Article Text

Abstract
The current coronavirus pandemic is an ongoing global health crisis due to COVID-19, caused by severe acute respiratory syndrome coronavirus 2. Although COVID-19 leads to little or mild flu-like symptoms in the majority of affected patients, the disease may cause severe, frequently lethal complications such as progressive pneumonia, acute respiratory distress syndrome and organ failure driven by hyperinflammation and a cytokine storm syndrome. This situation causes various major challenges for gastroenterology. In the context of IBD, several key questions arise. For instance, it is an important question to understand whether patients with IBD (eg, due to intestinal ACE2 expression) might be particularly susceptible to COVID-19 and the cytokine release syndrome associated with lung injury and fatal outcomes. Another highly relevant question is how to deal with immunosuppression and immunomodulation during the current pandemic in patients with IBD and whether immunosuppression affects the progress of COVID-19. Here, the current understanding of the pathophysiology of COVID-19 is reviewed with special reference to immune cell activation. Moreover, the potential implications of these new insights for immunomodulation and biological therapy in IBD are discussed.
- inflammatory bowel disease
- inflammation
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
The current COVID-19 pandemic is an ongoing global health crisis due to severe acute respiratory syndrome coronavirus 2 and provides major challenges for patient care in IBD.
Severe patients with COVID-19 may suffer from progressive pneumonia, acute respiratory distress syndrome and multiorgan failure due to hyperinflammation and a cytokine storm syndrome; the interleukin-6R antibody tocilizumab is currently tested for clinical therapy of severe COVID-19.
The receptor ACE2 is highly expressed in ileal and colonic tissue and ACE2 levels are induced in patients with IBD.
There is currently no evidence for an increased frequency of COVID-19 cases in IBD; a recent study suggesting decreased disease activity in IBD requires further investigations.
Based on the currently available limited data, immunomodulatory and biological therapies can be continued in patients with IBD in remission; however, close attention to new results should be paid in the dynamic pandemic situation.
Future cases of patients with IBD suffering from COVID-19 should be documented in the new SECURE registry (https://covidibd.org/).
Introduction
The current coronavirus pandemic is an ongoing global health crisis due to COVID-19. This infectious disease represents a key challenge for gastroenterologists. One of the major questions relates to the clinical handling of COVID-19 in the presence of inflammatory disorders of the GI tract such as in patients with IBD, particularly in the presence of immunosuppressive or immunomodulatory therapy. Therefore, this review will address the following questions:
What is the evidence for intestinal COVID-19 disease and how does COVID-19 induce mucosal immune cell activation and a cytokine release syndrome?
Is there an increased or a decreased risk for patients with IBD to suffer from COVID-19?
