Article Text

Abstract
Objective Familial adenomatous polyposis (FAP) is characterised by the development of hundreds to thousands of adenomas at different evolutionary stages in the colon and rectum that will inevitably progress to adenocarcinomas if left untreated. Here, we investigated the genetic alterations and transcriptomic transitions from precancerous adenoma to carcinoma.
Design Whole-exome sequencing, whole-genome sequencing and single-cell RNA sequencing were performed on matched adjacent normal tissues, multiregionally sampled adenomas at different stages and carcinomas from six patients with FAP and one patient with MUTYH-associated polyposis (n=56 exomes, n=56 genomes and n=8,757 single cells). Genomic alterations (including copy number alterations and somatic mutations), clonal architectures and transcriptome dynamics during adenocarcinoma carcinogenesis were comprehensively investigated.
Results Genomic evolutionary analysis showed that adjacent lesions from the same patient with FAP can originate from the same cancer-primed cell. In addition, the tricarboxylic acid cycle pathway was strongly repressed in adenomas and was then slightly alleviated in carcinomas. Cells from the ‘normal’ colon epithelium of patients with FAP already showed metabolic reprogramming compared with cells from the normal colon epithelium of patients with sporadic colorectal cancer.
Conclusions The process described in the previously reported field cancerisation model also occurs in patients with FAP and can contribute to the formation of adjacent lesions in patients with FAP. Reprogramming of carbohydrate metabolism has already occurred at the precancerous adenoma stage. Our study provides an accurate picture of the genomic and transcriptomic landscapes during the initiation and progression of carcinogenesis, especially during the transition from adenoma to carcinoma.
- colon carcinogenesis
- colorectal adenomas
- Familial adenomatous polyposis (FAP)
- MUTYH-associated polyposis (MAP)
- Field cancerization
- Single-cell transcriptome profiling
- Tumor heterogeneity
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- colon carcinogenesis
- colorectal adenomas
- Familial adenomatous polyposis (FAP)
- MUTYH-associated polyposis (MAP)
- Field cancerization
- Single-cell transcriptome profiling
- Tumor heterogeneity
Significance of this study
What is already known on this subject?
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome primarily caused by inherited mutations in APC (adenomatous polyposis coli).
Patients with FAP will develop hundreds to thousands of adenomas in the colon and rectum that will inevitably progress to adenocarcinomas if left untreated.
Early prevention of FAP is challenging, and the standard of care for classical FAP is prophylactic colectomy or proctocolectomy.
What are the new findings?
Spatially separated tumours can originate from the same cancer-primed cell in patients with FAP, indicating that the pathogenic events may happen long before the appearance of clinically identifiable adenomas, even in a macroscopically normal epithelium.
By comparing the transcriptomic signatures of the epithelial cells between the normal colon tissues from patients with FAP and the normal colon tissues from patients with sporadic colorectal cancer, we found that the normal epithelium of patients with FAP already exhibits enhanced metabolic processes and proliferative activity.
Reprogramming of carbohydrate metabolism has already happened in precancerous adenomas.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Although the adjacent normal epithelium of patients with FAP patents has not accumulated potential driver mutations and copy number alterations (CNAs), at the transcriptomic level, these tissues have already been primed for carcinogenesis, suggesting the necessity of targeting the ‘“normal’” mucosa for the prevention, diagnosis, and therapy of patients with FAP patients.
Reprogramming of carbohydrate metabolism has already occurred at early stages during the carcinogenesis of lesions in patients with FAP patients, suggesting that carbohydrate metabolism may be a target for early prevention of adenomas.
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome primarily caused by inherited mutations in adenomatous polyposis coli (APC).1 2 Patients with FAP develop hundreds to thousands of adenomas in the colon and rectum that will inevitably progress to adenocarcinomas if left untreated.3–5 Colectomy is the main prophylactic treatment for FAP.3 4 6 APC is a tumour suppressor gene regulating cell adhesion and migration, maintenance of genome stability and apoptosis. The most important role of the APC gene is negatively regulating the WNT signalling pathway by mediating the degradation of CTNNB1, a key component of the WNT signalling pathway. Enhanced WNT signalling pathway activity in colon cells can lead to epithelial hyperplasia.7–9
Approximately 85% of non-FAP sporadic colorectal cancers (CRCs) have somatic APC mutations, and APC was shown to be the gatekeeper gene during the adenoma–carcinoma sequence of CRC.7 10 Unlike sporadic CRCs, from which only one lesion can be collected, the lesions at multiple evolutionary stages in FAP make this condition an ideal natural model for tracing colorectal carcinogenesis.
Previous studies have applied bulk whole-exome sequencing (WES) and whole-genome sequencing (WGS) approaches to investigate the differences in genomic alterations between adenomas and carcinomas, aiming to uncover the crucial events during the progression of CRC.11 12 But the differences among lesions obtained from a large cohort may be confounded by the interindividual variations, such as genetic background, eating habits and intestinal flora. In this study, we simultaneously collected adjacent normal tissue, adenomas at different stages and carcinomas from the same patient. A multiregional sampling strategy was employed to investigate intratumour heterogeneity. For each lesion, three sets of data were simultaneously obtained, including WES, WGS and single-cell RNA-seq data. The genomic landscapes and clonal architecture of lesions at different evolutionary stages from the same patient with FAP were comprehensively investigated. In addition, single-cell RNA-seq data were used to investigate the transcriptomic heterogeneity among multiregionally sampled lesions from the same patient with FAP and to explore the transcriptome dynamics in tumour cells during the initiation and carcinogenesis of colorectal adenomas.
