate cholestasis-induced liver fibrosis by injecting AAV.GDF11 in BALB/c mice fed with a DDC diet (figure 2G). After confirming GDF11 overexpression in the liver (figure 2H and online supplementary figure 3e–f), we analysed tissues for fibrosis markers. Similar to the CCl4 model, we observed reduced levels of fibrosis in the livers of AAV.GDF11-injected mice (figure 2I–L). Thus, AAV-based overexpression of Gdf11 attenuates cholestasis-induced and toxin-induced liver fibrosis.
To investigate whether hepatic Gdf11 overexpression in fibrotic mice would cause any profibrogenic effects in other organs, we additionally analysed heart, lungs, kidneys, muscles, brain, intestine and the spleen (online supplementary figure 3c–h). Both, Sirius Red staining and Acta2 expression ruled out induction of fibrosis in above-mentioned organs.
We next examined whether administration of recombinant (r)GDF11 mimics the antifibrotic effects obtained by AAV.GDF11 in both the CCl4-induced and the DDC-induced fibrosis model (online supplementary figure 4a and f). In fact, the hydroxyproline assay, H&E, Sirius Red and desmin staining, as well as qPCR analysis (online supplementary figure 4b–j) for fibrogenic genes showed reduced levels of fibrosis in mice injected with rGDF11.
Supplemental material
The hepatic Gdf11 overexpression promotes LPCs
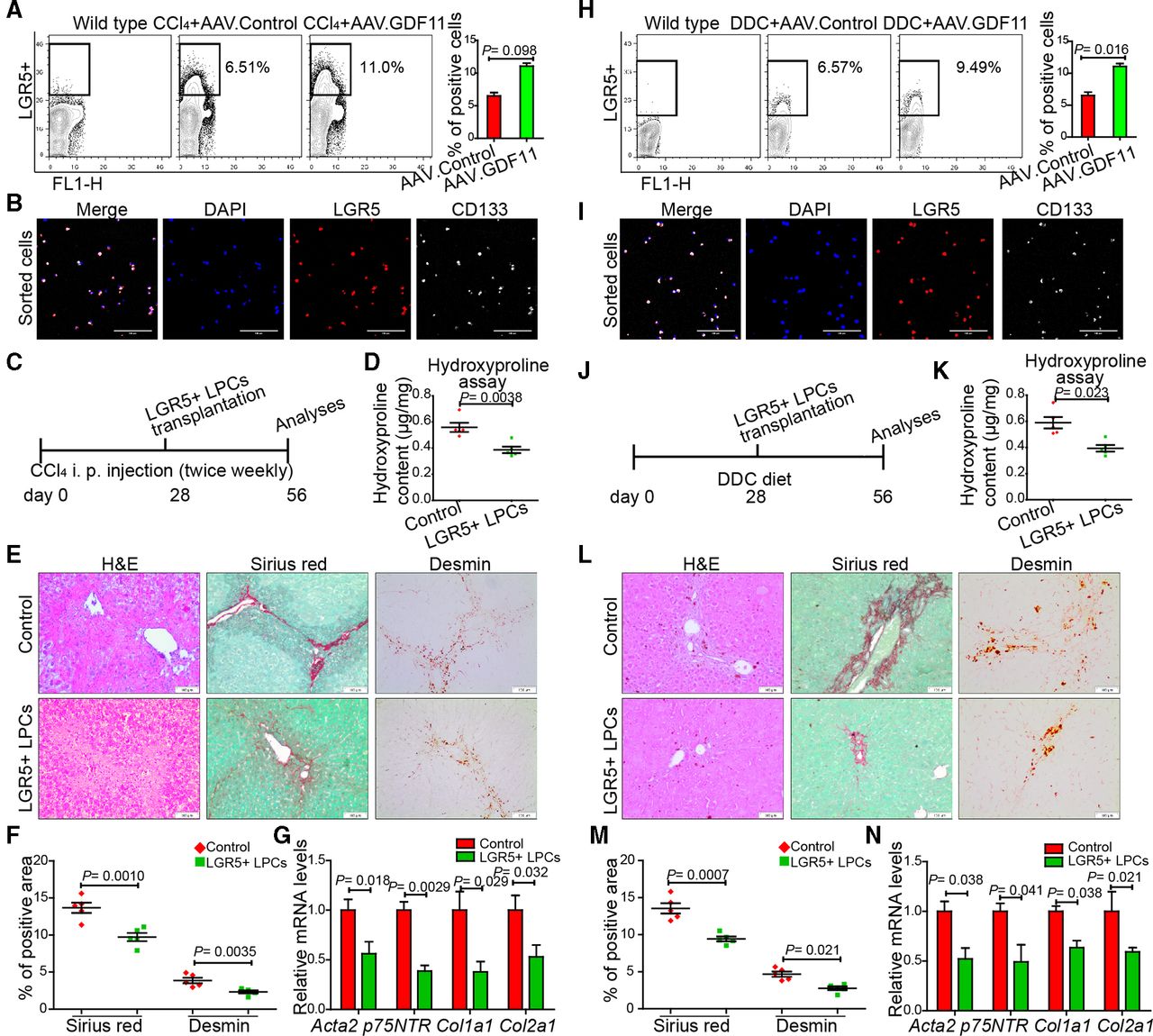
LGR5+ LPCs in the healthy liver are virtually undetectable; however, they emerge in response to chronic injuries.15 16 Since GDF11 has also been demonstrated to promote progenitor cell fates in other organs,17 19 we reasoned that GDF11 administration in fibrotic liver expands the hepatic LGR5+ progenitor pool. We tested this possibility by fluorescence-activated cell sorting (FACS) analysing LGR5+ cells in CCl4-injected (figure 3A) as well as in DDC-fed mice (figure 3H). We observed increased numbers of LGR5+ cells in AAV.GDF11-injected fibrotic mice compared with the LGR5+ cells either in AAV.control-injected fibrotic mice or in wild-type healthy controls (figure 3A and H). To validate that the isolated cells are indeed progenitor cells, we stained them for the presence of CD133, another characteristic marker of LGR5+ epithelial progenitors.30 The co-expression of LGR5 with CD133 confirmed the epithelial progenitor phenotype of the sorted cells (figure 3B and I). The mRNA expression analyses revealed that isolated LGR5+ cells express higher levels of Lgr5, Prom1, Epcam and Krt19 and lower levels of Hnf4a and Alb (online supplementary figure 6a).