What is the effect of corticosteroids and classical immunosuppressive agents such as methotrexate (MTX) and azathioprine on COVID-19?
What is the effect of biological agents and cytokine blockers on COVID-19?
COVID-19
A newly identified beta-coronavirus, denoted severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), caused a cluster of pneumonia cases in Wuhan, China, in December 2019.1–5 The disease caused by SARS-CoV-2 was subsequently named by WHO as COVID-19. Genome sequencing of SARS-CoV-2 showed that the virus is closely related (with 88% identity) to two bat-derived SARS-like coronaviruses, but displays more distant similarities to SARS-CoV (about 79%) and Middle East respiratory syndrome coronavirus (MERS-CoV) (about 50%).6 Structural analyses of SARS-CoV and SARS-CoV-2 identified 380 amino acid substitutions between these coronaviruses, which may cause functional and pathogenic divergence between these viruses.7 Another recent study suggested that SARS-CoV-2 uses mutations and recombination in different genomic regions including the membrane, envelope, nucleocapsid and spike glycoprotein regions to become a novel infectious agent.8 In fact, nine putative recombination patterns were identified in the spike glycoprotein, RdRp, helicase and ORF3a regions as well as six recombination regions in the S gene that may control evolutionary survival and antigenicity thereby allowing SARS-CoV-2 to cross borders from its original host (bats) to humans1 2 6 and to adapt to a human host.8
Several studies have analysed the mechanisms by which SARS-CoV-2 may enter the human host. These studies showed that the surface spike glycoprotein (S protein) on the surface of the virus binds to the receptor ACE2,9 10 a monocarboxypeptidase controlling cleaving of several peptides within the renin-angiotensin system that is highly expressed on pneumocytes (type 2) in the lung, particularly in smokers11 (figures 1–2). In this context, the cleavage of the S protein regulates viral uptake and is regulated by the cell surface-associated transmembrane protease serine protease 2 (TMPRSS2), a key enzyme for S protein cleavage and priming.12 13 Subsequently, the receptor binding domain of the S protein is recognised by the extracellular peptidase domain of ACE2 mainly through polar residues thus providing a molecular basis for coronavirus recognition and infection.9 10 Bioinformatical studies indicated that sequence mutation of SARS-CoV-2 with methylation of the 5’-cap structure may affect ACE2 binding and facilitate viral RNA escape recognition by the host innate immune system.14
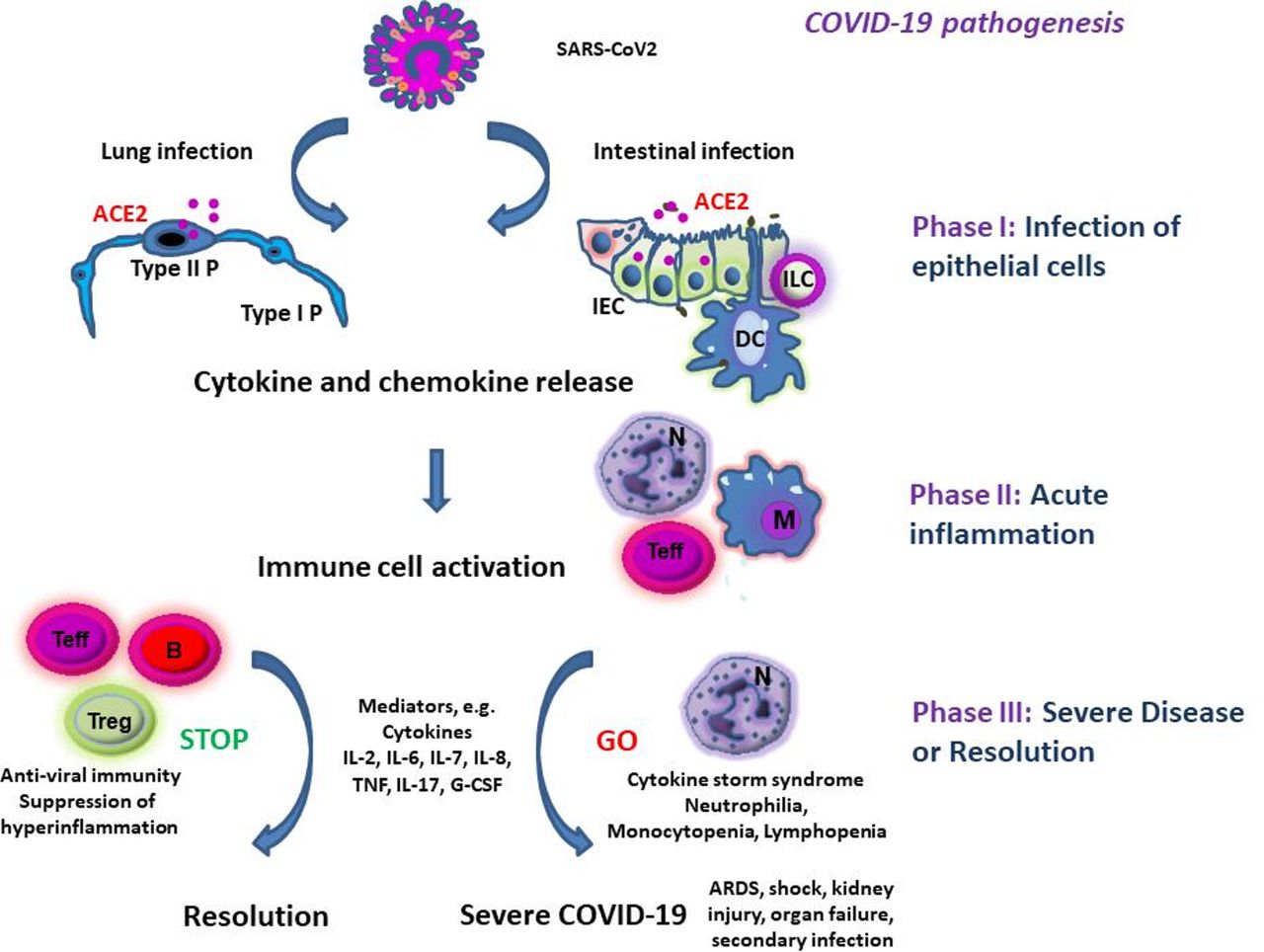
Hypothetical pathogenesis of COVID-19. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infects ACE2 expressing epithelial cells in the lung and/or the intestine. This is followed by production of mediators causing immune cell activation. Overwhelming immune cell activation may lead to severe complications including acute respiratory distress syndrome (ARDS), shock and kidney or multiorgan failure. B, B lymphocytes; IEC, intestinal epithelial cell; ILC, innate lymphoid cell; M, monocyte/macrophage; N, neutrophils; Teff, effector T cells; Treg, regulatory T cell; Type I P, type I pneumocytes; Type II P, type II pneumocytes.