Materials and methods
Tumour collection and preparation
Tissues were sampled immediately after surgical resection of the specimens. For each patient, the normal mucosa, small adenomas, medium adenomas, large adenomas and carcinomas were sampled. For most adenocarcinomas larger than 10 mm in diameter, two to five regions were sampled. Each region was divided into two parts—one for haemotoxylin and eosin (H&E) and immunohistochemical staining and the other for single-cell collection and genomic DNA extraction.
Whole-exome sequencing
Extracted genomic DNA (200 ng) was fragmented by sonication into fragments of 150–200 bp. After end repair, a predesigned code adaptor with an extra 3 bp barcode was ligated to the fragmented DNA. Then, the ligated DNA was subjected to four amplification cycles with NEB index primer, NEB universal primer and 2× KAPA HiFi HotStart ReadyMix (Kapa Biosystems, cat. KK8054). Next, libraries with different barcode adaptors were pooled for whole-exome sequence capture using SureSelectXT Human All Exon v5 and SureSelectXT Human All Exon v6 kits (Agilent Technologies, cat. G7530-9000).
Modified STRT-Seq
We modified the STRT-Seq13–15 protocol for application to a large number of single cells, as we mentioned in previously published papers.16–18 See online supplementary materials and methods for more details.
Supplemental material
Sanger sequencing
All APC germline mutation sites and somatic mutation sites, as well as the germline mutation sites in MUTYH, were verified by Sanger sequencing. The primers used for Sanger sequencing are summarised in online supplementary table 3.
Supplemental material
Data analysis
For further details, please see the online supplementary materials and methods.
Results
Overview of the cohort
The cohort in this study comprised six patients with FAP, one patient with MUTYH-associated polyposis (MAP) and two patients with sporadic CRC. A total of 56 samples were collected: 6 peripheral blood samples, 12 adjacent normal tissues, 23 low-grade adenomas (LGIN, including grade I and grade II), 5 high-grade adenomas (HGIN, grade III) and 10 carcinomas (online supplementary table 1 and online supplementary figure 1A). The smallest adenomas were 2 mm in diameter, and the largest carcinomas were 50 mm in diameter. For most lesions larger than 10 mm in diameter, two to five regions were sampled separately (figure 1A). In total, 86 regions from the 56 samples were collected. The sequencing data for each region were summarised (online supplementary table 2).
Supplemental material
Supplemental material
Supplemental material

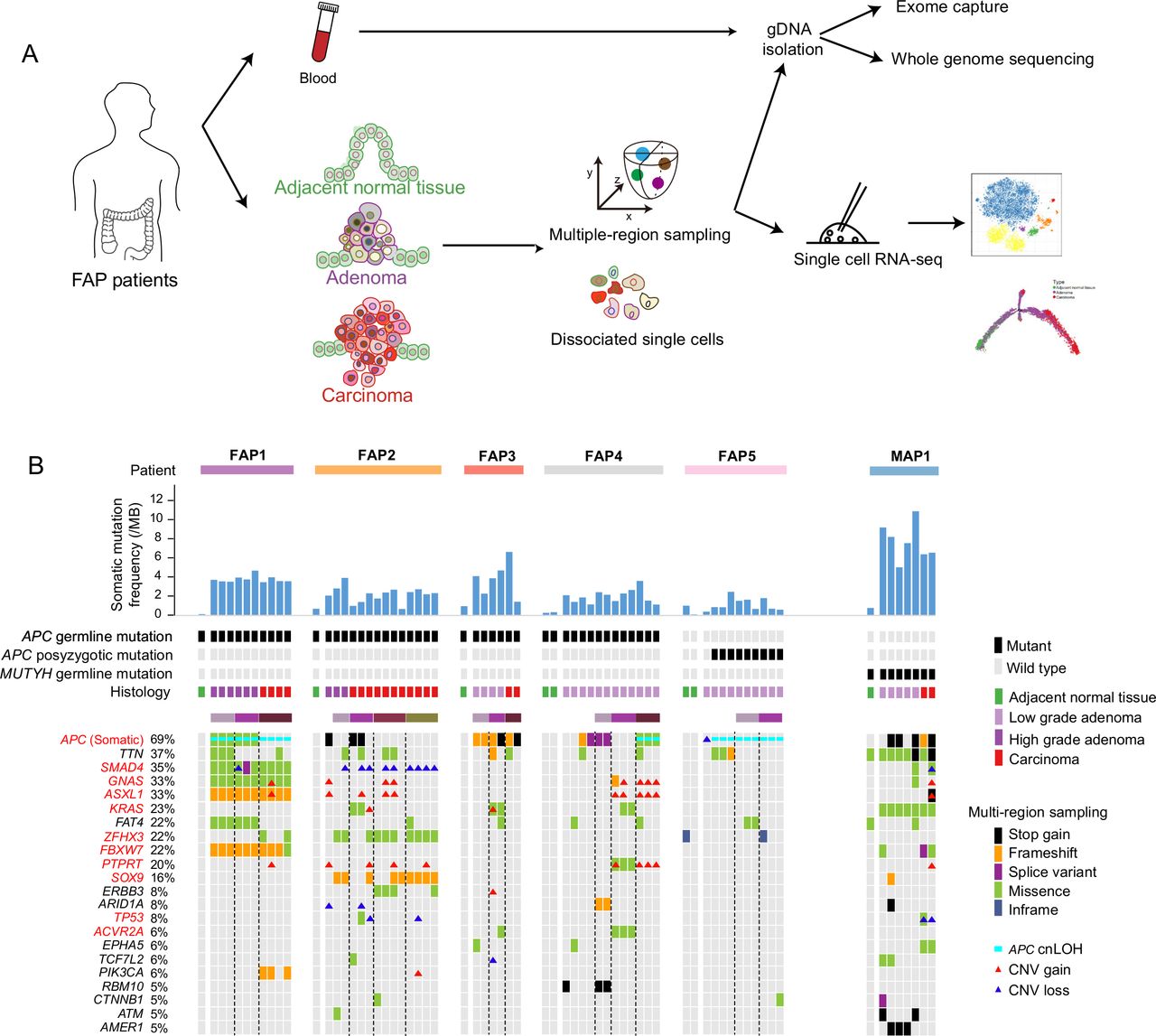
Landscapes of genomic alterations in FAP. (A) Workflow. Adjacent normal tissue, adenomas and carcinomas from colon and rectum of FAPs were obtained for dissociation into single cells. Multiple-region sampling was used for adenocarcinomas larger than 10 mm. Parts of the dissociated cells were used for single-cell RNA-seq, and the remaining parts and matched peripheral blood were used for whole-exome sequencing and whole-genome sequencing. (B) Potential driver events in the five patients with FAP and one patient with