Article Text

Abstract
The pathophysiological background of decompensated cirrhosis is characterised by a systemic proinflammatory and pro-oxidant milieu that plays a major role in the development of multiorgan dysfunction. Such abnormality is mainly due to the systemic spread of bacteria and/or bacterial products from the gut and danger-associated molecular patterns from the diseased liver triggering the release of proinflammatory mediators by activating immune cells. The exacerbation of these processes underlies the development of acute-on-chronic liver failure. A further mechanism promoting multiorgan dysfunction and failure likely consists with a mitochondrial oxidative phosphorylation dysfunction responsible for systemic cellular energy crisis. The systemic proinflammatory and pro-oxidant state of patients with decompensated cirrhosis is also responsible for structural and functional changes in the albumin molecule, which spoil its pleiotropic non-oncotic properties such as antioxidant, scavenging, immune-modulating and endothelium protective functions. The knowledge of these abnormalities provides novel targets for mechanistic treatments. In this respect, the oncotic and non-oncotic properties of albumin make it a potential multitarget agent. This would expand the well-established indications to the use of albumin in decompensated cirrhosis, which mainly aim at improving effective volaemia or preventing its deterioration. Evidence has been recently provided that long-term albumin administration to patients with cirrhosis and ascites improves survival, prevents complications, eases the management of ascites and reduces hospitalisations. However, variant results indicate that further investigations are needed, aiming at confirming the beneficial effects of albumin, clarifying its optimal dosage and administration schedule and identify patients who would benefit most from long-term albumin administration.
- liver cirrhosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Advances in the pathophysiology of decompensated cirrhosis
Many complications of cirrhosis, such as renal dysfunction and ascites formation, have long been attributed to effective hypovolaemia due to peripheral arterial vasodilation.
It is now clear that decompensated cirrhosis is characterised by a systemic proinflammatory and pro-oxidant milieu playing a major role in the pathogenesis of multiorgan dysfunction.
The systemic spread of bacteria and/or bacterial products secondary to abnormal translocation from the gut and danger-associated molecular patterns from the diseased liver trigger the release of proinflammatory mediators by activating immune cells.
The abrupt exacerbation of these abnormalities leads to the development of acute-on-chronic liver failure (ACLF), characterised by hepatic and extrahepatic organs/systems failure and high short-term mortality but with potential for reversibility.
Systemic cellular energy crisis secondary to mitochondrial dysfunction likely contributes to multiorgan failure in this syndrome.
The pathophysiological mechanisms underlying the clinical manifestations of decompensated cirrhosis and ACLF represent novel targets for mechanistic treatments.
Key messages
Potential role of albumin as disease-modifying agent in decompensated cirrhosis
The well-established indications to the acute or short-term use of albumin in decompensated cirrhosis mainly aim at improving effective volaemia or preventing its deterioration.
Serum albumin in decompensated cirrhosis undergoes structural and functional abnormalities endangering its non-oncotic properties such as antioxidant, scavenging, immune-modulating and endothelium protective functions. As a result, due to both quantitative (hypoalbuminaemia) and qualitative changes, the amount of circulating ‘effective’ albumin can be dramatically reduced.
The pleiotropic non-oncotic properties of albumin make it a potential multitarget agent for a mechanistic treatment of decompensated cirrhosis.
It has recently provided evidence that long-term albumin administration to patients with decompensated cirrhosis improves survival, prevents complications, eases the management of ascites and reduces hospitalisations, thus being cost-effective.
However, variant results indicate that further investigations are needed, aiming at confirming the beneficial effects of albumin, clarifying its optimal dosage and administration schedule and identify patients who would benefit most from long-term albumin administration.
Introduction
The concept of compensated and decompensated cirrhosis
Cirrhosis is characterised by a prolonged asymptomatic period of persistent liver inflammation, extracellular matrix remodelling and relentless deposition of collagen in the liver tissue. This fibrosis together with focal hyperplasia and proliferation of hepatocytes eventually disrupts the hepatic architecture leading to characteristic regenerative nodules.1 During this period, the disease remains undiagnosed unless patients seek medical attention for another reason or are screened for liver disease because of risk factors.2 Besides the development of hepatocellular carcinoma, complications develop when the disruption of the liver architecture is severe enough to markedly increase portal pressure.3 The most common clinical presentation is ascites/oedema or, less frequently, bleeding from gastro-oesophageal varices. Late complications include jaundice, coagulopathy, hepatic encephalopathy, bacterial infections and acute kidney injury, particularly hepatorenal syndrome (HRS).4 5 Complications, after their first appearance, recur with increasing frequency and most patients die over a median period of about 2 years unless liver transplantation is performed.6 The disease stage preceding the occurrence of complications is known as compensated cirrhosis, as opposed to the decompensated stage during which patients develop complications. In addition to the array of complications related to portal hypertension, complexity of cirrhosis is amplified by the development of an acute-on-chronic liver failure (ACLF). ACLF is a clinical syndrome characterised by rapid onset of failure of the liver and/or extrahepatic organs, including kidneys, systemic circulation or brain, within the context of an acute decompensation (AD) of the disease as defined by the development of ascites, encephalopathy, gastrointestinal bleeding and/or bacterial infection.7 ACLF often occurs in patients with prior history of decompensated cirrhosis but also develops without of previous decompensation. It is associated with high short-term (1 month) mortality rate, being the most frequent cause of death of cirrhosis.8
The peripheral arterial vasodilation concept: the initial rationale for the use of albumin in cirrhosis
A major pathogenic event in the development of complications of cirrhosis is an impairment of circulatory function related to portal hypertension.9 The dysfunction in the arterial circulation consists of a reduction in systemic vascular resistance due to primary vasodilation of the splanchnic arterial circulation (figure 1). This vasodilation is most likely due to an enhanced production/activity of vasodilator factors, particularly nitric oxide, endogenous cannabinoids and/or carbon monoxide, likely related to bacterial translocation mediated through portal hypertension, as well as dysfunctional contractile pathways.10–12 In decompensated cirrhosis, arterial pressure must be maintained by an increased activity of the systemic vasoconstrictor factors, namely the renin-aldosterone and the sympathetic nervous systems, and the non-osmotic secretion of antidiuretic hormone.9 At this stage, cardiac output may also be impaired owing to the impairment in cardiac function related to cirrhotic cardiomyopathy.13

Peripheral arterial vasodilation hypothesis. In patients with cirrhosis and portal hypertension, a peripheral arterial vasodilation, mainly occurring in the splanchnic circulatory area, endangers effective volaemia.9 Qualitative and quantitative changes in the intestinal microbiota and impairment in intestinal mucosal barrier, translocation of bacteria or bacterial products, and local inflammation and release of inflammatory vasoactive mediators is likely the initial sequence of events leading to splanchnic arterial vasodilation in cirrhosis. With the progression of the disease, bacterial translocation increases, inflammation become systemic and effective hypovolaemia worsens. Activation of the hypothalamic–pituitary–adrenal axis, widespread release of norepinephrine (NE) in the sympathetic nervous system terminals, increased adrenal secretion of epinephrine (E), activation of the renin–aldosterone system and increased release of antidiuretic hormone (ADH) are the main homeostatic responses to restore arterial pressure, leading to renal fluid retention, which accumulates as ascites, dilutional hyponatraemia and hepatorenal syndrome (HRS) as the most relevant consequences. The well-established indications for the use of albumin in patients with decompensated cirrhosis rely on this pathophysiological background and mainly aim at restoring effective volaemia. No, CO, H2S, PGs and BDK are the abbreviations of nitric oxide, carbon oxide, hydrogen sulfide, prostaglandins and bradykinin, respectively. SNS, sympathetic nervous system.