The ACE/ACE2 receptor system (modified according to Zhang et al 52). The classical renin-angiotensin-aldosterone system ACE/angiotensin II/angiotensin type 1/2 receptor (AT1-R) and ACE2/Ang 1–7/MAS-1 receptor (MAS1-R) systems are shown.49–52 The ACE/AT1-R system has been mainly implicated in pro-inflammatory immune responses and tissue injury. In contrast, the ACE2/MasR system appears to play a key role in many anti-inflammatory pathways controlling tissue protection.
After membrane fusion between the virus and the host cell, the viral genome RNA reaches the cytoplasm followed by the translation of accessory and structural proteins. Newly generated envelope and nucleocapsid proteins as well as genomic RNA then form viral particle buds followed by the formation of virion-containing vesicles that may fuse with the cell membrane resulting in the release of viruses in the local microenvironment.10
Clinical symptoms in COVID-19 vary between patients but most individuals have a mild form of the disease with no or flu-like symptoms including dry cough, fever, a runny nose and fatigue. Additional symptoms may comprise shivering, throat pain, anosmia, headache, joint pain, nausea and diarrhoea. In more severe forms of the disease, marked inflammation and progressive pneumonia occur leading to difficulties in breathing. Finally, respiratory failure due to pneumonia, acute respiratory distress syndrome (ARDS) and multiorgan failure may occur in severe cases and are associated with high mortality in patients with COVID-191–4 (figure 1). The severity of the disease is modulated by co-factors such as the age and comorbidities.15 COVID-19 is more severe in older patients as well as in patients suffering from diabetes, hypertension or obstructive lung disorders. Finally, smokers were suggested to be 1.4 times more likely to have severe symptoms of COVID-19 and approximately 2.4 times more likely to be admitted to an intensive care unit, need mechanical ventilation or die compared with non-smokers. Thus, smoking is most likely associated with progression of COVID-19.16
The infection with SARS-CoV2 is currently diagnosed by SARS-CoV2 nucleic acid amplification tests using oropharyngeal swabs.1–4 However, the combination of the amplification test with CT scanning (figure 3) has been shown to improve the diagnosis of COVID-19. CT scans can detect disease even in the presence of a negative nucleic acid amplification test and a subgroup of patients with COVID-19 has pneumonia in spite of absent clinical symptoms.

{kind=link}
{kind=link}
{kind=link}
COVID-19 pneumonia in a medical doctor without known comorbidities detected by chest CT imaging. Two representative images demonstrating ground-glass opacities with consolidations are shown.
COVID-19 and the digestive system
ACE2 is expressed on pneumocytes of the lower airways possibly explaining the high frequency of pneumonia in affected patients.17–19 Although initial studies suggested that GI symptoms, such as diarrhoea (2%–10.1%), nausea and vomiting (1%–3.6%), are not very common in COVID-19,1 17–19 a recent study showed that GI symptoms are more frequently present than initially thought. In fact, analysis of 651 patients revealed that 11% of patients present with nausea, vomiting or diarrhoea. Severe types of COVID-19 were more frequently seen in patients with GI symptoms (23%) as compared with patients without GI symptoms (8%) consistent with the possibility that intestinal manifestation of COVID-19 may augment mucosal cytokine production and clinical outcome of the disease. A subgroup of patients with COVID-19 with GI symptoms also had significantly higher rates of fever, shortness of breath and headache, respectively.14 It was suggested that GI symptoms may cause patients with COVID-19 to be more prone to electrolyte disturbances including decreased serum sodium levels that may aggravate the disease.