MAP(including 8 adjacent normal tissues, 31 regions from 20 low-grade adenomas, 9 regions from 4 high-grade adenomas and 19 regions from 7 carcinomas). Top, the patient origin, somatic mutation frequency per mega-base, inherited or postzygotic mutations on APC or MUTYH, and tumour grade for each sample are indicated. Middle, graph shows detailed information about the mutations of potential driver genes. The samples from the same patient were arranged together. The genes were sorted by the frequency of genomic alterations in the cohort. Different types of genomic alterations, including stop gain, frameshift, splice variant, missense, inframe, APC cnLOH and copy number alterations, are shown by different colours. Lesions using multiregion sampling strategy are indicated. The black dot line separates different multiregion sampled lesions. APC, adenomatous polyposis coli; cnLOH, copy neutral loss of heterogeneity; CNV, copy number variation; FAP, familial adenomatous polyposis; MAP, MUTYH-associated polyposis.
Four of the patients with FAP had signatures of family inheritance (online supplementary figure 1B–G). Inherited germline mutation sites in the APC gene were identified in FAP1 (p.E1309fs), FAP2 (p.E1397fs), FAP3 (p.W699*) and FAP4 (c.834+1G>C, a splice donor variant) (online supplementary figure 2A and online supplementary table 3). The clinical phenotype of FAP5 was suspected to be caused by postzygotic mutations in APC because no family history was found, and three tandem APC mutations (p.V1377fs, p.Y1376fs and p.E1374_H1375delinsD) were shared by nine regions (13 regions in total) from FAP5 (online supplementary figure 2A,B and online supplementary table 3). The case of polyposis in MAP1 was suspected to be MAP. Two germline stop-gain mutations (p.R19* and p.Q267*) were found in MUTYH (online supplementary figure 2C) in all lesions from MAP1. Consistent with the findings of previous studies,4 6 19–23 all lesions from MAP1 had somatic mutations in KRAS and had a higher percentage of C to A mutations than lesions from the patients with FAP (figure 1B and online supplementary figure 2D). Although both FAPs and MAPs are associated with multiple colorectal adenomas, their molecular pathogenesis and somatic genetic alterations are distinct.20 All germline and postzygotic mutations in APC in the patients with FAP, as well as the germline mutations in MUTYH in MAP1, were verified by Sanger sequencing (online supplementary table 3).
Genomic alteration landscape in the cohort
The mean depth of the exomes was 64 reads, and about 85% of the covered regions were mapped by more than 30 high-quality reads (online supplementary table 3). In total, 8,327 somatic mutations were found across the 67 samples with WES data. After stringent filtering, 4,220 potential pathogenic mutations were identified, including 3,570 single nucleotide variants and 650 indels (online supplementary table 4 and online supplementary materials and methods). Many genes were reported to be recurrently mutated in CRC,12 and the mutational landscapes of these potential driver genes in our cohort were summarised (online supplementary table 5 and figure 1B).