Understanding circulatory abnormalities in cirrhosis led to development of several therapeutic strategies, which are currently used in clinical practice, to improve/prevent kidney complications, including blockade of sodium retaining factors and/or treatment with vasoconstrictor drugs.14–16 Some of these strategies include the intravenous administration of albumin with the objective of improving arterial underfilling. Currently, accepted indications of albumin include prevention of circulatory dysfunction following large-volume paracentesis, prevention of HRS in patients with spontaneous bacterial peritonitis (SBP), and management of HRS in association with vasoconstrictor drugs.17
On this background, the current article reviews the new concepts on pathogenesis of cirrhosis progression, in particular the role of systemic inflammation in the development of complications of cirrhosis and ACLF, and the potential role of albumin to prevent disease progression and reduce the inflammatory status.
Pathophysiology of AD and ACLF
The clinical course of decompensated cirrhosis is characterised by repeated episodes of AD, which may (30% of cases) or may not be associated with organ failure(s) (ACLF or ‘mere’ AD, respectively).18
Systemic inflammation
Systemic inflammation progressively increases with the progression of cirrhosis, arising in the compensated stage and becoming more marked in stable decompensated cirrhosis.19–21 Systemic inflammation further increased when an episode of AD develops and culminates with ACLF. In fact, patients with AD show more evident features of systemic inflammation (plasma levels of C reactive protein (CRP); soluble inflammatory signals (proinflammatory and anti-inflammatory cytokines and chemokines)) and oxidative stress (irreversibly oxidised form of albumin and human non-mercaptalbumin 2 (HNA2)) compared with healthy subjects and stable cirrhotics (table 1).18 22 23 In turn, patients with ACLF show much higher blood levels of CRP, inflammatory molecules and HNA2 than those with AD.18 23 Moreover, while patients with AD have a peripheral blood white cell count within the normal range, those with ACLF exhibited leucocytosis. Together, these findings indicate that ACLF is characterised by full-blown systemic inflammation.18 Systemic inflammation in AD of cirrhosis and ACLF may overlap systemic changes occurring in sepsis; however, complications and outcome of patients with cirrhosis are very distinct from that of patients with sepsis without cirrhosis due to the presence of portal hypertension and liver failure, which are a hallmark of the liver disease.24
Plasma concentrations of cytokines, C reactive protein and irreversible oxidised albumin in healthy subject and patients with cirrhosis with and without ACLF (data gathered from Clària et al 22)
Little is known about the inducer(s) of systemic inflammation in AD.25 Bacterial infections are involved in 33% of cases of ACLF,18 even though the true prevalence is likely underestimated. Recent findings from the Predicting Acute-on-Chronic Liver Failure in Cirrhosis (PREDICT) study indicate that ACLF occurred in two distinct contexts: in one, ACLF was associated with bacterial infection, even if the reason for hospitalisation was severe alcoholic hepatitis or gastrointestinal haemorrhage, while in the second, ACLF developed without any clear trigger such as infection, trauma, burns, pancreatitis or hypovolaemic shock (Trebicka J. Predicting acute-on-chronic liver failure in cirrhosis (PREDICT) study. 2019). In sepsis-related ACLF, the systemic inflammation already present in decompensated cirrhosis is likely accentuated by infective, live bacteria, while acute bursts of translocation of bacterial products (but not viable bacteria) from the gut to the bloodstream, a phenomenon related to systemic inflammation, are likely involved in ACLF without identifiable triggers.26 27 This interpretation was recently supported from a 3 months longitudinal investigation showing transient peaks of serum interleukin (IL)-6 in patients with decompensated cirrhosis.28 Another factor driving the onset of accentuated systemic inflammation is the severity and type of hepatocyte cell death induced by both infection and liver-directed precipitating events such as alcoholic hepatitis, superimposed viral infections such as hepatitis B or E virus infection or drugs. The main form of cell death in AD seems to be apoptosis.29 However, more immunogenic forms such as necroptosis-(regulated necrosis induced by stimuli including death receptors, interferons, toll-like receptors (TLRs), intracellular RNA and DNA sensors and mediated by receptor interacting protein kinase-3 and its substrate mixed lineage kinase like) and pyroptosis (programmed, non-apoptotic, lytic form of cell death occurring as a result of cleavage of the effector protein Gasdermin-D) are observed in ACLF.30–32 This releases proinflammatory substances collectively referred to as damage-associated molecular patterns (DAMPs). Therefore, sepsis-related and sepsis-unrelated ACLF would share mechanistic commonalities, that is, the immune system response to pathogen-associated molecular patterns (PAMPs), either released by infecting bacteria or spread from the intestinal lumen, or DAMPs (figure 2).8 However, it is still unclear why, in a given context (eg, SBP), ACLF exhibits a stronger inflammatory response than AD. The intensity of the inflammatory response to PAMPs and DAMPs may depend on genetic host factors; single-nucleotide variants might modulate the amount of inflammatory molecules produced by innate immune cells or changes in the expression of pattern recognition receptors (PRRs) such as TLR.7 33 The quantity of exogenous or endogenous ‘invaders’ may also play a role; the higher bacterial burden or concentration of PAMPs, DAMPs or PRRs, the more intense the inflammatory response.