Supplemental material

GDF11 promotes the expansion of LGR5+ liver progenitor cells (LPCs), which are capable of attenuating liver fibrosis in mice. (A,H) Representative FACS analysis of LGR5+ LPCs in AAV8-GDF11 or AAV control-injected mice in the CCl4 (n=8) (A) and the DDC (n=8) (H) fibrosis model. (B,I) The confocal immunofluorescence images of sorted LGR5+ cells, which were subsequently co-stained with CD133 antibody. Scale bars, 100 µm. (C,J) Schematic overview of the experimental set-up for LGR5+ LPC transplantation in fibrotic mice (n=5 mice per group for both CCl4 and DDC model). The control mice were injected with saline via spleen. (D,K) Measurement of the total collagen content by the hydroxyproline assay in LGR5+ LPC transplanted mice. (E,L) Representative images of H&E, Sirius Red and desmin staining. Scale bars, 100 µm. (F,M) Quantifications of Sirius Red and desmin staining in LGR5+ LPC transplanted and control mice. For each mouse, 6 liver sections were stained in batches and pictures from 12 random fields per section were captured and quantified in a blinded manner using Image-J. (G,N) The qPCR analyses of fibrosis-related genes in LGR5+ LPC transplanted mice. Experiments were repeated twice for A, H and three times for C–G and J–N. Data are mean±SEM; two-tailed Student’s t-test. AAV8, adeno-associated virus serotype 8; CCl4, carbon tetrachloride; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FACS, fluorescence-activated cell sorting; GDF11, growth differentiation factor 11; DAPI, 4′,6-diamidino-2-phenylindole; LGR5, leucine-rich repeat-containing G-protein-coupled receptor 5.
To further examine the effect of GDF11 on LPCs, we isolated mouse LGR5+ cells by FACS and treated LGR5+ cell-derived liver organoids with rGDF11 (online supplementary figure 5a). We confirmed formation of organoids, which exhibited expression of cholangiocyte markers such as KRT19 and SOX9 as well as HC markers such as HNF4A (online supplementary figure 5b–c). The treatment of LGR5+ cell-derived mouse liver organoids with 40 ng/mL rGDF11 led to an increase in organoids number and LGR5+ cell numbers and further elevation of Lgr5 and Prom1 mRNA expression (online supplementary figure 5d–e). A dose higher than 40 ng/mL did not increase Lgr5 and Prom1 expression further (online supplementary figure 5f).
Supplemental material
Transplantation of LGR5+ LPCs inhibits liver fibrosis
We then examined whether GDF11-treated LGR5+ LPCs are capable of resolving liver fibrosis. We sorted LGR5+ cells from AAV.GDF11-injected fibrotic mice and transplanted 5×105 of these cells into another set of BALB/c mice that were either injected with CCl4 (figure 3C) or fed with DDC diet (figure 3J). We tested transplantation efficacy in a subset of fibrotic mice that were transplanted with 5×105 LGR5+ cells, which were in turn transduced with a lentiviral vector encoding for green fluorescent protein (GFP) under the transcriptional control of a cytomegalovirus (CMV) promoter (online supplementary figure 6b). We sacrificed mice 7 days after transplantation and observed the presence of GFP-positive LGR5+ cells in fibrotic livers (online supplementary figure 6c), indicating successful transplantation. The histological analyses, Sirius Red and desmin staining and qPCR for fibrogenic markers showed amelioration of both CCl4-induced (figure 3D–G) and DDC-induced liver fibrosis (figure 3K–N), indicating that transplantation of LGR5+ cells leads to reduction in liver fibrosis.
GDF11 promotes human LGR5+ progenitor cells in organoid culture
To address whether GDF11 is able to enhance the expansion of human liver progenitors, we established a human liver organoid culture and subsequently supplemented it with rGDF11 (figure 4A). Human liver organoids expressed markers of both cholangiocytes such as KRT19, SOX9 and HCs such as HNF4A (figure 4B–C) and also upregulated expression of mature HC markers in the presence of HC differentiation medium (figure 4D). Also, on day 4 following the treatment of human liver organoids with GDF11, we observed an increase in their numbers (figure 4E) as well as elevation of LGR5 and PROM1 expression (figure 4F) compared with controls. Similar to mouse organoids, the treatment of human liver organoids with GDF11 did not change HNF4A expression (figure 4F). In addition, the number of LGR5+ cells within the organoids increased after treatment with rGDF11 (figure 4G). Of note, GDF11 did not inhibit hepatic differentiation of the LGR5+ cells in organoids since characteristic marker of HC differentiation such as secreted albumin exhibited no significant difference compared with respective controls (figure 4H). Together, these results indicate that GDF11 promotes the expansion of LGR5+ cells in human liver organoids.