A recent study in >100 outpatients with mild courses of COVID-19 demonstrated the presence of diarrhoea in approximately 30% of patients20 suggesting that diarrhoea may be a frequent hallmark of mild disease. The presence of diarrhoea could be due to direct infection of GI cells.21 In this regard, gastric, duodenal and rectal epithelial cells rather than cells in the oesophagus were shown to express the SARS-CoV2 receptor ACE2.22 COVID-19 led to infection of these cells followed by expression of the viral nucleocapsid protein indicating that SARS-CoV-2 may spread from infected to uninfected cells in the GI tract. Infection was not associated with marked macroscopic inflammation on endoscopy. However, in the lamina propria of stomach, duodenum and rectum, numerous infiltrating plasma cells and lymphocytes with interstitial oedema were seen in a COVID-19 patient22 indicating mucosal immune cell activation. In addition to local enteric infection, viraemia following lung infection may occur in few patients (approximately 1%) and may lead to a secondary attack of SARS-CoV2 on ACE2 target organs such as the kidney and the intestine.23
RNA from SARS-CoV-2 was detected in samples from faeces of patients with COVID-19,22 24 suggesting a possible faecal-oral transmission route. Moreover, infectious SARS-CoV-2 was isolated from stool of these patients indicating the presence of infectious virions in the GI tract that may contribute to viral spread. Thus, these observations suggest that SARS-CoV-2 may actively infect and replicate in the GI tract. Interestingly, in a subgroup of patients with SARS-CoV-2 (>20%), viral RNA in faeces could be detected even after negative conversion of the viral RNA in the respiratory tract.22 24 These findings are consistent with a model in which faecal-oral transmission may even occur after viral clearance in the airways. Future studies will have to confirm this pilot study and to determine whether testing of stool samples for SARS-CoV-2 is essential in COVID-19. The potential presence of SARS-CoV-2 in stool is critical for our understanding of COVID-19, as a recent study did not find any evidence for the presence of infectious virus in stool samples from affected patients in spite of the presence of SARS-CoV-2 RNA.25
Immune cell activation in COVID-19
There is overwhelming evidence that COVID-19 results in innate and adaptive immune cell activation in the infected host.23 26 The release of mediators and chemokines by infected cells rapidly leads to a local accumulation of neutrophils at the site of the infection. While such neutrophils may exert important antiviral effector functions, they also secrete cytokines and chemokines that attract further immune cells such as monocytes and T lymphocytes (figure 1). Thus, they may contribute to exaggerated immune responses and COVID-19 non-survivors had higher levels of neutrophils than survivors.19 Also T-cell activation is present in patients with COVID-19. While CD4+ T cells are important in providing B cells help and mediating protective humoral immunity by stimulating B cells to produce virus-specific antibodies, CD8+ T cells have been suggested to provide key cytotoxic function and to limit local infection. Insufficient clearance of the infection, however, leads to progressive hyperinflammation. In fact, many patients with COVID-19 with progressive pneumonia, ARDS and multiorgan failure suffer from hyperinflammation and a cytokine storm syndrome.27–29 It has been suggested that this cytokine storm in COVID-19 may lead to secondary haemophagocytic lymphohistiocytosis, a hyperinflammatory disorder triggered by viral infections that is associated with multiorgan failure and high lethality.27 30 31 This hyperinflammation may be particularly deleterious in older patients with T-cell immunosenescence; a chronic low-grade proinflammatory state of the immune system that affects many older individuals, even when they are apparently healthy and free of risk factors.32
Various recent studies described the immune cell dysregulation in COVID-19 by using peripheral blood mononuclear cells. A study in 286 patients with severe COVID-19 demonstrated that affected individuals exhibit lower lymphocyte counts, higher neutrophil counts, an elevated neutrophil-lymphocyte ratio as well as decreased percentages of monocytes, eosinophils and basophils.33 Neutrophilia has been suggested as risk factor for the development of ARDS and progression from ARDS to death in COVID-19.15 Additionally, a reduction of NK cell percentages was noted in COVID-19, while B cells were within the lower level of normal range.4 34 Lymphocytopenia was a hallmark finding in patients with severe COVID-19 cases and present in 83.2% of the patients on admission.4 Both CD4+ and to a lesser extent CD8+ T-cell numbers were reduced in severe COVID-19 cases, although the CD4+/CD8+ T-cell ratio remained normal. Moreover, within the CD4+ T-cell subset, decreased numbers of effector memory T cells (CD45RO+) and regulatory T cells (CD25+CD127 low) were noted, while the percentage of naive T cells (CD45RA+) increased.33 The higher naïve/memory T-cell ratio in severe as compared with mild COVID-19 cases suggested marked immune cell alteration in these patients. Moreover, the suppression of regulatory T cells in severe cases indicated insufficient counterregulation of proinflammatory immune responses that may further aggravate hyperinflammation and tissue injury. With regard to B-cell activation, total levels of IgA, IgG and IgM were similar between severe and mild COVID-19 cases suggesting no major general impairment in B-cell activity. However, no tests for protective antibodies in serum were made in this study. Finally, no differences in complement levels were noted between both groups.33