Working hypothesis for the mechanisms of organ failures in ACLF. Pathogen-associated molecular patterns (PAMPS; eg, lipopolysaccharide (LPS)) are specifically recognised by pattern-recognition receptors (PRRs; eg, Toll-like receptor (TLR) 4 for LPS) expressed in innate immune cells and epithelial cells.4 7This process is called structural feature recognition. Damage-associated molecular patterns (DAMPs; eg, high mobility group box 1 (HMGB1), S100A8 (MRP8, calgranulin A) and S100A9 (MRP14, calgranulin B)), are molecules resulting from stressed cells that, once released, follow a similar path. PAMPs are either released by an infecting alive bacterium or ‘translocated’ from the gut lumen to blood, while DAMPs are released from tissues where necroptosis and pyroptosis take place (liver and extrahepatic organs (not shown)). Whichever the origin of these molecular patterns, their recognition by PRRs results in the production of a broad variety of inflammatory molecules (cytokines, chemokines and lipids), vasodilators and of reactive oxygen species, particularly in phagocytes. Intense systemic inflammation may cause collateral tissue damage (a process called immunopathology) and subsequently organ failures (hypothesis 1). Extrahepatic tissue damage can result in the release of DAMPs, which may perpetuate or accentuate PAMP-initiated and DAMP-initiated systemic inflammatory response (not shown).4 7 Systemic inflammation is energetically expensive, and the immune tissue can be prioritised for nutrient allocation at the expense of vital non-immune tissues. These may adapt to nutrient scarcity by reducing mitochondrial oxidative phosphorylation (OxPhos) and therefore ATP production, which would contribute to the development of organ failures (hypothesis 2). Of note, the two hypotheses are not mutually exclusive. ACLF, acute-on-chronic liver failure.
Immune paralysis and increased risk of bacterial infections
As reported above, AD and particularly ACLF are frequently precipitated by bacterial infections. Furthermore, nosocomial infections are the most frequent and severe complication of ACLF unrelated to bacterial infections at diagnosis, favouring progression to the most severe grade.34 The reasons of this high risk for bacterial infections is largely unknown.
Although peripheral blood leucocytosis is a hallmark of ACLF and a component of prognostic scores for ACLF and AD,35 36 its cell subsets are largely undefined and would merit investigation. Some studies showed that circulating monocytes in ACLF exhibit features of ‘immune paralysis’ in response to PAMPs. Namely, the frequency of CD14+ monocytes expressing the tyrosine-protein kinase Mer (hereafter called Mer) is increased.37 This protein, encoded by MERTK and belonging to TAM receptors family, inhibits lipopolysaccharide (LPS)-associated, TLR4-mediated, induction of inflammatory signals in immune cells. Consistently, Mer-expressing monocytes from ACLF patients had a depressed inflammatory response to LPS reverted by Mer pharmacological inhibition.37 In another study, the frequency of mononuclear CD14+ CD15-HLA-DR-myeloid-derived suppressor cells was increased in ACLF (representing half of CD14+ cells) and exhibited immunosuppressive properties. Indeed, T cell proliferation, tumour necrosis factor-alpha (TNF-α) and IL-6 production in response to TLR stimulation and uptake of Escherichia coli were reduced.38 A third study showed that monocytes from patients with ACLF had elevated frequencies of IL-10-producing cells, reduced HLA-DR expression and impaired phagocytic and oxidative burst capacity.39 Transcriptomic data revealed a profile of alternatively polarised, tolerant monocytes. Interestingly, the pharmacological inhibition of glutamine synthesis favouring glutaminolysis improved the monocytes phagocytic and inflammatory.39 Similarly, neutrophils from patients with ACLF present severe defects of phagocytosis and ability to kill ingested bacteria despite marked activation. Due to the importance of neutrophils in bacterial clearance, it is not surprising that the severity of this dysfunction define the risk of infection and mortality.40 Immune-suppressing signals to monocytes may also be involved; for example, decreased serum albumin in patients with AD (including ACLF) binds lower amounts of prostaglandin E2 (PGE2). The resulting increased bioavailability of PGE2 dampens the macrophage response to LPS.41 Innate immune cells (monocytes and neutrophils) play a major role in the host resistance against infection; in response to bacterial cues, monocytes produce a variety of molecules with to reduce the bacterial burden.42 Together, these studies show that ACLF is associated with increased frequency of certain subsets of neutrophils and immune-suppressed monocytes, which may decrease host resistance capacity and increase susceptibility to bacterial infection. Notably, immune paralysis features are observed in less severely decompensated cirrhosis. Endotoxinaemia and metabolic disturbances (eg, hyperammonaemia and hyponatraemia) may contribute to immune paralysis and be reversed by albumin administration.43–45
Pathophysiology of end-organ alterations in AD and ACLF: immunopathology and cellular energetic crisis
The mechanisms underlying organ dysfunction in AD and organ failure(s) in ACLF are poorly understood. There is a close correlation between the intensity of systemic inflammation and end-organ dysfunction. Patients with AD and mild systemic inflammation often develop kidney dysfunction.25 In ACLF, the intensity of systemic inflammation is closely associated with the number of organ failures (figure 2).18 In the general population, cytokines released in response to bacterial cues in patients with sepsis and severe sepsis activate innate immune cells that further release cytokines, proteases and reactive oxygen species within vital organs. All these effects cause collateral tissue damage, a process called immunopathology.46 Similarly, systemic inflammation in AD and ACLF may cause tissue damage via immunopathology resulting in organ dysfunction or failure, respectively (hypothesis 1, figure 2).7 8 47 However, to date, no immunopathological studies performed in patients with AD or ACLF providing robust support to this hypothesis are available. Still, some studies showed that liver-derived systemic inflammatory markers are associated with the development of organ failures and especially cardiac dysfunction.48 49
In patients or animals with sepsis but without liver disease, systemic inflammation is energetically expensive because of the production of inflammatory molecules, respiratory burst, acute-phase response, cellular migration and proliferation, requiring reallocation of nutrients to fuel the inflammatory response.42 50 Because systemic inflammation induces sickness behaviour of anorexia and asthaenia, which decrease food intake, mobilisation of stored fuels is critical for mounting the inflammatory response.42 50 This elicits proteolysis (skeletal muscles), lipolysis (adipose tissue) and glycogenolysis (liver), releasing amino acids, fatty acids and glucose to be used as fuels. This metabolic demand can be so high to exceed resources, so that nutrients are prioritised to immune tissues. Thus, non-immune tissues adapt by decreasing their energetic demand and reducing mitochondrial oxidative phosphorylation (OxPhos).50 Results of high-throughput metabolomics and lipidomics performed in the sera from a large cohort of patients with AD or ACLF showed that ACLF was distinct from AD by a blood signature witnessing intense proteolysis, and lipolysis, and marked inhibition of mitochondrial OxPhos.51 Although these findings were similar to those reported in sepsis in the general population,50 it is relevant that the ACLF-associated blood metabolite signature was unrelated with the presence or absence of detectable bacterial infection, indicating that such a signature can be seen in both sepsis-related and ‘spontaneous’ ACLF. Moreover, in ACLF with single organ failure, the metabolite signature was detected whichever the failing organ (liver, brain and kidney), indicating that cellular metabolism abnormalities were very similar irrespective of the type of organ affected. Because the ACLF-associated blood metabolite signature is consistent with the existence of mitochondrial inhibition in various organs, this mechanism may play a crucial role in the development of organ failures (hypothesis 2, figure 2). However, these results require a cautious interpretation as, to date, they were reported in a single study and were based on the assumption that plasma metabolic alterations reflected what happened in tissues. As occurs with immunopathology, why a generalised metabolic dysregulation would affect different organs in different patients is unknown. It is noteworthy that hypothesis 1 (role of immunopathology) and hypothesis 2 (role of competition for metabolic resources) are similarly plausible and not mutually exclusive.