GDF11 promotes human liver progenitor cell expansion in ex vivo cultured organoids. (A) Schematic overview of the experimental setup. (B) Serial images showing the growth of human liver organoids. (C) Representative confocal images of human liver organoids stained for KRT19, SOX9, HNF4a. Nuclei were counterstained with DAPI. Scale bars, 100 µm. (D) Analysis of the hepatocyte differentiation by immunofluorescence staining for hepatocyte markers such as albumin (ALB) and major urinary protein (MUP). (E) Representative images of organoids treated with rGDF11 (bottom) and untreated (top) and quantification of organoid numbers at day 4. In each well, all areas with organoids were imaged by serial pictures and quantified in a blinded manner (n=10 wells). (F) qPCR-based analysis of human LGR5, PROM1 and HNF4A mRNA expression in organoids either treated with rGDF11 or untreated. (G) FACS analysis for LGR5+ cells on rGDF11-treated or rGDF11-untreated organoids and dissociated to single cells. (H) The mRNA expression (left) and ELISA for albumin performed with culture medium collected (right) from human liver organoids that were either treated with rGDF11 or remained untreated. (I) Schematic overview of experiments showing human primary hepatic myofibroblasts transfected with GDF11 siRNA and subsequently co-cultured with human liver organoids. (J) qPCR analysis of human GDF11 in human myofibroblasts. (K) Representative images of organoids co-cultured with human myofibroblasts transfected either with siGDF11 (bottom) or scramble (top) and quantification of organoid numbers at day 4. For each well, all areas with organoids were imaged by serial pictures and quantified in a blinded manner (n=10 wells). (L–M) qPCR analysis of human LGR5, PROM1, HNF4A and FACS analysis for LGR5+ cells on human liver organoids co-cultured with human myofibroblasts transfected with siGDF11 (right) or scramble (left). Experiments were repeated twice for B–D, and three times for E–M. Data are mean±SEM; two-tailed Student’s t-test. DAPI, 4′,6-diamidino-2-phenylindole; FACS, fluorescence-activated cell sorting; GDF11, growth differentiation factor 11; LGR5, leucine-rich repeat-containing G-protein-coupled receptor 5.
We next addressed the question whether suppression of GDF11 expression in human primary hepatic myofibroblasts affects the expansion of human progenitor cells. At first, we confirmed the activation phenotype of myofibroblasts by immunofluorescence staining of α-smooth muscle actin (α-SMA) and Ki67. The presence of α-SMA but absence of Ki67 staining at day 9 in culture confirmed their activation phenotype and primary nature, respectively (online supplementary figure 7a–c). Next, we inhibited the expression of GDF11 in human primary hepatic myofibroblasts by siRNAs and subsequently co-cultured them with human liver organoids (figure 4I–J). The expansion of human liver organoids was hampered, when they were co-cultured with human hepatic myofibroblasts that were transfected with GDF11 siRNAs (figure 4K). Furthermore, LGR5 and PROM1 mRNA levels as well as the number of LGR5+ cells within the organoids decreased in the presence of co-cultured hepatic myofibroblasts transfected with GDF11 siRNAs (figure 4L–M). HNF4A mRNA expression remained unchanged in the presence of GDF11 siRNAs. We confirmed these results by co-culturing human primary hepatic myofibroblasts and LGR5+ cells and examined organoids formation in the presence of GDF11 neutralising antibody. We observed significant reduction in the number of organoids and expression of LGR5 as well as PROM1 when co-culture was supplemented with GDF11 antibody (online supplementary figure 8a–d). Albumin secretion however remained unchanged, suggesting that GDF11 is unlikely to affect the HC differentiation (online supplementary figure 8e). Thus, taken together the loss of GDF11 in hepatic myofibroblasts abrogates expansion of progenitor cells in human liver organoids.
Supplemental material
Supplemental material
Because LGR5+ cells are present in fibrotic livers and their transplantation is able to reduce liver fibrosis, we asked if presence of GDF11 renders a fibrolysis activity to LGR5+ cells. To address this, we first co-cultured myofibroblasts with LGR5+ cells, which reduced the expression of fibrogenic genes of myofibroblasts in vitro, further confirming that LGR5+ cells reduce liver fibrosis in vivo (online supplementary figure 8f). Then, in a separate experiment, we pretreated LGR5+ cells with rGDF11 for 24 hours. We then co-cultured those GDF11-pretreated LGR5+ cells with human primary hepatic myofibroblasts and analysed the expression of fibrogenic genes such as ACTA2 and COL1A1. Our qPCR analyses revealed that GDF11-pretreated LGR5+ cells reduced the expression of fibrogenic genes (online supplementary figure 8g). Thus, GDF11 treatment not only enhances the number of LGR5+ cells but also renders them with a higher antifibrotic activity.