Supplemental material
Supplemental material
Consistent with previous reports that APC is a gatekeeper gene in human colorectal epithelial cells, APC had the highest mutation frequency, including somatic mutations, copy neutral loss of heterogeneity (cnLOH) and copy number alterations (CNAs), as inferred by using all lesions from patients with FAP (mutated=36, all=52; mutation frequency=69%) (figure 1B).22 The previously reported adenoma–carcinoma transition model is characterised by sequential accumulation of mutations in APC, KRAS and TP53. 22 For FAP patients with APC germline mutations, potential pathogenic genomic alterations in KRAS and TP53 were found in only seven samples (11%) and two samples (3%), respectively (figure 1B), which may due to the difference of pathological grade of the lesions that were used. The most frequently mutated signalling pathway was the WNT signalling pathway (online supplementary figure 3A).12 Then we analysed the mutation burdens (MBs) (number of mutations per mega-base) of multigrade lesions. The results showed that the low-grade adenomas had already shown significantly more somatic mutations compared with the adjacent normal tissue. The carcinoma had relatively heavier somatic MBs, although not statistically significant due to the limited sample size, than the low-grade adenomas. Interestingly, the high-grade adenomas had higher MB than the carcinomas (not significant, p=0.075) (online supplementary figure 3B).11 12 24 Previously reported recurrent CNAs in CRCs (copy number gain on chromosomes 7, 8, 13 and 20; copy number loss on chromosomes 5q, 15q and 18q) were also found in our cohort (online supplementary figure 4).
Clonal architecture of lesions at different evolutionary stages from the same patient with FAP
It is generally assumed that different lesions originate from different cells, and this was the case in FAP3, FAP4, FAP5 and MAP1 (online supplementary figures 5–8). Phylogenetic analysis inferred from the somatic mutation profile indicated that 172 somatic mutations were shared by spatially separated lesions in FAP1, including potential pathogenic mutations in GNAS (p.R844H), SMAD4 (p.C115W), FBXW7 (p.S668fs and p.G687V) and ASXL1 (p.P808fs) (figures 2A,B). The mutation frequencies of the lesion-shared mutations in FAP1 were comparatively higher than those of nonshared mutations, indicating that these spatially separated lesions from FAP1 are monoclonal and originated from the same cell (online supplementary figure 9A). The same pattern was also found in FAP2 (online supplementary figures 10 and 11). A total of 136 somatic mutations were shared by spatially separated lesions in FAP2, including potential pathogenic mutations in SOX9 (p.Q312fs) and ZFHX3 (p.P2116L). The mutation frequencies of the lesion-shared mutations in FAP2 were comparatively lower than those in FAP1, implying a polyclonal origin of the lesions in FAP2 (online supplementary figure 10C). Since these two patients had previously undergone total colectomy due to severe polyposis, all lesions collected from these two patients were obtained from the residual rectum resected in the second surgery for rectal cancer. Thus, these lesions were spatially close to each other and were at the late stage of carcinogenesis (figure 2C and online supplementary figure 10D). Combining all of the previously mentioned information, we propose that the process described in the previously reported field cancerisation model also occurred in these two patients and contributed to physically separated lesions.25–28 In this model, a precancerous cell accumulates potential pathogenic mutations that endow it with a proliferative advantage, and its daughter cells then extend to adjacent areas by crypt fission. Next, with the accumulation of other pathogenic events, spatially separated lesions are formed (figure 2C and online supplementary figure 10D). This phenomenon emphasises that pathogenic events may occur long before the appearance of clinically identifiable adenomas, even in macroscopically normal epithelium.

Clonal architectures of lesions at different evolutionary stages from FAP1. (A) Heatmap showing the regional distribution of somatic mutations in all samples from FAP1. All mutations were classified into three types: lesion-shared mutations (dark blue), branched/truncal mutations (deep orange) and private mutations (light green). The number of private mutations for each sample is shown. Samples from different lesions are separated by a black line. (B) Phylogenetic tree of lesions at different evolutionary stages from FAP1 by using maximum parsimony algorithm. The colours of the lines in the phylogenetic tree correspond to the mutation types as mentioned previously. Potential driver mutations and cnLOH of APC genes are shown. (C) Schematic diagram indicating that all the lesions of FAP1 originated from the same cell. (D) Clonal or individual CNAs and cnLOHs were presented on the phylogenetic tree of lesions from FAP1 constructed from somatic mutations. Ade, adenoma; APC, adenomatous polyposis coli; Car, carcinoma; cnLOH, copy neutral loss of heterogeneity; FAP, familial adenomatous polyposis; HGIN, high-grade intra-epithelial neoplasia; Nor, adjacent normal tissue; R, region.