The concept of albumin as a drug
Biosynthesis and metabolism
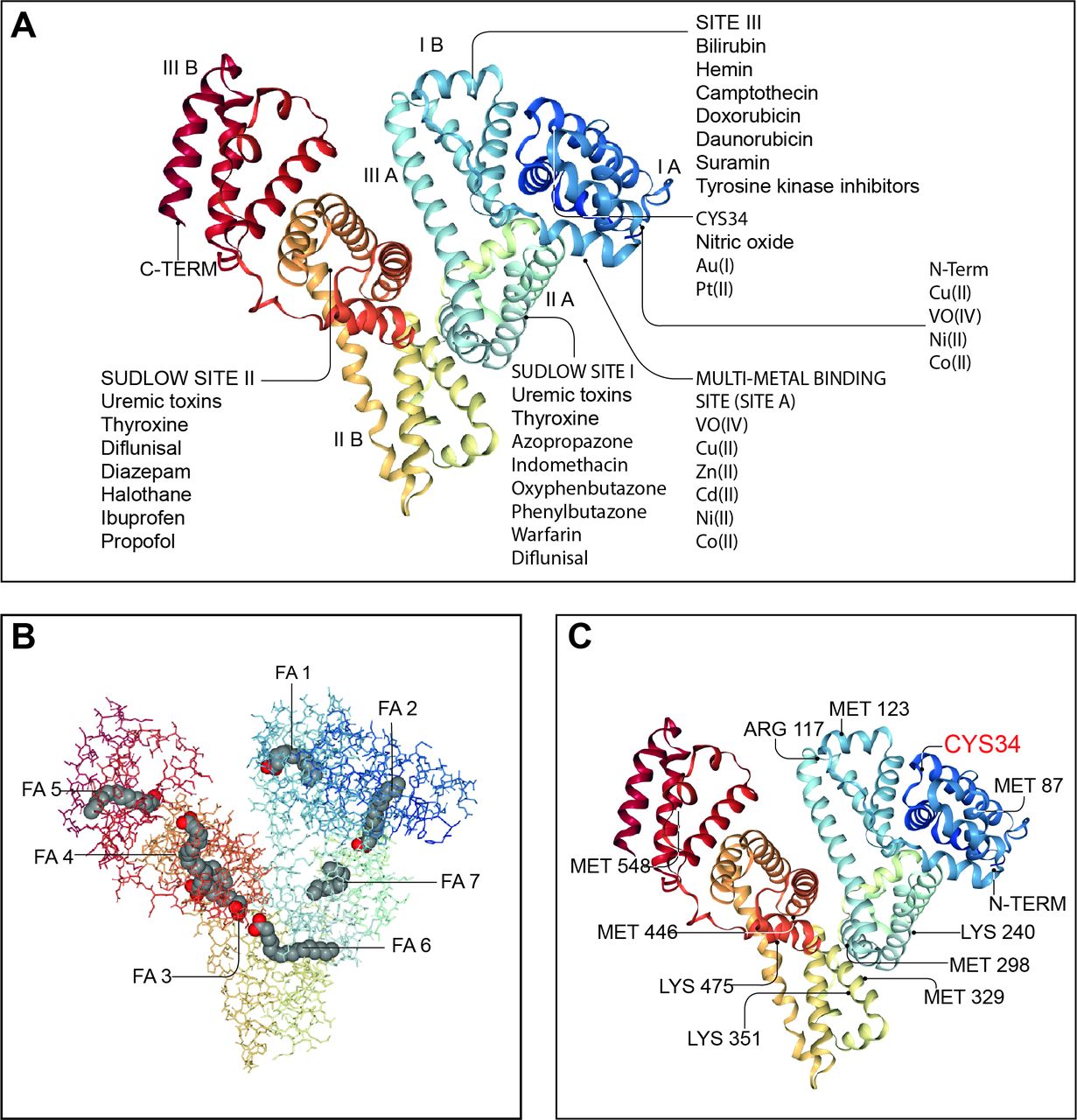
Albumin is a globular, water-soluble 67 kDa protein, negatively charged at neutral pH due to the abundance of aspartate and glutamate residues in its 609 amino acid sequence (figure 3).52 53 Albumin is the most abundant protein in serum (3.5–5.0 g/dL, which represents approximately half of all serum proteins) and in extracellular fluids.52 Albumin is exclusively synthesised by hepatocytes, where it is translated from a single gene as preproalbumin, imported into the endoplasmic reticulum for cleavage of its N-terminal prepropeptide by a serine protease, transported to the Golgi and continuously secreted into the bloodstream.54 Serum albumin has a prolonged half-life of approximately 20 days.55 Its persistence in the circulation derives from its constant uptake and recycling by hepatocytes, a process regulated in a pH-dependent manner by the neonatal crystallisable fragment receptor (FcRn) (figure 4).56 FcRn binds albumin on the surface of hepatocytes and endothelial cells, redirecting albumin into the vascular space and away from bile and extracellular space.56 57 Although renal tubular epithelial cells also express FcRn, the main contribution to albumin recycling and circulating levels steadiness derives from FcRn expression by hepatocytes and endothelial cells, since its absence leads to hypoalbuminaemia.57

Endogenous and exogenous binding sites in the albumin molecule. Many of the physiological functions of human serum albumin rely on its ability to reversibly bind to an extremely wide range of ligands to increase their solubility in plasma, to transport them to specific tissues or organs or to dispose of them when they are toxic. Since ligands are frequently biologically active, albumin also modulates their pathophysiological effects. Panel A: molecular structure of human serum albumin with an indication of its subdomains (IA, IB, IIA, IIB, IIIA and IIIB), of the N and C termini and the main endogenous and exogenous compounds binding sites including the Sudlow’s sites I and II, the site III, the multimetal binding site (site A) and the CYS34 site. Panel B: albumin molecule shows seven long-chain fatty acid (FA) binding sites (FA1–FA7). In addition to transporting FAs, these sites also transport and inactivate biological active lipids involved in systemic inflammation including prostaglandins and PAMPs (lipopolysaccharide and lipoteichoic acid). Panel C: albumin is the most important regulator of extracellular oxidative stress and presents many binding sites for reactive oxidative species. The most important binding site is the Cys-34 residue, which can be reversible or irreversible oxidised giving rise to two molecular forms of oxidised albumin (HNA1 and HNA2). Albumin images from: RCSB PDB (rcsb.org) of PDB ID 1E7I.53 PAMPs, pathogen-associated molecular patterns.

