Article Text

Abstract
Objectives Lipid mediators in the GI tract regulate satiation and satiety. Bile acids (BAs) regulate the absorption and metabolism of dietary lipid in the intestine, but their effects on lipid-regulated satiation and satiety are completely unknown. Investigating this is challenging because introducing excessive BAs or eliminating BAs strongly impacts GI functions. We used a mouse model (Cyp8b1–/– mice) with normal total BA levels, but alterations in the composition of the BA pool that impact multiple aspects of intestinal lipid metabolism. We tested two hypotheses: BAs affect food intake by (1) regulating production of the bioactive lipid oleoylethanolamide (OEA), which enhances satiety; or (2) regulating the quantity and localisation of hydrolysed fat in small intestine, which controls gastric emptying and satiation.
Design We evaluated OEA levels, gastric emptying and food intake in wild-type and Cyp8b1–/– mice. We assessed the role of the fat receptor GPR119 in these effects using Gpr119–/– mice.
Results Cyp8b1–/– mice on a chow diet showed mild hypophagia. Jejunal OEA production was blunted in Cyp8b1–/– mice, thus these data do not support a role for this pathway in the hypophagia of Cyp8b1–/– mice. On the other hand, Cyp8b1 deficiency decreased gastric emptying, and this was dependent on dietary fat. GPR119 deficiency normalised the gastric emptying, gut hormone levels, food intake and body weight of Cyp8b1–/– mice.
Conclusion BAs regulate gastric emptying and satiation by determining fat-dependent GPR119 activity in distal intestine.
- appetite
- bile acid
- gastric emptying
- obesity
- small intestine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hydrolysed intestinal lipids induce satiety and satiation.
The subset of bile acids that are 12α-hydroxylated are synthesised at a higher rate in obesity and insulin resistance.
Deleting Cyp8b1, the enzyme required to produce 12α-hydroxylated bile acids, impairs intestinal lipid absorption in mice.
What are the new findings?
Lowering 12α-hydroxylated bile acids in mice slows lipid hydrolysis and allows lipid access to the lower intestine, which slows gastric emptying and reduces food intake.
The fat receptor GPR119 mediates this lipid-sensing pathway.
The effects of this pathway on gastric emptying require intact signalling through the gut hormones GLP-1 and PYY.
How might it impact on clinical practice in the foreseeable future?
Cyp8b1 inhibition is a potential target for obesity and diabetes treatment.
Introduction
Feeding behaviour is governed in the brain and in the GI tract. Infusion of fat directly into duodenum potently decreases food intake,1–4 indicating that fat sensing in the GI tract is a vital element to induce hypophagia. There are at least two mechanisms by which intestinal fat suppresses food intake. One is initiated within enterocytes of the jejunum, where dietary fat is converted into fatty acid ethanolamides (FAEs), such as oleoylethanolamide (OEA). Jejunal OEA is a bioactive signalling lipid that induces satiety—defined as an increase in the time interval between meals—via PPARα activation and the vagally mediated gut–brain axis.1 4–6 A second mechanism is initiated by the presence of dietary fat in the intestine, particularly the lower intestine, which slows gastric emptying. This results in gastric distention, enhances the perception of fullness and results in satiation—defined as a decrease in meal size.5 This satiation is expected to be mediated partly by enteroendocrine cells of the lower intestine, which secrete gut hormones such as GLP-1 and PYY in response to the products of dietary triglyceride hydrolysis: free fatty acids (FFA) and 2-monoacylglycerol (2-MAG). Hormone secretion in response to FFA and 2-MAG is expected to be carried out by G protein–coupled receptors (GPCRs).7–9 GPR119 is one candidate receptor, and it can be activated by 2-MAG.7 8 Thus, intestinal lipids derived from dietary fat have the potential to induce at least two pathways—one in the jejunal intestine via OEA-PPARα and one in the lower intestine via 2-MAG-GPR119—to promote both satiety and satiation. Existing experimental paradigms cannot easily distinguish which of these pathways is dominant in the regulation of total food intake.