The antifibrotic function of hepatic GDF11 is abolished upon in vivo depletion of LGR5+ cells
We next ascertained whether amelioration of fibrosis on Gdf11 overexpression indeed requires enhanced numbers of LGR5+ cells. To address this, we ablated LGR5+ cells in Gdf11 overexpressing fibrotic mice using AAV encoding diphtheria toxin A (DTA) under the transcriptional control of Lgr5 promoter (figure 5A and G). Administration of AAV.LGR5.DTA led to more than 55% reduction in number of LGR5+ cells in livers of AAV.GDF11-injected mice (4.79% for CCl4 model in figure 5B and 4.22% for DDC model in figure 5H) compared with number of LGR5+ cells (11% for CCl4 model in figure 3A and 9.49% for DDC model in figure 3H) observed in mice that were injected with AAV.GDF11 only. On ablation of LGR5+ cells, mice injected with AAV.GDF11 exhibited similar level of fibrosis as respective controls in both CCl4-induced (figure 5C–F) and DDC-induced (figure 5I–L) liver fibrosis. Thus, our in vivo data obtained on ablation of LGR5+ cells indicate that Gdf11 overexpression attenuates liver fibrosis via expansion of LGR5+ cells. Notably, we observed that ablation of LGR5+ cells in fibrotic livers without AAV.GDF11 injection leads to increased fibrosis (figure 5C–F), indicating that LGR5+ cells may have a beneficial role in recovery from chronic injury in the wild-type condition.
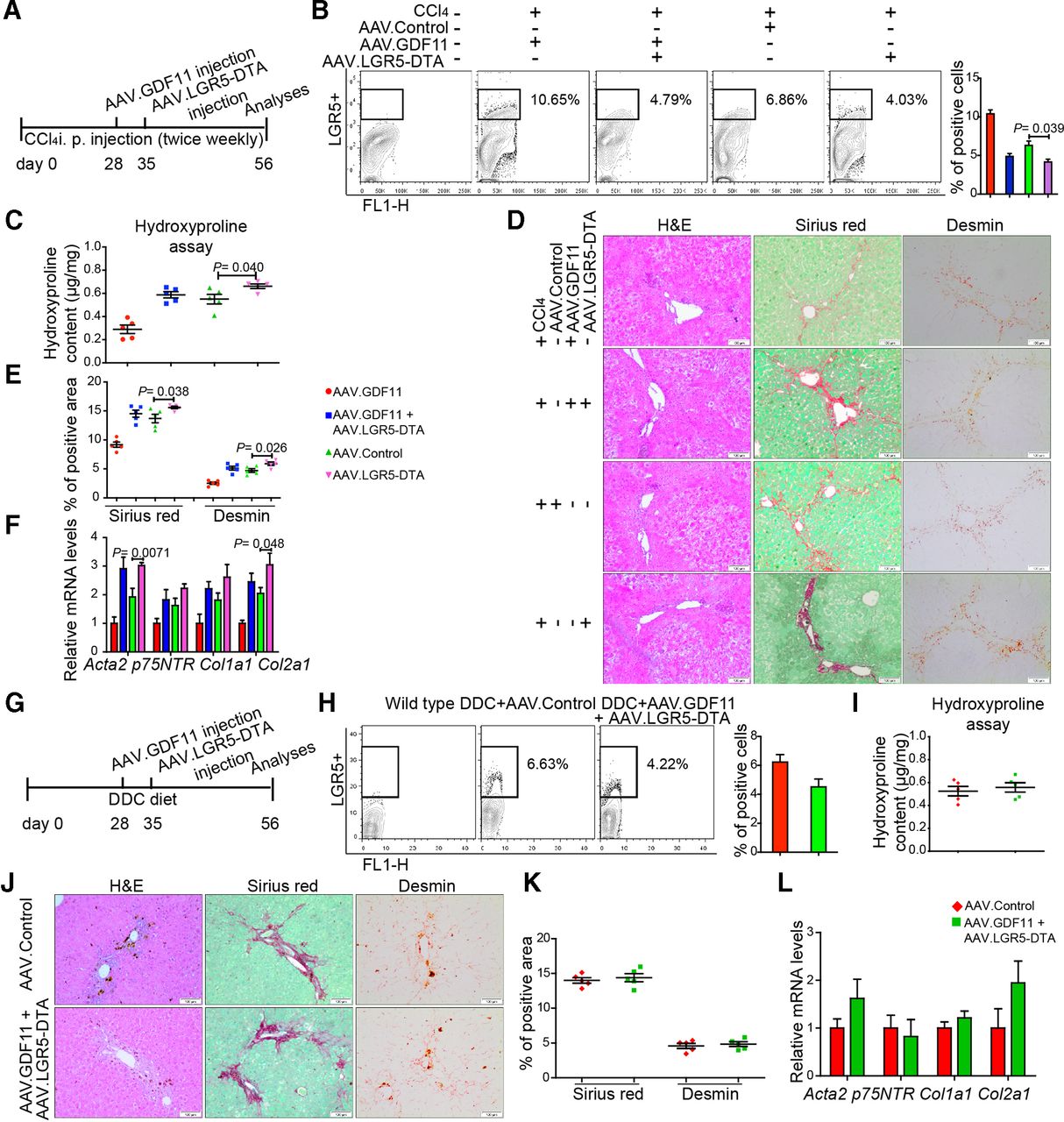
The depletion of LGR5+ LPCs diminishes GDF11-mediated attenuation of liver fibrosis. (A,G) Schematic overview of the experiments in CCl4 (B–F) and DDC (H–L) fibrosis mouse models (n=5 mice per group for both CCl4 and DDC model). (B,H) Representative FACS analysis of LGR5+ LPCs in AAV.GDF11 and AAV.LGR5.DTA or AAV control-injected mice in the CCl4 (B) and the DDC (H) fibrosis model. (C,I) Measurement of total collagen content by hydroxyproline assay. (D,J) Representative pictures of H&E, Sirius Red and desmin staining. Scale bars, 100 µm. (E,K) Quantifications of Sirius Red and desmin staining shown in panels D and J. For each mouse, 6 liver sections were stained in batches and pictures from 12 random fields per section were captured and quantified in a blinded manner using Image-J. (F,L) The qPCR analyses of fibrosis-related genes such as Acta2, p75Ntr, Col1a1 and Col2a1. All experiments shown in this figure were repeated twice. Data are mean±SEM; two-tailed Student’s t-test. AAV, adeno-associated virus; CCl4, carbon tetrachloride; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; DTA, diphtheria toxin A; FACS, fluorescence-activated cell sorting; GDF11, growth differentiation factor 11; LGR5, leucine-rich repeat-containing G-protein-coupled receptor 5; LPC, liver progenitor cell.