Albumin recycling by endothelial cells. Endothelial cell recycling is the mechanism maintaining high concentrations of healthy albumin within the systemic circulation. Albumin is synthesised by the liver and rapidly released to the intravascular compartment.52 The total amount of albumin in humans is approximately 360 g, 120 g being in the intravascular and 240 g in the extravascular compartment. intravascular albumin is constantly being exchanged (4%–5%/hour through the endothelium with the extravascular pool. In organs having sinusoids or capillaries with fenestrated endothelium, albumin can pass through the large capillary gaps. In the remaining capillaries with continuous endothelium, albumin is transported by an active transcytotic mechanism mediated by the gp60 receptor albondin (not shown). There is a second group of receptors (gp18 and gp30) expressed in many tissues that governs degradation of albumin. These surface cell receptors (not drawn in the figure) show 1000-fold higher affinity for chemically modified albumin (ie, oxidised albumin). Once internalised, this modified albumin is degraded in the lysosomes. Finally, a third type of albumin receptor (FcRn) that rescues albumin from lysosomal degradation contributes to extend the albumin half-life. The low endosomal pH promotes the link of healthy albumin and FcRn in the acidified endosome. When the recycling endosome contacts the higher plasma pH, healthy albumin is released to the systemic circulation.
Albumin as plasma expander, pleotropic scavenger, antioxidant and immunomodulatory molecule
Albumin accounts for approximately 75% of plasma oncotic pressure. A large evidence exists for its use as plasma expander in patients with cirrhosis, but recent data also support other indications.28 58 Indeed, albumin exerts several homeostatic functions as potent scavenger, antioxidant and immunomodulatory molecule. For example, albumin reversibly binds many molecules, permitting solubilisation and transport.58 This occurs principally at sites I/II but may also occur electrostatically through its negative charge (figure 3). Therefore, it modulates the activity of many drugs and binds a range of metallic ions, toxic metabolites and inflammatory mediators potentially affecting systemic inflammation, antioxidant and endothelial function.58 In particular, bilirubin, a toxic insoluble metabolite of haem breakdown, is bound and exclusively transported by albumin to hepatocytes for conjugation with glucuronic acid and elimination via the biliary–intestinal system.59 Moreover, albumin binds bile acids, maintaining their plasma concentration within normal levels and lower than in the portal circulation.60 In liver cirrhosis, serum bile acid concentrations increase to the levels seen in the splanchnic circulation.61 In this context, enhanced serum bile acid binding by albumin supplementation may decrease their possible adverse effects. Importantly, albumin possesses about seven binding sites for fatty acids with moderate to high affinity, enhancing the concentration of fatty acids in blood by several orders of magnitude.62 Finally, a number of metabolites with well-known effects on the immune system, such as those belonging to the tryptophan–kynurenine pathway, mostly circulate bound to albumin. Therefore, their free serum levels in decompensated cirrhosis can be increased due to hypoalbuminaemia, structural changes of albumin or binding competition with endogenous and exogenous substances.63
Albumin is also antioxidant and its infusion reduces oxidative stress during the systemic inflammatory response. Albumin predominantly circulates in a reduced state with a free thiol group in the Cys-34 residue acting as a free radical scavenger for reactive oxygen and nitrogen species. Albumin also restricts oxidative stress damage by neutralising free copper and iron.58 Finally, albumin displays immunomodulatory properties. For example, albumin modulates innate immune responses to sepsis, and cirrhosis-associated PGE2-mediated immune dysfunction improved following albumin infusion.41
Effects on cardiocirculatory function and systemic inflammation
Albumin is frequently used in clinical practice as plasma expander, since its administration increases circulating blood volume. Studies performed in the 1980s demonstrated that albumin administration prevents paracentesis-induced cardiocirculatory dysfunction, an alteration characterised by a systemic haemodynamics impairment leading to effective hypovolaemia. Albumin prevents the decrease in cardiac output and the increase in plasma renin activity.64 Alternative plasma expanders are less effective and, therefore, are not recommended.65