Bile acids (BAs) emulsify dietary lipid, which allows it to be efficiently hydrolysed and absorbed in the proximal intestine. Data from mice demonstrate that among endogenous BA species, this lipid absorption function is most effectively carried out by cholic acid (CA).10–13 We reasoned that the composition of endogenous BAs determines whether fat is absorbed efficiently in the upper small intestine or whether unabsorbed lipid species, such as 2-MAG, enter the distal intestine where they may activate enteroendocrine cell–induced satiation. BAs also have an allosteric function in which they can bind within the substrate binding pocket of, and stabilise the activity of, the enzyme N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD), which produces appetite-regulating lipid mediators such as OEA and arachidonolyethanolamide (AEA).14 15 This stabilising function is most effectively carried out by deoxycholic acid (DCA). Thus, BAs may contribute to intestinal lipid sensing in multiple ways, but their roles are incompletely defined.
Others and we have previously reported that mice lacking the 12α-hydroxylase Cyp8b1 have no decrease in the total levels of BAs, but an altered BA composition, with a lack of the 12α-hydroxylated BAs CA and DCA, and their conjugated forms.12 13 These mice also have impaired absorption of dietary cholesterol and triglycerides.10–13 Because Cyp8b1–/– mice have normal total levels of BAs, this impairment in lipid absorption is due to the lack of 12α-hydroxylated (12-OH) BAs and the inefficiency of the remaining non-12-OH BAs to promote lipid absorption. 12-OH BAs comprise CA, DCA and their conjugates; non-12-OH BAs include muricholic acids (MCAs), chenodeoxycholic acid (CDCA) and their conjugates. The consequence of this impairment is that a small amount of dietary fat travels the full length of the GI tract, ultimately appearing in the faeces. We reasoned that Cyp8b1–/– mice may be a new tool to (1) dissect the role(s) of BAs on intestinal lipid sensing and (2) investigate the relative contributions of different intestinal lipid sensing pathways on food intake.
Materials and methods
Animal experiments
Cyp8b1–/– and GPR119–/– mice are on the C57BL/6 genetic background (Taconic #11784 and #TF1293). We crossed Cyp8b1–/– mice with GPR119–/– mice to obtain double knockout mice, and used littermate male mice between 8 and 15 weeks of age for experiments. Mice were fed a normal chow diet (3.4 kcal/g, Purina 5053, 24.7% kcal from protein, 62.1% carbohydrate and 13.2% fat), a fat-free diet (3.3 kcal/g, Envigo TD 03314, 24.2% kcal from protein and 75.8% from carbohydrate) or a high-fat diet (5.21 kcal/g, Research Diets D12492, 20.0% kcal from protein, 20.0% carbohydrate and 60.0% fat). Mice were provided with the diet and water ad libitum and maintained on a 12-hour light/dark cycle, set with lights on at 7:00. For food intake measurements, mice were individually housed, and the food dispenser was located inside the cage.
Detailed methods can be found in supplementary materials and methods.
Supplemental material
Statistics
Results are presented as mean±SEM. Data were analysed by one-way and two-way ANOVA with Tukey’s multiple comparisons test or Student’s t-test.
Results
Reduced food intake induces low body weight of Cyp8b1–/– mice
Cyp8b1–/– mice maintained on standard chow diet showed a ~10% reduction in body weight compared with wild-type (WT) mice, which was attributable to small but statistically significant decreases in both adiposity and lean mass (figure 1A–F).10 11 13 Ad libitum chow intake of Cyp8b1–/– mice was slightly decreased (figure 1G) relative to WT controls. Indirect calorimetry experiments showed no differences in respiratory exchange ratio, oxygen consumption, heat production or activity between genotypes (online supplementary figure 1A–D). To test whether this level of food intake was sufficient to induce the low body weight of Cyp8b1–/– mice, we performed a pair-feeding experiment. Body weights of pairfed WT mice decreased to the level of the Cyp8b1–/– mice (figure 1H). Taken together, these data support the suggestion that reduced food intake contributed to the lower body weight of Cyp8b1–/– mice. Accordingly, we then examined the putative mechanisms underlying this reduced food intake in Cyp8b1–/– mice.