Although all lesions from FAP1 originated from the same cell, highly intertumour and intratumour heterogeneities were observed (figure 2B). APC (p.R1858Q) and FAT4 (p.R555Q) were shared by the two adenomas, while CNAs were only observed in the carcinoma. Moreover, two lineages developed in the carcinoma tissue from FAP1. SMAD4 (p.G386D) was obtained only in Car R3, while PIK3CA (p.*1069fs) was shared by the other three regions (figure 2B). In addition to somatic mutations, Car R3 also had distinct CNAs and loss of heterogeneity mutations compared with the other three regions (figure 2D and online supplementary figure 9B,C).29 Clonal architecture analysis of other multiregionally sampled lesions from FAP4 and FAP5 supported the punctuated evolution model; most of the potential driver mutations and recurrent CNAs were clonal (online supplementary figures 6 and 7).30
Transcriptome profiling of patients with FAP
To trace the transcriptome dynamics during the initiation and carcinogenesis of colorectal adenomas, 8,757 single cells were collected from the six patients with FAP described earlier, as well as from two patients with sporadic CRC. After stringent filtering, 7,583 (86.6%) cells were kept for further analyses (online supplementary materials and methods and online supplementary table 6). Unsupervised clustering using the regulation network signatures of transcription factors grouped the cells into six clusters. According to the expression patterns of well-known cell type markers, we defined environmental cells (endothelial cells, fibroblasts, macrophages, mast cells, T cells and B cells) and epithelial cells (online supplementary figure 12 and online supplementary table 6). The proportions of immune cells (mast cells, macrophages, T cells and B cells) and fibroblasts were significantly increased in carcinomas compared with adjacent normal tissues and adenomas, implying enhanced immune infiltration in carcinomas (online supplementary figure 12E). Then, we focused on the transcriptome dynamics of the epithelial cells during carcinogenesis.
Supplemental material
Enhanced metabolic processes and proliferative activity in the normal epithelium of patients with FAP
Since patients with FAP are born with germline mutations in the APC gene, we speculated that the normal colon epithelium of patients with FAP may show transcriptomic signatures different from those of the normal colon epithelium of patients with sporadic CRCs. The transcriptomes of 707 single epithelial cells from adjacent normal tissues of six patients with FAP and 152 epithelial cells from adjacent normal tissues of two sporadic CRC specimens were profiled. These normal tissues did not share potential pathogenic mutations or CNAs with their corresponding adjacent lesions (figure 2 and online supplementary figures 5–7, 10, 11 and 13).
Differentially expressed genes between these two types of adjacent ‘normal’ epithelium were analysed. Genes located on the sex chromosomes were excluded to avoid gender bias. We found that 2,569 genes were upregulated in the normal colon epithelium of patients with FAP (online supplementary table 7). The most significantly upregulated gene in FAP epithelium was OLFM4, which was reported as a marker for intestinal stem cells and subsets of CRC cells (figure 3A).31 Another upregulated gene, ATF3, is reported to be a repressor transcription factor for carcinogenesis and can be induced by hypoxia (figure 3A).32 Gene ontology analysis of these upregulated genes in FAP revealed strong enrichment in metabolic processes, including peptide biosynthetic, nucleotide metabolic, amino acid metabolic, lipid metabolic and carbohydrate metabolic processes (figure 3B). In addition, the genes involved in the cell cycle were also enriched, indicating that the proliferative potential of the cells from adjacent normal epithelium of patients with FAP has already been enhanced (figure 3B). The enhanced proliferative activity of the normal epithelium from patients with FAP was verified by immunohistochemical staining for MKI67 (figure 3C,D).
Supplemental material

Transcriptome differences between the ‘normal’ epithelium of patients with FAP and the normal epithelium of sporadic patients with CRC. (A) Heatmap showing the top DEGs with the highest p-value between epithelial cells from adjacent normal tissue of patients with FAP (30 genes) and epithelial cells from adjacent normal tissue of patients with sporadic CRC (15 genes). The full list of DEGs is summarised in online supplementary table 7. (B) Gene ontology analysis of genes that show higher expression levels in FAP colon epithelium compared to CRC normal colon epithelium. (C) Immumohistochemical staining of MKI67 in adjacent normal tissue of FAP2 and adjacent normal tissue from patient with CRC (non-FAP). Scale bar, 100 µm. (D) Five views (×400) were randomly chosen from immumohistochemical staining to calculate the percentage of MKI67-positive epithelial cells (MKI67-positive epithelial cells/all epithelial cells) for each normal mucosa from three patients with sporadic CRC (patients without FAP) and three patients with FAP. Significance analysis was also done between the normal mucosa from patients with FAP and from the normal mucosa of patients without FAP. CRC, colorectal cancer; DEG, differentially expressed gene; FAP, familial adenomatous polyposis.
Transcriptomic heterogeneity among lesions at different evolutionary stages from the same patient with FAP
Since all lesions from FAP1 originate from the same cell, we sought to investigate the transcriptomic heterogeneity of these lesions. By using tSNE analysis, all epithelial cells from FAP1 were grouped into four clusters. Cluster 1 and cluster 2 were composed mainly of cells from three regions of adenoma #1 (Ade1 region1, Ade1 region2 and Ade1 region3) and the second region of the carcinoma (Car region2). Cluster three was mainly composed of cells from adenoma #3 (Ade3) and the third region of the carcinoma (Car region3). Cluster 4 was mainly composed of cells from the first and fourth regions of the carcinoma (Car region1 and Car region4) (figure 4A). ANG, ANPEP and FGL1 exhibited high expression specifically in cluster 2 (figure 4B). ANG was reported to enhance the formation of new blood vessels.33 ANPEP is a marker gene for enterocytes, and FGL1 was reported to be highly expressed in colorectal adenocarcinomas.34–36 The genes specifically expressed in cluster 3 were enriched in gene ontology (GO) terms such as lymphocyte activation, adaptive immune response and defense response to bacterium, implying that adenoma #3 and the third region of the carcinoma may exhibit an enhanced immune response (figure 4C). Immunohistochemical staining of CD8-positive T cells also indicated more infiltrated immune cells in Car region1 compared with the other three regions (figure 4D). The cells of cluster 4 showed distinct transcriptomic signatures compared with cells from other clusters. The genes specifically expressed in cluster 4 were enriched in the GO terms WNT signalling pathway, negative regulation of cell proliferation, morphogenesis of an epithelial sheet and cell junction assembly (figure 4E). These results indicate that the transcriptomic heterogeneity within tumours may be shaped by their spatial location and affected by the surrounding microenvironment. A similar pattern was identified in FAP2 (C1 R1 and C1 R2) and FAP3 (A1 R1 and C R2), as well as in patients without FAP (CRC1 Car R3 and CRC2 Car R2) (online supplementary figures 14 and 15 and online supplementary table 8). However, a spatial region-specific transcriptomic signature was not found in low-grade adenomas from FAP4 and FAP5 (online supplementary figure 16 and online supplementary table 8).