Albumin also prevents renal dysfunction and death in patients with SBP.66 Its administration improves effective blood volume by attenuating peripheral arterial vasodilation and endothelial dysfunction (plasma von Willebrand factor reduction) and enhancing cardiac work, effects not observed with hydroxy-ethyl starch.67 Albumin administration also improves effective blood volume, thus leading to vasoconstrictor systems deactivation, and increases arterial pressure in patients with HRS treated with terlipressin, effects not observed with terlipressin alone.68 Experimental studies have also shown that albumin improves cardiac inotropism in cirrhotic rats with ascites independent of volume expansion and related to the reversal of the negative effects of TNF-α and oxidative stress on cardiac contractility.69
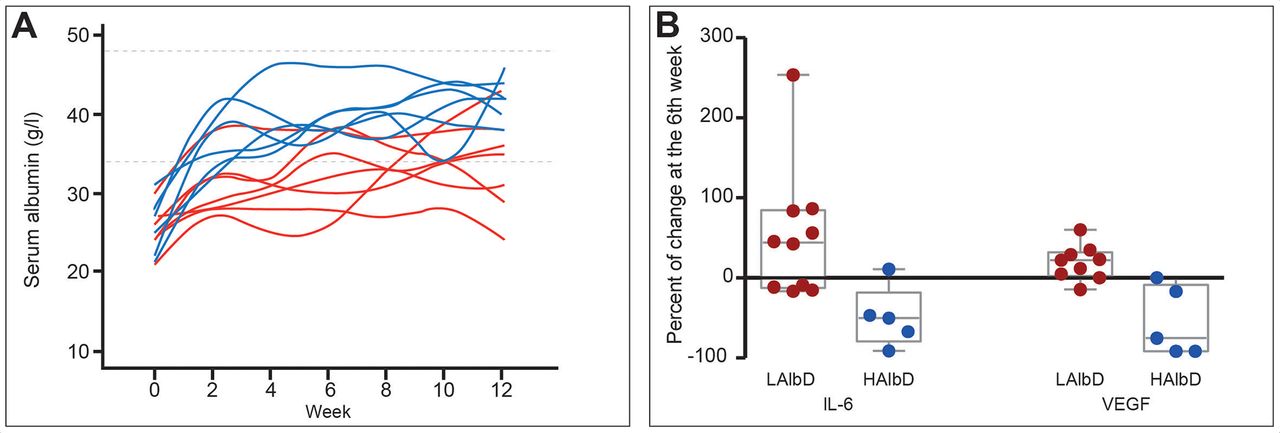
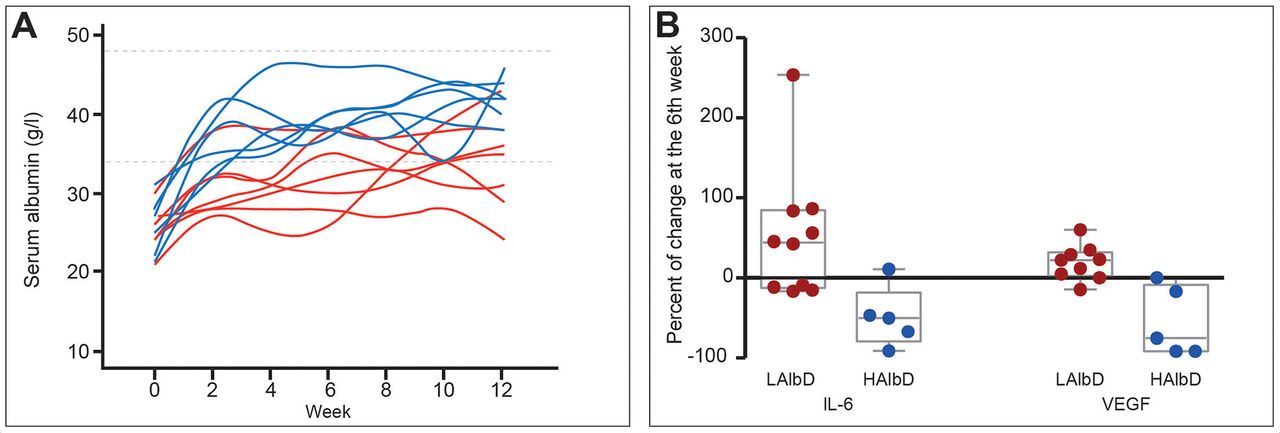
Albumin also attenuates systemic inflammation. Its administration (1.5 g/kg/week) for 12 weeks improved markers of systemic inflammation (IL-6, granulocyte colony-stimulating factor (G-CSF), IL-1ra and vascular endothelial growth factor) in patients with decompensated cirrhosis, an effect confirmed in a short-term investigation in patients with bacterial infections (figure 5).28 Notably, recent in vitro studies attribute the endothelial effects and anti-inflammatory properties of albumin to its uptake by endothelial and immune cells.70 The effect of albumin on endothelial cells is mediated through its antioxidant properties, while its ability to block endosomal TLR signalling acts in the immune cells (Clària J. The immunomodulation effects of albumin. 2019).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of long-term albumin treatment on serum albumin concentration and inflammatory cytokines. Albumin was given at high dosage (HAlbD: 1.5 g/kg body weight (BW) every week) or low dosage (LAlbD: 1 g/kg BW every 2 weeks) during 12 weeks to two groups of patients with decompensated cirrhosis. High albumin but not low albumin dosage was associated with normalisation of serum albumin concentration and significant decrease in the plasma levels of the inflammatory cytokines interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF). This figure was published in gastroenterology, vol 157, Fernández J et al, Effects of albumin treatment on systemic and portal haemodynamics and systemic inflammation in patients with decompensated cirrhosis, page 149, Copyright Elsevier (2019).28
Pharmacokinetics and pharmacodynamics of albumin: towards a personalised use of treatment in cirrhosis
The pharmacokinetics of infused albumin are principally determined by blood volume, serum albumin concentration and amount infused. In healthy volunteers, 50% of a 25 g dose of albumin was lost from the vascular bed 240 min after infusion compared with 65% of 50 g. Conversely, after a rapid withdrawal of 900 mL of blood followed by albumin infusion, 80% of a 50 g dose and 65% of 100 g remained at 240 min. Hypovolaemia and hypoproteinaemia, similar to decompensated cirrhosis, produced a third set of results: after 25 or 50 g albumin infusion, 40% remained in the vascular bed at 240 min.71
The structural integrity of the albumin molecule is essential for its biological functions. Unfortunately, such a structure is quite vulnerable to damaging toxins in a proinflammatory and pro-oxidant circulatory microenvironment, as occurs in decompensated cirrhosis.22 Oxidative damage of the Cys-34 residue, the most common alteration, correlates to severity of cirrhosis and extent of systemic inflammation.22 72 73 Even more important, it independently predicts mortality up to 1 year of follow-up.22 72 73 Besides the Cys-34 residue, post-transcriptional damage can simultaneously involve several other molecular sites (ie, truncation of the N-terminal and C-terminal portions, glycosylation and dimerisation).73 74 With the accumulation of damage in the total albumin pool, the portion maintaining a fully preserved structure (the so-called reduced native albumin) decreases in parallel.72 Moreover, oxidised forms of albumin enhance proinflammatory cytokine synthesis by peripheral human leucocytes, providing a rationale for replacement with reduced native albumin to limit systemic inflammation in cirrhosis.75 In parallel, impairment of albumin functions, such as binding and detoxification capacity, antioxidant function and the ability to chelate metal ions, develops and progresses in patients with increasing severity of cirrhosis.76
The assumption that the global function of albumin is related to its circulating amount and to its structural integrity leads to the concept that the ‘effective albumin concentration’. The proportion of albumin maintaining fully preserved structure and function is much lower than the serum albumin concentration routinely measured in clinical practice. Indeed, effective albumin concentration in decompensated cirrhosis was independently associated with disease severity and albumin function.77 Moreover, it was a better predictor of ACLF and short-term mortality than total serum albumin concentration.77 Based on these observations, future research on albumin should determine whether increasing effective albumin concentration represents a clinically relevant target of therapy and identify novel biomarkers of effective albumin concentration to be used in clinical practice.