Analysis of inflammation markers in serum showed elevated levels of procalcitonin, C reactive protein, D-dimer and ferritin in COVID-19.15 33 Moreover, in severe cases, significantly higher serum levels of tumour necrosis factor (TNF), interleukin (IL)-2R, IL-6, IL-8 and IL-10 were detected as compared with milder cases, while IL-1beta levels and the number of IFN-gamma producing CD4+ and CD8+ T cells remained unchanged.33 A comparison of cytokine and chemokine levels among COVID-19 intensive care unit (ICU) patients (n=13), non-ICU patients (n=28) and healthy controls further defined the plasma changes associated with the cytokine storm syndrome.35 ICU patients had significantly higher levels of IL-1beta, IL-1ra, IL-2, IL-4, IL-6, IL-7, IL-8, IL-9, IL-10, IL-13, IL-17, fibroblast growth factor, granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage-colony stimulating factor, interferon (IFN)-gamma, TNF, vascular endothelial growth factor, IP-10, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1alpha, platelet-derived growth factor (PDGF) and MIP-1beta than healthy controls, while levels of eotaxin, regulates upon activation normal T cell expressed and secreted (RANTES), IL-5, IL-12 and IL-15 were unchanged. This finding is probably due to production of cytokines and chemokines by numerous cells including epithelial cells, neutrophils, monocytes and T cells and mirrors the broad overwhelming hyper-responsiveness of the immune system in COVID-19 (figure 1). Moreover, ICU patients had significantly higher levels of IL-2, IL-7, IL-10, TNF, G-CSF, MCP-1 and MIP-1a than non-ICU patients indicating that these cytokines with known immunoregulatory and/or chemotactic functions may regulate the progression of the disease. Although IL-6 levels were not significantly higher in the ICU group versus the non-ICU group, another study in 43 patients suggested that the combination of D-dimer and IL-6 differentiates between mild and severe COVID-19 cases. Based on these findings, IL-6 was suggested as potential biomarker of disease activity.36
COVID-19 and IBD
The COVID-19 receptor ACE2 is particularly highly expressed in intestinal epithelial cells from the terminal ileum and to a lesser extent in the colon, where mucosal inflammation in patients with IBD (Crohn’s disease (CD); UC) is frequently detected.37 38 In this context, ACE may act as a co-receptor for nutrient uptake, in particular for amino acid resorption from food.39 Moreover, immunohistochemical analyses revealed higher ACE2 protein expression in inflamed IBD samples from the terminal ileum and colon as compared with controls (no differences between inflamed and non-inflamed regions). Furthermore, ACE2 activity in the colon was elevated in non-inflamed colon in IBD as compared with controls and active IBD. Finally, the average expression of soluble ACE2 was shown to be increased in patients with IBD (mainly in CD) and a higher ACE2:ACE ratio in plasma was noted in patients with IBD as compared with controls without IBD.38 40 Cytokines expressed in IBD, such as IFN-gamma, can potentially induce ACE2 expression by cytokine signalling events driving ACE2 promoter activity consistent with the idea that mucosal inflammation may increase expression of ACE2. A study using tandem mass tag-based shotgun proteomics additionally showed higher levels of ACE2 in inflamed intestinal areas in patients (CD) as compared with patients with UC.41 Finally, the fusion of SARS-CoV2 with the host cell membrane is critical for uptake in cells and is modulated by the S protein. Activation of the S protein via proeolytic cleavage is controlled by host trypsin-like proteases, whose activity is upregulated in IBD, and this effect might facilitate infection in patients with IBD.42 Collectively, these findings suggested the possibility that patients with IBD might be particularly susceptible to COVID-19. However, there is no evidence so far that patients with IBD are highly susceptible to COVID-19. In contrast, a recent study from Wuhan studied 318 patients with IBD (204 UC and 114 CD) during the local outbreak of the disease and did not report any COVID-19 cases.43 The reasons for this observation are not entirely clear but might relate to the local adjustment of protective measures to prevent infection, the particular awareness of the IBD patient cohort to hygiene and infection prevention and the modulation of immunosuppressive therapy (eg, stop of treatment with immunomodulators and biologicals). Alternatively, patients with IBD might be less susceptible to COVID-19 and further studies in this regard are highly warranted.