We then investigated whether GDF11 overexpression exerts any major profibrogenic effects in normal healthy liver. In fact, we did not detect histological abnormalities, signs of fibrosis, induction of LGR5+ cells or hepatocellular carcinoma in mice 14 weeks after AAV.GDF11 injection (online supplementary figure 9a–e). Thus, the data presented in figure 3 and online supplementary figure 9 indicate that GDF11 is able to promote the number of existing LGR5+ cells but unable to cause induction of LGR5+ cells.
Supplemental material
Modulation of GDF11 in hepatic myofibroblasts
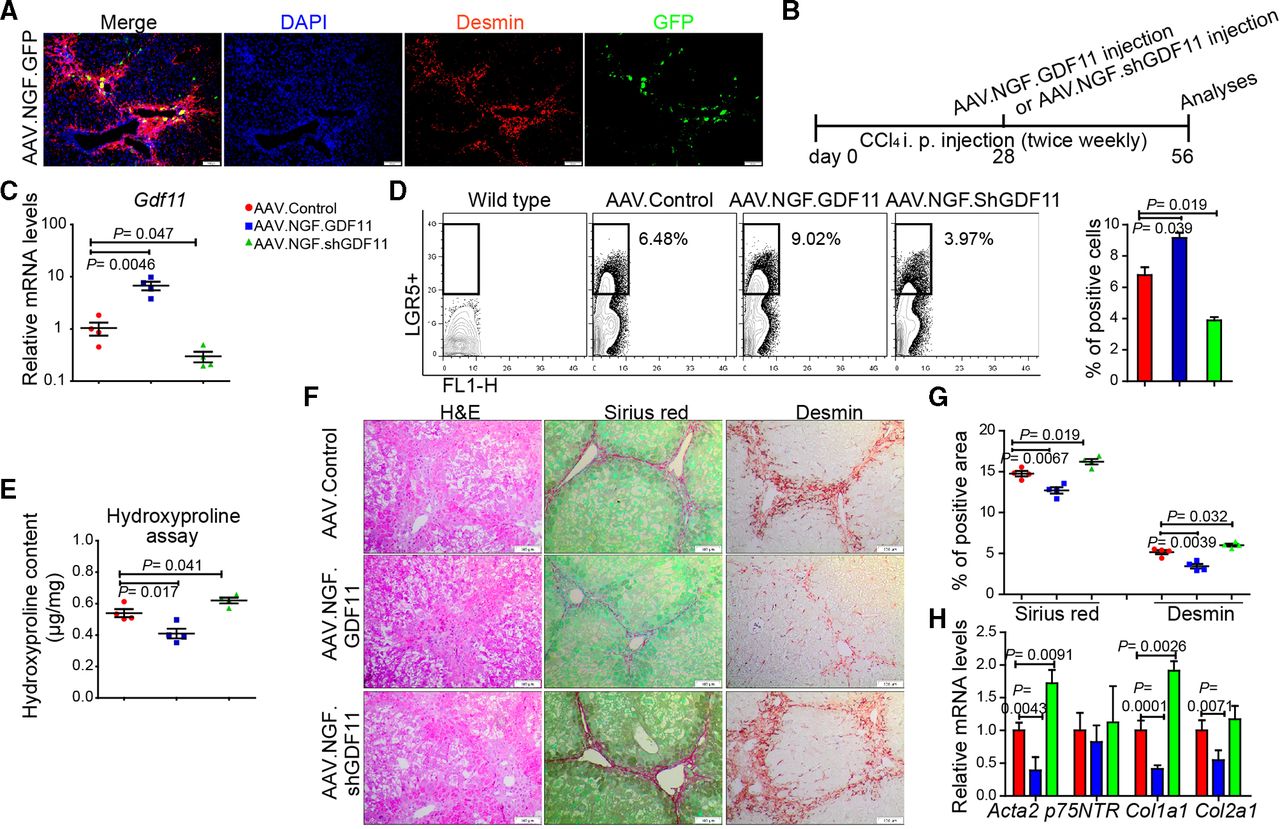
Since hepatic myofibroblasts possess highest Gdf11 expression among the examined liver cell types (figure 1hH) and LGR5+ cells (online supplementary figure 5g), we asked whether modulation of Gdf11 expression in myofibroblasts affects fibrogenesis. The loss or gain of GDF11 in cultured primary human (online supplementary figure 10a–d) or mouse hepatic myofibroblasts (online supplementary figure 10e–h) neither affected viability nor fibrosis markers such as Acta2 and Col1a1. These results indicate that GDF11 modulation in myofibroblasts itself had little, if any, role in fibrogenesis. We then sought to investigate if in vivo modulation of Gdf11 in mouse hepatic myofibroblasts affects liver fibrosis. To address this, we constructed an AAV variant that preferentially targets hepatic myofibroblasts. The capsid of this AAV variant is designed to express a peptide that binds to p75NTR, a receptor that is highly expressed on hepatic myofibroblasts. We confirmed preferential targeting of myofibroblasts in fibrotic mouse liver on injecting mice with AAV.NGF.GFP. We observed GFP expression in desmin-positive areas in the fibrotic livers, suggesting targeting of hepatic myofibroblasts (figure 6A).