Supplemental material
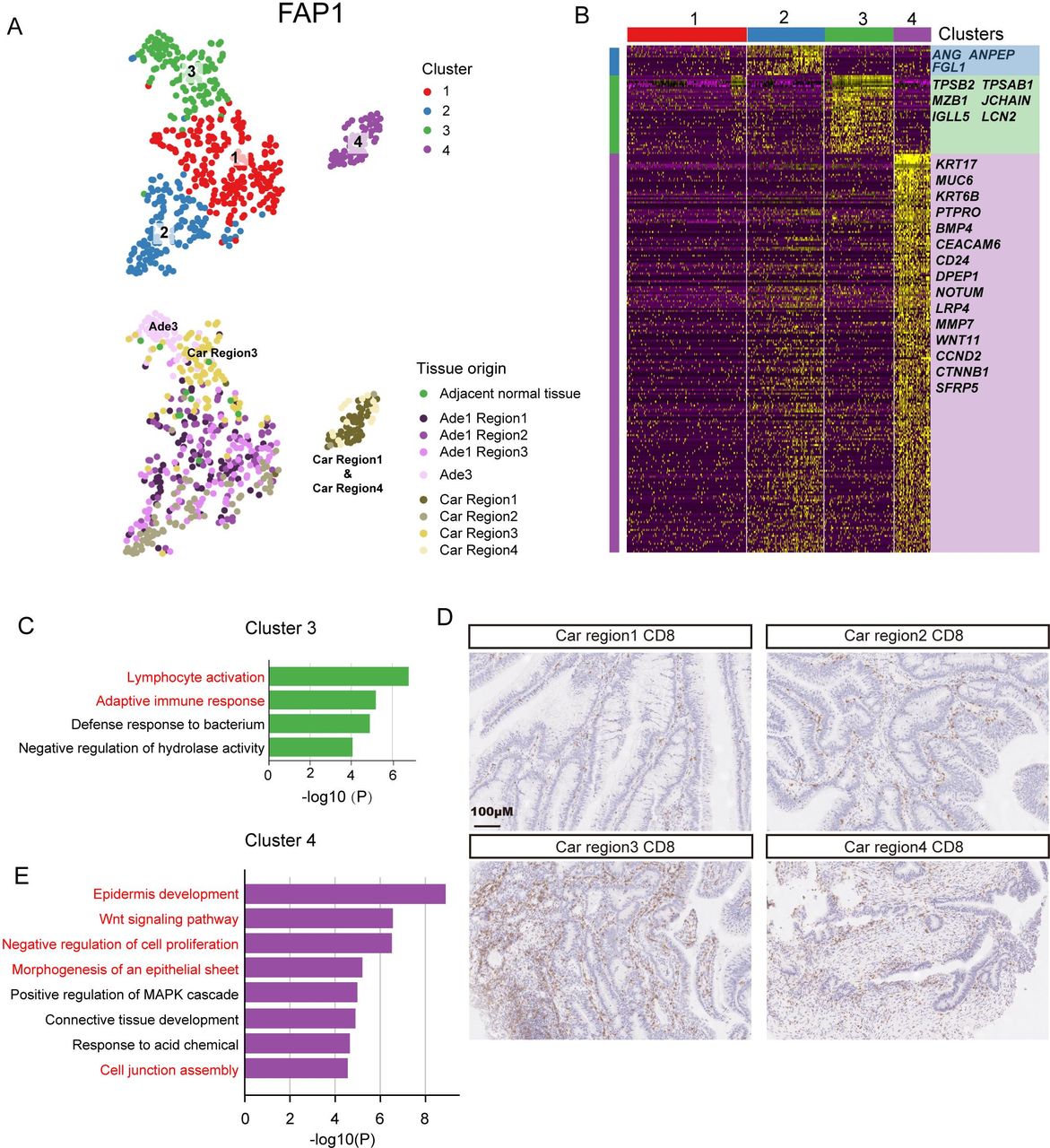
Transcriptome heterogeneity of lesions at different evolutionary stages from FAP1. (A) Clustering analysis of all epithelial cells from FAP1 by using tSNE. Four clusterswere identified (top) and the tissue origin for each single cell was indicated (bottom). (B) Heatmap showing the specifically highly expressed genes in each cluster. (C) GO analysis of genes highly expressed in cluster 3. (D) Immumohistochemical staining of CD8 in four regions of carcinomas from FAP1. Scale bar, 100 µm. (E) GO analysis of genes highly expressed in cluster 4. Car, carcinoma; Ade, adenoms; FAP, familial adenomatous polyposis; GO, gene ontology; MAPK, mitogen-activated protein kinase.