Is there a theoretical rationale to assume that patients with IBD might be less susceptible to inflammation? There is currently no direct evidence that altered expression of ileal and colonic ACE2 and TMPRSS2 directly regulates viral entry in IBD and expression levels of these proteins may depend on the local inflammatory activity and the degree of epithelial damage. Further studies are essential to address these points. However, in addition to the full-length, transmembrane ACE2 protein on the surface of epithelial cells, a soluble form of ACE2 can be generated by proteolytic cleavage that lacks the membrane anchor and circulates in small amounts in the blood.44 It has been suggested that the soluble form of ACE2 acts as a competitive binding partner for SARS-CoV and thus sequestrates viruses and prevents their binding to the cellular full-length ACE2 protein.45 46 Interestingly, the cleavage of membrane ACE2 into the soluble form is regulated by the TNF-α convertase ADAM17, a protease upregulated in patients with active IBD.47 Consistently, elevated levels of ACE2 were seen in plasma from patients with IBD,38 40 suggesting the possibility that increased levels of soluble ACE2 may limit infection progression and modulate susceptibility to infection.48 Additionally, expression of ACE2 in IBD may control anti-inflammatory immune responses, as discussed in the next paragraph.
The ACE2 receptor has been shown to be part of the renin-angiotensin-aldosterone system (RAAS) that comprises the classical RAAS ACE/angiotensin II/angiotensin type 1/2 receptor (AT1-R) and ACE2/Ang 1–7/MAS-1 receptor (MAS1-R).49–52 While the ACE/AT1-R system has been mainly implicated in pro-inflammatory immune responses and tissue injury, the ACE2/MasR system plays a crucial role in many anti-inflammatory pathways controlling tissue protection (figure 2). Functional studies in murine experimental colitis models indicated that modulation of ACE2 expression may affect the severity of colitis activity. In fact, deficiency in the ACE2 cleavage controlling protein ADAM17 aggravated dextran sulphate sodium (DSS) colitis activity.53 Moreover, ACE2 expression was shown to be elevated in the colon during experimental DSS colitis as compared with controls.50 A chemical inhibitor of ACE2 (GL1001) reduced DSS colitis severity54 suggesting that ACE2 plays a pathogenic role in colitis. In contrast, ACE2 deficiency caused enhanced susceptibility to DSS colitis probably due to altered immune cell trafficking, changes in innate immunity and cytokine production as well as alterations of the gut microbiota.39 In this study, dietary tryptophan primarily absorbed via an ACE2-dependent epithelial transport pathway regulated the expression of antimicrobial peptides via mammalian target of rapamycin (mTOR), either directly through nutrient sensing and/or through the tryptophan-nicotinamide pathway. The findings suggested a direct link between dietary amino acid metabolism and innate immunity, the composition of the intestinal microbiota, the susceptibility to colitis and malnutrition. Collectively, these results were consistent with a model in which ACE2 is protective in experimental colitis. Consistently, angiotensin (Ang 1–7) ameliorated colitis activity in the DSS colitis model, while blockade of the MAS-1 R aggravated disease, indicating a protective role of the ACE2/Ang1-7/MAS-1 R pathway.
Immunotherapy in IBD: potential effects on COVID-19
Several immunotherapies in patients with IBD have been associated with an increased risk of infections as they may inhibit intracellular signalling cascades needed for the host to fight pathogens.55–57 Corticosteroids, for instance, are potentially associated with an increased risk for infections and have been widely used in studies from China for treatment of ARDS and hyperinflammation in COVID-19. Although these studies were not designed to evaluate the effects of steroids, one study even suggested that treatment with methylprednisolone decreases the risk of death in COVID-19.15 However, as corticosteroids suppress lung inflammation but may also inhibit antiviral immune responses and pathogen clearance, the use of steroids during COVID-19 is controversial.58 Studies during previous coronavirus outbreaks in MERS and SARS suggested delayed viral clearance and potential complications such as diabetes and psychosis in patients receiving high doses of corticosteroids. In the absence of controlled clinical trials on the use of corticosteroids, interim guidelines from WHO advised against the use of steroids unless indicated for another reason.58 In agreement with this concept, a recent uncontrolled study in COVID-19 suggested that patients with high steroid use (n=9) have significantly worse clinical outcomes (ARDS, shock, kidney injury, continuous renal replacement, secondary infection) than patients without steroid use (n=32).35 However, no data on low-dose and short-term steroid use are currently available. Thus, the potential advantages and disadvantages of steroid use should be carefully considered in individual patients until more data become available. Thiopurine (azathioprine, 6-mercaptopurine) and JAK1/3 inhibitor (tofacitinib) treatment can potentially reduce the number of activated T cells and affect T-cell activation and effector function. Although no data are currently available in this context in IBD, this might affect the course of COVID-19, as lymphopenia was associated with worse prognosis in this disease.15 Moreover, tofacitinib has an increased risk for certain viral infections (eg, herpes zoster infection).59–61 Thus, treatment indication needs to be discussed in individual cases. At this point, no evidence for stopping these treatments in patients with remission exists. Similarly, there are no specific data available on MTX.