Supplemental material
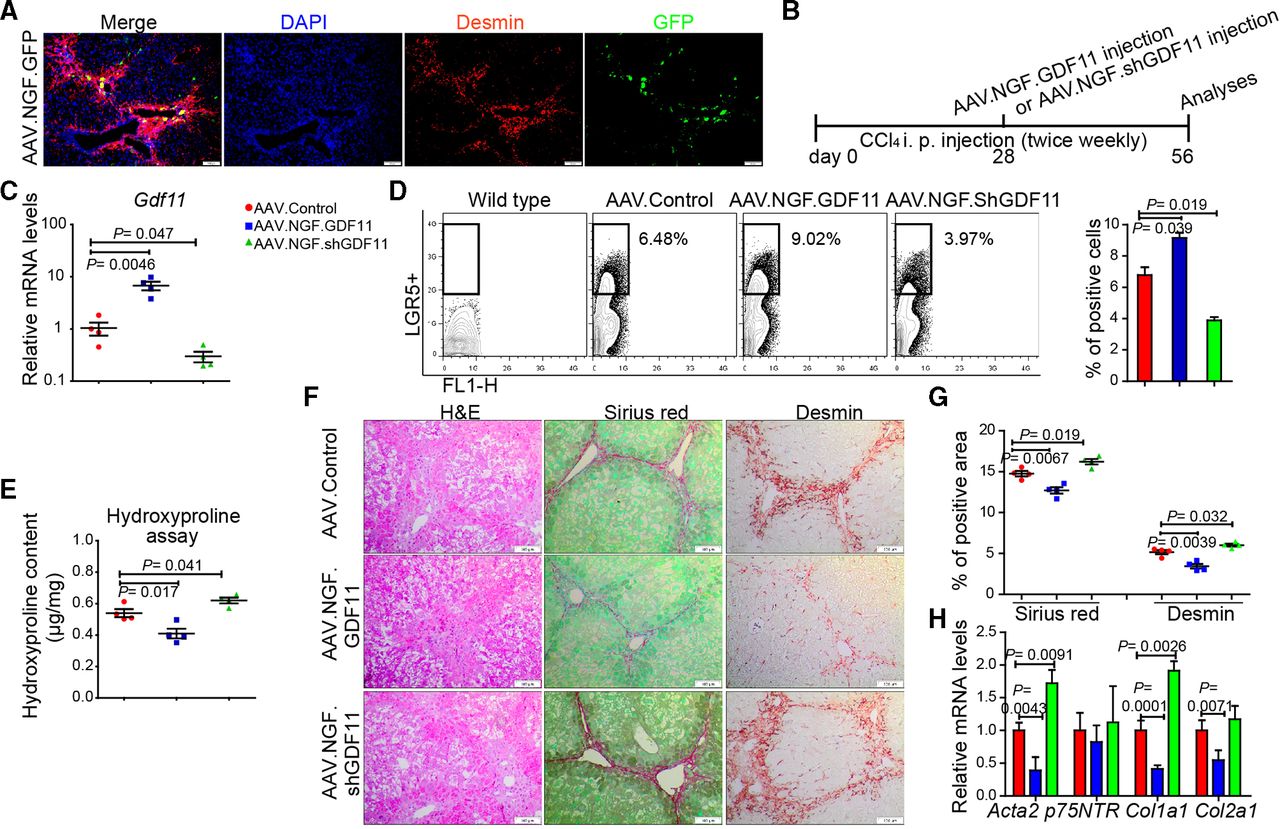
In vivo modulation of Gdf11 in hepatic myofibroblasts. (A) Preferential targeting of hepatic myofibroblasts is shown by co-staining of GFP and desmin in livers of recipients of AAV.NGF.GFP virus. (B) Schematic overview of the experiments in CCl4-induced fibrosis mouse models (n=4 mice per group). (C) The qPCR analyses of Gdf11 expression in murine livers after AAV.NGF.GDF11 or AAV.NGF.shGDF11 injection. (D) Representative FACS analysis of LGR5+ LPCs in AAV.NGF.GDF11 or AAV.NGF.shGDF11-injected mice in the CCl4 fibrosis model. (E) Measurement of total collagen content by hydroxyproline assay. (F) Representative pictures of H&E, Sirius Red and desmin staining. Scale bars, 100 µm. (G) Quantification of Sirius Red and desmin staining is shown in panel F. For each mouse, 6 liver sections were stained in batches and pictures from 12 random fields per section were captured and quantified in a blinded manner using Image-J. (H) The qPCR analyses of fibrosis-related genes such as Acta2, p75NTR, Col1a1 and Col2a1. Data are mean±SEM; two-tailed Student’s t-test. AAV, adeno-associated virus; CCl4, carbon tetrachloride; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; FACS, fluorescence-activated cell sorting; GDF11, growth differentiation factor 11;LGR5, leucine-rich repeat-containing G-protein-coupled receptor 5; LPC, liver progenitor cell.