The metabolic signature of cancer has already been established in precancerous adenomas
Monocle237 was used to construct a pseudotime map of carcinogenesis that could reflect the major transcriptomic changes during progression from normal epithelium to carcinoma, and only epithelial cells were used to perform the pseudotime map construction. Cells from the adjacent normal epithelium and grade I adenomas were mainly distributed at the beginning of the pseudotime trajectory, while cells from carcinomas were mainly distributed at the end of the pseudotime trajectory (online supplementary figure 17A,B). This pattern was preserved across all patients with FAP (online supplementary figure 17C). Then, genes that changed dramatically along the pseudotime trajectory were analysed. Four groups of genes that showed different dynamic patterns along the pseudotime trajectory were analysed (figure 5A,and online supplementary table 9). The first group of genes showed increased expression levels in the adjacent normal epithelium. During carcinogenesis, these genes were first downregulated at the early phase of pseudotime and were then upregulated near the end of the pseudotime trajectory. Gene ontology analysis revealed that these genes participated in the citric acid (tricarboxylic acid cycle (TCA)) cycle, respiratory electron transport, gluconeogenesis, mitochondrial activity, and so on (figure 5B). This result was consistent with previous findings that cancer cells mainly use glycolysis instead of the TCA pathway for energy production and generation of intermediate precursors for metabolite biosynthesis, a phenomenon called the Warburg effect.38–44 Interestingly, most of the genes involved in the TCA cycle were first downregulated from the adjacent normal epithelium to adenomas and were then slightly upregulated from adenomas to carcinomas, indicating that the TCA pathway is already strongly repressed in precancerous adenomas; although this repression was later alleviated in carcinomas, TCA cycle activity was still lower in carcinomas than in adjacent normal epithelium (figure 5C). The genes in group 2 were also downregulated at the early stage of carcinogenesis, while the genes in group 3 were not downregulated until the late stage of carcinogenesis. The genes in group 2 were also enriched in metabolic processes, while the genes in group 3 were enriched in ion hemostasis and apoptotic signalling pathways. The genes in group 4 were upregulated at the late stage of carcinogenesis and enriched in the GO terms myeloid leucocyte activation, wound healing and cellular response to acid chemical, indicating multiple adaptive responses during cancerisation (online supplementary figure 17D).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transcriptome dynamics during carcinogenesis. (A) Heatmap showing scaled expression of dynamic genes along the pseudotime. The definition of dynamic genes was described in online supplementary Materials and methods.The full list of the dynamic genes is summarised in online supplementary table 8. Rows of the heatmap represent genes that show dynamic changes along the pseudotime, and these genes were clustered into four groups according to their expression pattern along the pseudotime. We sorted the cells along the pseudotime into 100 windows. The colour scheme represents the z-score distribution from −3 (blue) to 3 (red). The pie plot shows the percentages of cells from multigrade lesions at early, mid and late stage of the pseudotime. (B) Gene ontology analysis of genes from group 1 in (A). (C) Heatmap showing scaled expression of TCA-related genes along the pseudotime. The colour scheme represents the z-score distribution from −3 (blue) to 3 (red). TCA, tricarboxylic acid cycle.
The EMT programme is upregulated as early as the low-grade adenoma stage
The most significant difference between carcinomas and precancerous adenomas is the ability of carcinoma to invade and metastasise.10 Epithelial-to-mesenchymal transition (EMT) was reported to participate in multiple cancer behaviours, especially in cancer invasion and metastasis.40 45–49 We wanted to determine whether EMT-related programmes were upregulated in precancerous adenomas by analysing the expression dynamics of EMT-related genes along the pseudotime trajectory of carcinogenesis. The expression level of EPCAM (an epithelial marker gene) was first slightly upregulated and then downregulated during carcinogenesis, whereas the expression level of VIM (a mesenchymal marker gene) was gradually upregulated (online supplementary figure 18A). The EMT-related genes were generally upregulated along the pseudotime trajectory, indicating that the mesenchymal signature was rapidly enhanced during carcinogenesis even in early-stage adenomas. The expression level of TGFBI gradually increased during adenoma carcinogenesis (online supplementary figure 18A,B). This gene was reported to be the key regulator of partial EMT in head and neck cancer.50 By immunohistochemical staining, we found that the abundance of the TGFBI protein was also increased in adenomas and carcinomas (online supplementary figure 18C).