Anti-TNF antibodies are frequently used for IBD therapy.62–64 As TNF inhibition may potentially affect antiviral immunity and has been shown to affect hepatitis B virus reactivation, TNF blockade could regulate the susceptibility to COVID-19. However, analyses of TNF levels in COVID-19 led to different results. One study showed no effect on TNF levels in severe COVID-19 cases in spite of the regulation of other proinflammatory cytokines.65 In contrast, another study reported that COVID-19 ICU patients had significantly higher serum levels of TNF than non-ICU patients.35 TNF may also exert pathogenic effects in COVID-19 by augmenting the expression of ACE2 or by augmenting lymphopenia through induction of direct leucocyte death via TNF/TNFR1 signalling in T cells.66 These findings argue for a potentially protective effect of TNF inhibition in COVID-19 and further studies are needed to address this point.
The alpha4/beta7-specific antibody vedolizumab blocks immune cells homing by preventing the interaction between these integrins and MAdCAM-1, thereby preventing immune cell trafficking and suppressing mucosal inflammation in patients with IBD.67–69 Direct effects have been shown on homing of T helper (Th)1, Th2, Th9, Th17 and Treg cells to the inflamed intestine.70 71 No marked increase of viral infections was noted in patients with IBD receiving vedolizumab therapy.72 Moreover, no viral reactivation was seen in patients with hepatitis. Furthermore, vedolizumab treatment in addition to antiretroviral therapy led to sustained virological control in SIV-infected macaques.73 These findings encouraged a phase I clinical trial of vedolizumab in patients with HIV and this study did not show effects on the viral load in this disease on antibody therapy.74 75
Ustekinumab is a cytokine antibody blocking the p40 subunit of the two cytokines IL-12 and IL-23. This antibody has been approved for clinical therapy of patients with IBD.76–79 Mechanistically, this antibody prevents Th1 T-cell priming via IL-12 and simultaneously inhibits perpetuated activation of IL-23 responsive Th17 cells.80–84 No increase in viral infections in patients with IBD has been noted on ustekinumab therapy. It is tempting to speculate that IL-23 blockade in COVID-19 may suppress the pathogenic Th17 response in the cytokine storm syndrome, although no studies have addressed this concept so far. Thus, there are currently no major concerns on the use of this antibody for clinical therapy in patients with IBD due to the current COVID-19 risk situation.
Although cytokine blockers and Jak inhibitors have raised theoretical concerns with regard to IBD therapy, it should be noted that these agents are currently considered for clinical therapy of COVID-19 cases with hyperinflammation and ARDS.27 Jak inhibitors blocking Jak2 have been suggested to block viral entry and the Th17 part of the cytokine storm syndrome without altering interferon signalling (this has not been demonstrated for the Jak1/3 inhibitor tofacitinib so far).85 Additional studies suggested potential benefits of cytokine blockade in sepsis. Re-analysis of a phase III trial in sepsis suggested that blockade of IL-1 signalling via anakinra may result in significant survival benefits in patients with hyperinflammation and elevated cytokine production.86 Moreover, TNF inhibition has been suggested to be helpful in selected patients with hyperinflammation and high IL-6 levels in sepsis.87 Finally, the IL-6R antibody tocilizumab is currently considered for therapy of hyperinflammation and ARDS in COVID-19 and a prospective phase III trial has been initiated (www.clinicaltrialsarena.com/new/roche-actemra-covid-19-trial) to clarify its effects on cytokine storm syndromes in the disease. This antibody had been previously tested in a small phase II trial in CD and the results suggested potential efficacy and safety.88 However, blockade of IL-6 signalling has recently raised concerns in CD, as IL-6 antibodies (PF-04236921) led to reduction of inflammation but the formation of abscesses and perforation in a subgroup of patients with IBD.89
In summary, there is currently no evidence for an increased risk or aggravated outcomes in patients with IBD in the context of COVID-19. A special COVID-19 risk situation might be present in patients with IBD who are pregnant. However, no clinical cases have been published so far. In general, few studies on the effect of COVID-19 on pregnancy are available and it has been suggested that it is necessary to carefully monitor suspected pregnant women before and after delivery as well as newborn children in this context.90 91 Other COVID-19 risks situation comprise older patients with IBD with comorbidities as well as patients suffering from malnutrition who may be at risk for infections and severe courses of the disease, respectively.92–95 Furthermore, dependent on the local situation, there could be an increased risk for COVID-19 in the context of IBD surgery in the hospital. Finally, experimental COVID-19 treatment with hydroxychloroquine or remdesivir may increase the risks for drug-drug interactions with established IBD medications (www.drugs.com: potentially increased risk of combination therapy with hydroxychloroquine and adalimumab/infliximab for nerve damage). With regard to the effect of IBD on COVID-19, it should be pointed out that further studies are required in this highly dynamic situation. There is no evidence to suggest that patients with IBD should discontinue IBD-specific medications. However, older patients with IBD with comorbidities such as diabetes mellitus, obstructive lung disease, coronary heart disease and hypertension might have an increased risk for COVID-19 and further studies are urgently needed to address this point. In this context, there is an ongoing international programme initiated by the International Organization for the Study of IBD to register COVID-19 cases in patients with IBD in the SECURE registry (https://covidibd.org/) in order to obtain further insights into the disease susceptibility and effect of immunomodulatory therapy. Currently available recommendations for patients with IBD and medical doctors are provided in boxes 1 and 2.