To overexpress or knockdown GDF11 in hepatic myofibroblasts, we injected fibrotic mice with AAV.NGF.GDF11 encoding GDF11 under the transcriptional control of CMV promoter or AAV.NGF.ShGDF11 encoding shRNA against GDF11 (figure 6B). We first confirmed overexpression of GDF11 or knockdown of GDF11 on injection of AAV.NGF.GDF11 or AAV.NGF.ShGDF11 (figure 6C). We then analysed whether GDF11 modulation in hepatic myofibroblasts influences the expansion of LGR5-positive cells. Our FACS analyses showed that the gain of GDF11 in myofibroblasts enhances number of LGR5-positive cells (figure 6D). On the contrary, knockdown of GDF11 in myofibroblasts decreased the number of LGR5-positive cells (figure 6D). Furthermore, hydroxyproline assay, immunohistochemistry (IHC) for Sirius Red, desmin, H&E and gene expression analysis for fibrogenic genes revealed that overexpression of GDF11 in hepatic myofibroblasts suppresses fibrosis, whereas knockdown of GDF11 in hepatic myofibroblasts leads to increased fibrosis (figure 6E–H). Thus, modulation of GDF11 in hepatic myofibroblasts supports our findings that overexpression of GDF11 reduces liver fibrosis, as also seen on GDF11 overexpression in HCs (figure 2). This is most likely due to the fact that GDF11 is a secreted protein, which we found to not affect myofibroblasts and rather act on LGR5-positive cells.
Similar to GDF11, we found that LGR5 expression is also upregulated in patients with fibrosis (online supplementary figure 11a–b). We then investigated whether GDF11 expression is correlated with different grades of fibrosis and with LGR5+ expression. We therefore analysed their expression in RNA isolated from patients with fibrosis grades of F0, F2 and F4. We found that GDF11 expression correlates with increased fibrosis (online supplementary figure 11c). Importantly, LGR5 expression, similar to GDF11, was found to be higher in F4 than in F2 grade fibrosis (online supplementary figure 11d). These results along with our gain of function in vivo experiments suggest that elevation of GDF11 is a protective response against liver fibrosis; however, the mild increase in GDF11 is unable to exert relevant protection against injury unless overexpressed by AAV.
Supplemental material
The overexpression of hepatic GDF11 inhibits non-alcoholic steatohepatitis in mice
Finally, we examined the effects of GDF11 overexpression in non-alcoholic steatohepatitis (NASH),31 32 which is prevalent among 24% global population, and often results in fibrosis. To examine this, we administered AAV.GDF11 in male BALB/c mice fed with high-fat diet (HFD) for 14 weeks (figure 7A). We observed significant reduction in Non-alcoholic fatty liver disease Activity Score (NAS),26 lipid content and fasting glucose as well as insulin levels and the expression of genes involved in gluconeogenesis such as Pck1 and Gp6c in mice injected with AAV.GDF11 (figure 7B–F). Of note, mice injected with AAV.GDF11 had mild but significant reduction in their body weight compared with respective controls (figure 7G). Taken together, NAS, lipid content, expression of Pck1 and Gp6 and body weight analyses indicate that GDF11 overexpression in mice inhibits NASH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GDF11 inhibits NASH progression. (A) Schematic overview of the experiments (n=6 mice per group). (B) Representative images of HE and Oil red O-stained liver sections from AAV.control and AAV.GDF11-injected mice fed with high-fat diet (HFD) for 14 weeks. Scale bars, 100 µm. (C) The NAS score and Oil red O-positive area were assessed. For each mouse, 6 liver sections were stained in batches and pictures from 12 random fields per section were captured and quantified in a blinded manner. (D) Fasting glucose concentration in blood. (E) Fasting insulin concentration in blood. (F) The qPCR analysis for gluconeogenic genes Pck1 and G6pc. (G) The body weight of AAV.control and AAV.GDF11-injected mice. Data shown in this figure were repeated twice. AAV, adeno-associated virus; GDF11, growth differentiation factor 11; NAS, Non-alcoholic fatty liver disease Activity Score; NASH, non-alcoholic steatohepatitis.
We then measured GDF11 expression in patients with non-alcoholic fatty liver disease (NAFLD). We found that GDF11 is upregulated in NAFLD patients (online supplementary figure 12a). In contrast, mice fed with HFD but without AAV.GDF11 injection had no significant increase in GDF11 protein and mRNA compared with normal control mice (online supplementary figure 12b–c). One possible explanation for the absence of elevated levels of GDF11 could be that GDF11 increases in response to fibrosis, whereas in the case of HFD mice, we detected NASH but any significant level of fibrosis was absent (online supplementary figure 12d).