Discussion
Patients with FAP are born with germline mutations in the APC gene and will inevitably develop hundreds of multigrade precancerous adenomas in the colorectum. This characteristic makes FAP a natural model for investigating the adenoma–carcinoma transition process. Previous studies on multigrade precancerous adenomas in FAP or lesions at different evolutionary stages from a large cohort of patients with sporadic CRC reported higher mutational and CNA burdens in carcinomas than in precancerous adenomas. Interestingly, in our cohort, high-grade adenomas even have heavier MBs than carcinomas. However, since the sample size of the high-grade adenomas is relatively limited (n=9), the difference of MBs between high-grade adenomas and carcinomas is not statistically significant, which reduces the reliability of the results. In addition, Cross et al reported that advanced adenomas have higher intragenetic heterogeneity than carcinomas, implying that carcinomas have sharper fitness peaks due to stabilising selection.51 In our cohort, the intratumoural heterogeneity between adenomas and carcinomas was not significantly different (online supplementary figure 19). We suspect that the inconsistencies between these two studies may be due to the different cohorts studied. The adenomas and carcinomas included in the study by Cross et al were from patients with sporadic CRC, while the lesions included in our study were from patients with FAP.
Several potential driver genes, including APC, KRAS and TP53, were reported to be key factors promoting carcinogenesis. Even with the inherited germline mutation in APC, a second hit in the APC gene, including mutation and cnLOH, is still the most common recurrent event in patients with FAP. The mutation frequency of TP53 (8%) was relatively low in our cohort, as well as in a published FAP cohort (11%),51 compared with the 60% reported by the TCGA database.12 However, this conclusion is weakened by the limited sample size, and a larger cohort is needed to verify this hypothesis.
By analysing multiple lesions from the same patient with FAP, we found that adjacent lesions in patients with FAP can originate from the same cell. This result advances the findings of previous studies about field cancerisation by indicating that field cancerisation through crypt fission could give rise to multiple spatially adjacent tumours in patients with FAP.52 Gausachs et al also reported the possibility of a ‘genetic field effect’ in patients with FAP based on finding that physically separated crypts share hotspot mutations in KRAS. 29 However, the inference of the evolutionary relationship based on several mutation sites limits the reliability of this conclusion. Our analysis based on WES further confirmed the theory of field cancerisation in patients with FAP. For the four patients with FAP with germline mutations on APC, field cancerisation was observed in FAP1 and FAP2 but not in FAP3 and FAP4. One possible explanation is that the lesions from FAP3 and FAP4 were relatively far from each other (at least 5 cm away from each other) and in lower grade. The lesions from FAP1 and FAP2 were all spatially relatively close to each other and were at the late stage of carcinogenesis, resected in the second surgery from residual rectal cancer, leaving enough time for a precancerous clone to propagate. A larger sample size is needed to verify these hypotheses in the future. The consequence of field cancerisation is even worse in patients with FAP than in patients with sporadic CRC because every cell in patients with FAP has completed the first step in carcinogenesis via the inherited mutation in APC. From a clinical perspective, this observation implies that the initiation of cancerisation occurs long before the appearance of visible polyposis. Monitoring the rate of crypt fission events may help to prevent the initiation of potential lesions. In the future, quantitative investigation of the possible size of the field cancerisation zone will also help in the diagnosis and treatment of CRC. Investigating the normal regions interleaved between lesions in the field cancerisation zone will be helpful in identifying the important events in the initiation of early-stage adenomas.
Compared with the normal mucosa of patients without FAP, the normal mucosa of patients with FAP already exhibits enhanced metabolic processes and proliferative activity, which may be due to the long-term effects of inherited mutations in the APC gene. This result augments the necessity of monitoring the adjacent normal mucosa during the diagnosis and treatment of FAP. Metabolic reprogramming was reported to be one of the hallmarks of cancer, but whether metabolic reprogramming occurs at an early stage or a late stage during carcinogenesis has not been comprehensively analysed. By employing the scRNA-seq technique, we excluded the impact of nonepithelial cell types, such as immune cells, mesenchymal cells and endothelial cells, in tumours, and we found that metabolic reprogramming also occurs in adenomas and that the activity of the TCA pathway was even lower in adenomas than in carcinomas. The metabolic differences between adenomas and carcinomas imply that the reprogramming of metabolism, especially carbohydrate metabolism, may play important roles in tumour initiation as well as progression from adenoma to carcinoma.
In summary, we provided the first picture of the transcriptomic landscape of colorectal carcinogenesis in patients with FAP. The transcriptome dynamics during carcinogenesis were comprehensively investigated. In addition, we found that field cancerisation occurs in patients with FAP. These findings will advance our understanding of the pathogenesis of CRC.
Acknowledgments
This work was supported by a grant from Beijing Advanced Innovation Center for Genomics. Part of the analysis was performed on the High Performance Computing Platform of the Center for Life Science.
References
Footnotes
JL, RW, XZ and WW contributed equally.
Contributors FT, WF and XZ conceived the project. JL designed and performed the single cell RNA-seq, whole-exome sequencing and whole-genome sequencing experiments, with the help of XZ, SG, YM, XW, LG and HL. XZ performed the patient’s enrollment. XZ and WW performed tissue sampling and the H&E and immunohistochemical staining. RW conducted the bioinformatics analyses with the help of JL. HL and LG performed the histopathological reviews. JL and FT wrote the manuscript with the help from all authors.
Funding This study was funded by Beijing Advanced Innovation Center for Genomics.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Our study was approved by the Ethics Committee of Peking University Third Hospital (M2016170), and informed consent was signed by all involved patients before surgery.
Provenance and peer review Not commissioned; externally peer reviewed.