Supplemental material

Body weight, mass composition and pair feeding experiment in WT and Cyp8b1–/– mice. (A) Body weight of chow diet-fed mice (n=8 for each group). **p<0.01 (Student’s t-test). (B) Fat mass, (C) fat %, (D) lean mass, (E) lean % and (F) fluid mass (n=18 for each group). **p<0.01 (Student’s t-test). (G) Ad libitum food intake for 24 hours (n=8 for each group). (H) Body weight in pair feeding experiment (n=4 for each group). **p<0.01 WT vs Cyp8b1–/–, #p<0.05, WT vs paired-WT, ##p<0.01, WT vs paired-WT (two-way repeated ANOVA).
Cyp8b1-driven 12-OH BA production is required for jejunal but not hypothalamic OEA synthesis
Recent studies indicate that BAs stabilise and activate the enzyme NAPE-PLD.14 15 NAPE-PLD produces FAEs, such as OEA and AEA, that regulate food intake via upper small intestinal and hypothalamic signalling.4 6 16–19 To investigate the involvement of NAPE-PLD in the hypophagia of Cyp8b1–/– mice, we measured its products in jejunal and hypothalamic tissue in 16-hour fasted and 5-hour refed states. In jejunal epithelium, we found no differences between genotypes in AEA, docosahexaenoyl ethanolamide (DHEA), 2-arachidonyl glycerol (2-AG) or docosahexaenoylglycerol (DHG) (figure 2B–E). However, the robust induction of OEA after refeeding in WT mice was significantly blunted in Cyp8b1–/– mice (figure 2A). These data are consistent with the finding that DCA, a 12-OH BA, is better than other BA species at promoting NAPE-PLD activation.15

Effects of fasting and refeeding on fatty acid ethanolamides and cannabinoids in jejunal intestine and hypothalamus. Effect of 16-hour food deprivation (Fast) and 5-hour refeeding (Refed) on the levels of OEA (A, F), AEA (B, G), DHEA (C, H), 2-AG (D, I) and DHG (E, J) in jejunum (A–E) and hypothalamus (F–J) (n=4 for each group). *p<0.05, **p<0.01 (two-way repeated ANOVA).
In contrast to the jejunum, we found no differences between genotypes in levels of AEA, OEA or DHEA in mediobasal hypothalamus (figure 2F–H). We also found no differences in hypothalamic levels of 2-AG or DHG (figure 2I–J). Thus, we conclude that Cyp8b1 ablation does not affect NAPE-PLD activity in hypothalamus.
Jejunal OEA induces satiety by increasing the inter-meal interval in a manner dependent on PPARα activation.4 We evaluated the expression of PPARα and its target genes in the jejunum.20–22 There was no difference in the expression of Pparα, Abca1, Acox1, Apob, Col1a, Cpt1a or Dpt between the genotypes in jejunal mucosal and muscle tissue (online supplementary figure 2A,B). Next, to assess satiety and satiation, we evaluated meal patterning. Cyp8b1–/– mice showed significantly decreased meal size (online supplementary figure 3A). However, there was no difference in meal duration, frequency or post-meal interval (online supplementary figure 3B–D). Overall, these data do not support a role for the OEA-PPARα–satiety pathway in the hypophagia of Cyp8b1–/– mice.