Supplemental material
We also analysed LGR5+ cell numbers in mice fed with HFD and found that LGR5-positive cell numbers are elevated without AAV.GDF11 injection compared with normal mice (online supplementary figure 12e). These results suggest that LGR5+ cells emerged in HFD mice even in the absence of GDF11, indicating that other mechanisms may cause induction of LGR5+ cells in the absence of fibrosis, myofibroblasts and GDF11. Importantly, AAV.GDF11 injection in HFD mice increased LGR5+ cells up to 11%, confirming our findings that GDF11 promotes LGR5+ cells.
Discussion
Collectively, we show for the first time that GDF11 is elevated in fibrotic human and mouse liver and uncover a novel and potential role for GDF11 in tissue remodelling of chronic liver disease. Despite being a member of TGF-β superfamily, gain-of-function experiments show that GDF11 attenuates liver fibrosis by promoting the expansion of LGR5+ cells. This was confirmed by the expansion of LGR5+ progenitors in mouse and human liver organoids following treatment with recombinant GDF11 protein. Transplantation of LPCs in chronically injured liver mimicked the antifibrotic effect of GDF11. Our findings further revealed that GDF11 renders antifibrotic properties to LGR5+ cells.
Regeneration of the parenchymal liver cells after partial hepatectomy is maintained by remaining mature HCs and cholangiocytes and does not require progenitor cells. However, in case of injuries wherein inflammation is a key component, the emergence of LGR5+ cells14–16 was also observed in our experiments. Factors that regulate progenitor cells in chronic liver diseases have remained largely unknown. Recently, WNT,33 Notch,34 YAP35 and hedgehog36 signalling have been reported to regulate LPCs. The presence of LGR5+ cells and the concomitant emergence and expansion of myofibroblasts in fibrosis are often observed as a result of chronic inflammation and repeated toxic injury. GDF11, which is highly expressed in hepatic myofibroblasts, may thus serve as a growth factor to maintain proliferation of the LGR5+ progenitor pool. Furthermore, loss of antifibrotic effects of GDF11 when LGR5+ cells were ablated in vivo indicated that GDF11 requires LGR5+ cells for the manifestation of its antifibrotic activity.
A central question that arises from our study is how GDF11 induces expansion of LPCs and attenuates liver fibrosis. One possibility is that GDF11 facilitates the differentiation of LGR5+ cells into HCs because enhanced number of functional HCs mitigates fibrosis. Our in vitro data from human organoid argues otherwise: lack of significant elevation in albumin secretion on rGDF11 treatment indicates that HC differentiation from LGR5+ cells has only a limited role, if any. However, we cannot exclude the possibility of HC differentiation from LGR5+ cells or other facultative stem cells in liver injuries wherein HC proliferation is completely blocked, as convincingly shown previously.37 Second possibility is that GDF11-induced LGR5+ cells facilitate reversal of activated HSC state to a relatively quiescent HSC phenotype. Our in vivo date supports this notion as transplantation of LGR5+ cells reduced expression of Acta2, a marker of activated HSC. Whether such a feedback loop mechanism exists between LGR5+ cells and activated HSC remains to be determined.
The observation that LPCs do not contribute to hepatocellular carcinoma formation,38 39 thus, the GDF11-mediated expansion of LGR5+ cells in chronic injury would be safe and rather expected to suppress tumour development. Furthermore, in normal liver tissue, the GDF11 target cell population is absent and long-term overexpression did not induce LGR5+ cells. Overall, we did not observe any pathological effects of hepatic GDF11 overexpression in normal healthy liver or other organs. Based on the similar molecular network that governs fibrosis in organs such as liver and lungs, we speculate that GDF11 administration should be able to modulate fibrosis in other organs as well.
Taken together, we have discovered a protective function of GDF11 in liver fibrosis. Our findings provide a basic framework for further investigation of the therapeutic benefits of GDF11 in the treatment of liver fibrosis.
Acknowledgments
Authors thank Professor Hans R. Schöler, MPI Münster for providing helpful comments on this manuscript.
Footnotes
Contributors ADS conceived the idea, designed the study and provided the conceptual framework for the study. ZD, GS and QY performed all experiments and analysed the data. ADS wrote the manuscript with the help of ZD, MO, AB and AK. TY and SM helped with animal experiments and Western blot, respectively. A-CW and AK performed in situ hybridisation. GS, JZ, XJ and XS helped with collection and analysis of human fibrotic livers (China). HBantel and EJ provided human fibrotic liver samples (Germany). AS and AV generated human liver organoids. HBüning and MB cloned and provided AAV capsid variant for hepatic myofibroblasts. MM, TC and MO provided conceptual evaluation of the project. All authors commented on the manuscript and declared no conflicts of interest.
Funding The work was supported by the grants from Deutsche Forschungsgemeinschaft (DFG SH640/1-2, DFG EXC 62/2 and SFB-738) and Plus 3 Program of Boehringer Ingelheim. AB is supported from Deutsche Krebshilfe grant (111147). ZD and TY received stipends from China Scholarship Council.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Allexperiments were performed according to the regulations of the Institutional Animal Care and Use Committee of the Hannover Medical School, Germany (Protocol number: TVA 17/2658) and Zhongshan Hospital of Fudan University, Shanghai, China.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.