Recommendations for managing patients with IBD during the COVID-19 pandemic
Consider potential risk situations for severe acute respiratory syndrome coronavirus 2 infection
Patients with inflammatory bowel disease (IBD) on immunosuppressive agents.
Patients with active-stage IBD with malnutrition
Elderly patients with IBD
Patients with IBD frequently visiting medical clinic
Patients with IBD with underlying health conditions, such as hypertension and diabetes mellitus
Patients with IBD who are pregnant
Patients receiving experimental COVID-19 therapy plus IBD specific medications (potential drug drug interactions).
Medication for patients with IBD
Continue current treatment if disease is stable and discuss suitable medicine if disease has flared.
Use of mesalamine should be continued and should not increase the risk of infection.
Corticosteroid use can be continued, but be cautious of possible side effects.
A new prescription of immunosuppressant or increase in dose of an ongoing immunosuppressant is not recommended in epidemic areas.
Use of biologics such as the antitumour necrosis factors infliximab or adalimumab should be continued.
If infliximab infusion is not accessible, switching to adalimumab injection at home should be considered.
Vedolizumab use can be continued due to the specificity of the drug for the intestine.
Ustekinumab use can be continued, but starting ustekinumab requires infusion centre visits and therefore should be discussed before initiation of therapy.
Enteral nutrition might be used if biologics are not accessible.
Tofacitinib should not be newly prescribed in epidemic areas unless there are no other alternatives.
Surgery and endoscopy
Postpone elective surgery and endoscopy.
Consider screening for COVID-19 (complete blood count, nucleic acid detection and chest CT) before surgery.
Patients with IBD and fever
Fever is the most common reported symptom in COVID-19.
Contact your IBD doctor about potential option to visit outpatient clinic with personal protection provisions if temperature continues over 38°C.
Suspend the use of immunosuppressant and biological agents after consultation with your IBD doctor, and follow appropriate local guidance for suspected COVID-19 if COVID-19 cannot be ruled out.
Modified according to Mao et al.96
Recommendations for IBD centres in COVID-19 risk situations
Advise patients to strictly follow the WHO recommendations for COVID-19 prevention
Wash your hands regularly.
Keep potentially contaminated surfaces clean.
Respect social distancing.
Avoid to touch eyes, nose and mouth.
Stay home if you feel unwell.
Wear a mask to avoid infecting other people.
Reduce the risk of hand contamination (eg, by wearing gloves).
Avoid to use public toilets.
Consider to restructure your IBD patient care under consideration of the local situation
Strictly separate areas for care of patients with suspected/proven COVID-19 and patients with IBD in the hospital.
Assign-specific physicians to inpatient care, endoscopy and remote monitoring.
Convert multidisciplinary team meetings into virtual meetings.
Consider to avoid elective surgery.
Avoid crowding in the waiting area of the outpatient unit.
Consider to wear personal protective equipment and follow the WHO recommendations to prevent any contamination.
Consider to limit the number of patients in the outpatient clinic by focusing on infusion treatment.
Secure home delivery and adequate drug supply to all patients given subcutaneous and oral drugs.
Provide help in individual cases by using emails and telephone calls.
Send newsletters or information material by email.
Secure communication between local gastroenterologists and primary care doctors with your centre.
Consider alternative and safer ways of administration and remote monitoring of patients with IBD.
Modified according to Danese et al and Fiorino et al.97 98
References
Footnotes
Funding This study was funded by Deutsche Forschungsgemeinschaft (DFG SFB 643; SFB1181; TRR241; FOR2438; KFO257).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.