First studies on the long-term administration of albumin in cirrhosis
A therapeutic strategy able to modify the natural history of the disease by preventing the development of complications, thus improving survival, quality of life and healthcare costs is still an unmet need in the management of patients with decompensated cirrhosis.17 In this perspective, after the long-term use of albumin was shown to be associated with a better control of ascites in two pivotal studies,78 79 and in parallel with the advancement in our knowledge of the relevance of albumin non-oncotic properties in the treatment of decompensated cirrhosis, two randomised clinical trials and one observational study recently evaluated the effects of long-term albumin administration in this setting.80–82 Such a therapeutic approach attempts to interrupt the pathophysiological cascade responsible for the clinical manifestations of decompensated cirrhosis through a mechanistic treatment. In the complex network of many interacting and redundant pathophysiological pathways that underlie decompensated cirrhosis,83 long-term use of albumin could antagonise several mechanisms, including systemic inflammation and oxidative stress.
In the human Albumin for the treatmeNt of aScites in patients With hEpatic ciRrhosis (ANSWER) study, a non-profit, multicentre, randomised, open-label trial, 431 patients with non-complicated but persisting ascites despite diuretic administration were randomised to receive either standard medical treatment (SMT) or SMT plus albumin (40 g twice a week for 2 weeks and then 40 g/week) for 18 months.80 A significantly better 18-month overall survival was seen in patients receiving albumin (77%) with respect to those receiving SMT (66%), corresponding to a 38% reduction in the HR for mortality. The multivariable risk analysis for all-cause mortality considering transjugular intrahepatic portosystemic shunt placement and liver transplantation as competing events showed that albumin treatment was the sole protective variable. Long-term albumin use also improved the management of ascites, as the need of paracentesis and the incidence of refractory ascites declined by about 50%. Furthermore, the cumulative incidence of complications of cirrhosis, including SBP, non-SBP bacterial infections, episodes of renal dysfunction, as defined by serum creatinine above 1.5 mg/dL, HRS 1 and severe hepatic encephalopathy grade III or IV, as well as potential diuretic-induced side effects, such as hyponatraemia and hyperkalaemia, were reduced by 27%–70% in the albumin group. As a result, patients enrolled in the albumin arm had significantly less liver-related hospitalisations or days spent in hospital, so that the long-term albumin use proved to be also cost-effective. The core results of the ANSWER trial were confirmed by a prospective, non-randomised study that enrolled 70 patients with cirrhosis and refractory ascites.82 Patients who received SMT+albumin (20 g twice weekly) had a significantly lower 24-month mortality than those receiving SMT. Treatment with albumin was the sole independent protective factor against death and was associated to a lower cumulative incidence of rehospitalisations due to any complication of cirrhosis with the exception of GI bleeding.
However, a placebo-controlled clinical trial (Midodrine and Albumin for CirrHotic patients in the waiting list for liver Transplantation (MACHT) study), which enrolled patients with decompensated cirrhosis awaiting liver transplantation, challenged these results.81 In this study, 173 patients with ascites were randomised to receive either SMT plus albumin (40 g/15 days) and the α1-receptor agonist midodrine (15–30 mg/day according to pressor response) or SMT plus placebos. Treatment with midodrine and albumin was associated with a slight but significant suppression of plasma renin activity and plasma norepinephrine. However, neither arterial pressure nor plasma cytokine levels changed significantly. Finally, there were no significant differences between the two groups in the probability of developing complications of cirrhosis during follow-up or 1-year mortality.
The comparison of the characteristics of these studies can provide some relevant topics in order to interpret their contradictory results (table 2). The most striking difference is related with the amount of albumin administered, being about half in the MACHT trial compared with the two other studies. Moreover, a loading dose was only used in the ANSWER study. This was the likely reason for a significant and sustained increase of serum albumin (0.6–0.8 g/dL to a median value close to 4 g/dL) seen in the ANSWER study, while no changes occurred in the MACHT study. The dose of albumin and its effect on serum albumin level are likely to be crucial for the full development of all the biological properties of the molecule. In fact, a recent real-world implementation study confirmed the results of the ANSWER study showing that improvements in effective volaemia and systemic inflammation in patients with stable decompensated cirrhosis were only achieved with a high dose of albumin (1.5 g/kg of body weight (BW)/week vs 1.0 g/kg BW every 2 weeks).28 On the basis of these findings, it is likely that the positive results of the ANSWER study could be even improved using higher albumin therapeutic schedules or adjusting albumin dosage to therapeutic response. In this respect, preliminary results from a post hoc analysis of the ANSWER database showed that the level of serum albumin level reached after 1 month of treatment predicts the probability of 17-month overall survival, which was greater than 90% in those patients presenting a level greater than 4 g/dL.77 Thus, a relevant lesson from the most recent studies is that the serum albumin concentration should be used as guide for monitoring albumin dosage during long-term albumin treatment in cirrhosis.
Main differences among the answer and MACHT trials79 80
Future perspectives
The past 20 years have seen a huge change in our understanding, both of the biology of cirrhosis, its complications and also of albumin biology.84 The central feature, hitherto unrecognised is that cirrhosis is a state of systemic inflammation, which is heightened in patients with complications and in particular in those with ACLF.18 The above sections clearly highlight the importance of albumin therapy in patients with liver disease, which acts to correct both the circulatory dysfunction and also modulates inflammation. In addition to the use of albumin to prevent postparacentesis circulatory dysfunction,85 treat SBP66 and HRS,68 the recent suggestion that long-term outpatients albumin therapy may improve their survival provides the impetus to develop a personalised approach to albumin replacement, optimise the albumin solution that is being administered, better understand albumin biology and await results of ongoing clinical trials.
Personalised approach to albumin therapy: the concept of ‘effective albumin concentration’
At present, the target for albumin therapy is not clearly defined. The data from the two long-term infusion studies have shown different results partly explained by the amount of albumin that was administered. In the ANSWER study,80 40 g twice weekly for 2 weeks, and then 40 g weekly of 20% albumin was administered for up to 18 months whereas in the Barcelona study,81 40 g albumin was administered every 15 days for a median follow-up of 80 days. In the former case, a significant reduction in mortality was observed, but in the latter, no differences in survival was found. One could interpret the data as suggesting that the amount of albumin administered matters. An alternative hypothesis may be that the lack of survival benefit in the Barcelona study is related to the inclusion of sicker patients. It is well known that these patients have worse albumin function in all its domains, bringing into discussion about the concept of ‘effective albumin concentration’,86 which is based on measurement of function rather than just the concentration.76 84 All the three main functional domains, that is, the metal binding domain, the Cysteine-34 domain and the binding sites are now possible to be assessed easily using a cobalt binding assay, high-performance chromatography or electron paramagnetic resonance spectroscopy, respectively.70 It should therefore be possible to generate models whereby the ‘effective albumin concentration’ can be targeted rather than trying to achieve a given concentration. This sort of approach is likely to improve the efficacy and also significantly reduce the cost of albumin infusions.
Improving the quality of the commercially available human serum albumin
As albumin becomes more widely used as a therapeutic rather than just fluid, it is important to think how to improve the quality of the infused albumin. The process of production, maintaining stability and functionality of the commercially available albumin, has remained largely unchanged for many decades. For example, the albumin that is infused into patients contains caproylate and tryptophan, which have been suggested to have deleterious effects. In fact, a device has been developed to specifically remove these stabilisers (Albutec, Rostock, Germany). More significant is the observation suggesting that the commercially available albumins contains only about 50% albumin in the reduced, functionally active, mercaptalbumin form.87 Nearly 40% is present as HNA1.87 Not only is this form non-functional, but it may have deleterious consequences as it can activate inflammatory cells.75 New research is urgently needed to find ways to improve the quality of albumin used in clinical practice, which will reduce costs and may implicate on further improving the efficacy of the infused albumin.
Further understanding of the mechanisms of hypoalbuminaemia in cirrhosis, pathophysiological basis of structural modifications and mechanisms of its pleiotropic effects
In patients with cirrhosis and particularly in those with superimposed inflammation, albumin levels can decline rapidly. Better ability to measure albumin turnover and define exact mechanisms of hypoalbuminaemia may allow alternative approaches to increasing albumin concentrations. The exciting observation that albumin acts intracellularly both in the immune and endothelial cells is extremely interesting.70 75 Many aspects need clarification about the ultimate mechanism within the cell it targets, its fate, the mechanism by which it enters the cell and how this influences its pleiotropic actions.
Further clinical trials of albumin therapy in the acute and chronic situations
The better understanding of albumin biology and the contradictory results of the ANSWER and MACHT studies provides the rationale and the urgent need to perform further trials of long-term albumin. They should better define its potential role in clinical practice identifying patients who can benefit most from treatment and determining the criteria for selecting dose and frequency of albumin administration as well as for its discontinuation. The International PRECIOSA study has started to enrol and will provide the answer to this question (NCT03451292). The ATTIRE study, which explores the role of high dose albumin administration in patients with AD is being performed in the UK and has finished enrolling, randomising about 800 patients.88 The results are eagerly awaited.
Further clinical studies attempting to address albumin biology are using albumin in extracorporeal liver assist devices for patients with ACLF. The molecular adsorbent recirculating system (MARS) and Prometheus provided proof of concept that such a strategy could be successful but were not found to improve survival,89–91 but MARS reduced severity of hepatic encephalopathy. A novel device, DIALIVE, aims to remove and replace the damaged albumin while also removing DAMPs and PAMPs.92 A multicentre, randomised, controlled trial to assess safety and performance of DIALIVE in patients with ACLF versus standard of care is currently ongoing (NCT03065699). As an extension of the concept targeting replacement of the damaged albumin and removing DAMPs and PAMPs, a large phase III, multicentre, randomised, controlled, parallel-group, open-label study of plasma exchange using albumin 5% in ACLF patients has also recently been launched (NCT03702920). These clinical studies will provide further robust evidence for albumin therapy in patients with cirrhosis.
Footnotes
Contributors All the authors equally contributed to the writing of this manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests PG is recipient of an ICREA Academia Award. MB: personal fees from CLS Behring GmbH, personal fees from Grifols SA, personal fees from Takeda, personal fees from Martin Pharmaceuticals, personal fees from PPTA, personal fees from Octapharma, outside the submitted work. PA: personal fees from Grifols, grants from CLS Behring, outside the submitted work. PG: grants and personal fees from GILEAD, grants and personal fees from Mallinckrodt, grants and personal fees from Grifols, personal fees from Intercept, personal fees from Martin Phamaceuticals, personal fees from Sequana, personal fees from Promethera, outside the submitted work. RJ: other from Yaqrit Limited, grants from Takeda, other from Kaleido, from Akaza, from Mallinkrodt, other from Prometic, grants and other from Theoris, during the conduct of the study; other from Yaqrit Limited, grants from Takeda, other from Kaleido, other from Akaza, other from Prometic, other from Mallinkrodt, grants and other from Theoris, outside the submitted work. PC: personal fees from Grifols SA, grants and personal fees from Octapharma SA, personal fees from Kedrion SpA, personal fees from Takeda SA, personal fees from Alphasigma SA, outside the submitted work. JF: personal fees and other from Grifols, outside the submitted work. ALG: personal fees from CLS Behring, personal fees from Grifols, outside the submitted work. JT: personal fees from Gore, personal fees from Bayer, personal fees from Alexion, personal fees from MSD, personal fees from Gilead, personal fees from Intercept, personal fees from Norgine, personal fees from Grifols, personal fees from Versantis, personal fees from Martin Pharmaceutical outside submitted work. VA: personal fees from Grifols, outside the submitted work; VA has a patent 'method for diagnostic and/or prognostic of acute on-chronic liver failure syndrome in patients with liver disorders' pending.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.