Supplemental material
Supplemental material
Impaired dietary triglyceride absorption in Cyp8b1–/– mice is due to impaired hydrolysis
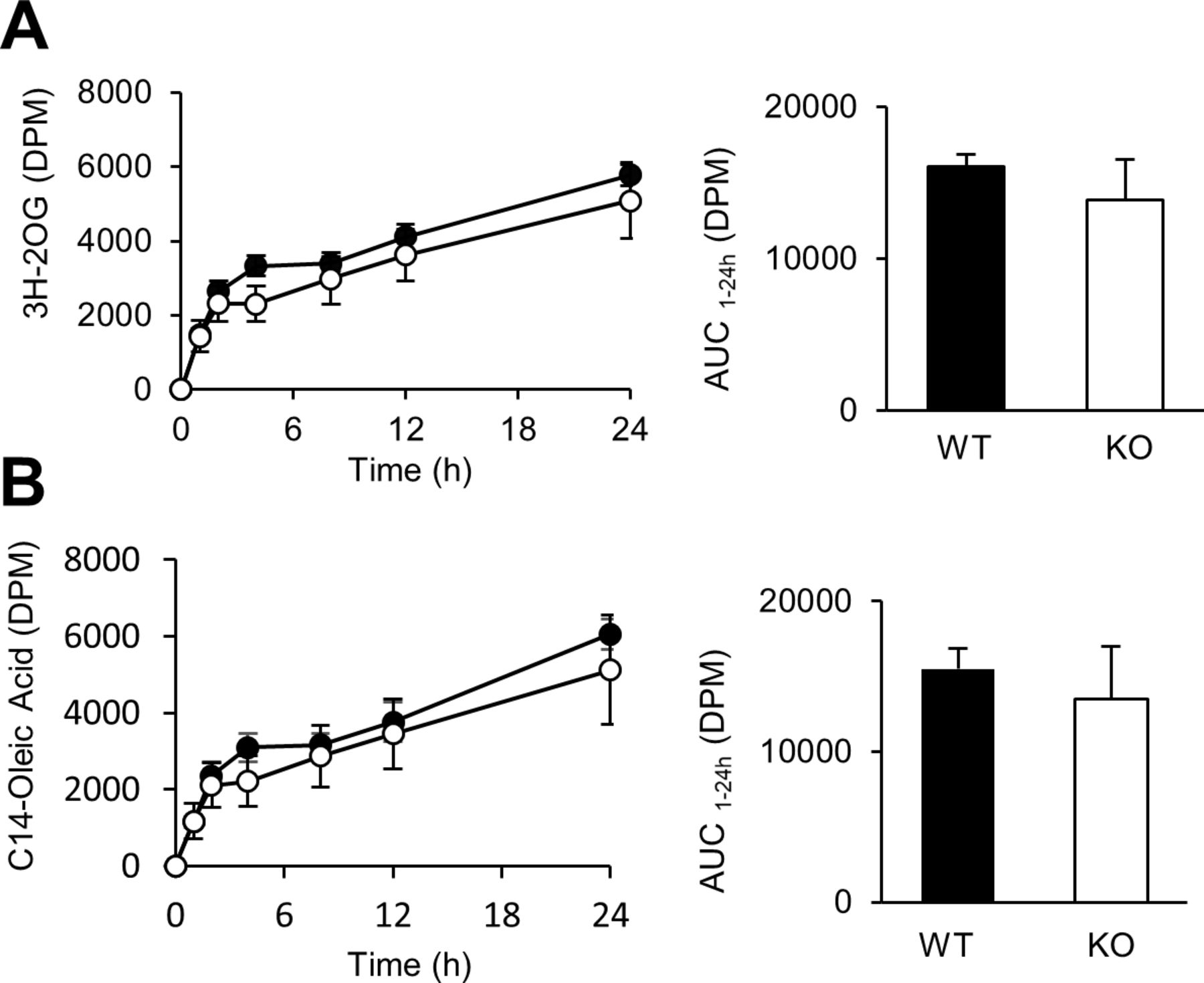
Our second hypothesis was that the hypophagia of Cyp8b1–/– mice was due to activation of lipid receptors in enteroendocrine cells of the lower intestine because of inefficient absorption of dietary lipids in proximal intestine. Previously, we and others have reported that dietary lipid absorption is impaired in Cyp8b1–/– mice.10–13 Our prior tracer analysis showed that in Cyp8b1–/– mice, absorption of an intact triglyceride ([3H]-triolein) is impaired and can be rescued by replacing the key missing BA, taurocholic acid.13 As a result of this impairment, small amounts of radiolabelled FFA and MAG travel the full length of the intestine, and are detected in faeces of Cyp8b1–/– mice.13 Lipidomics from our study13 confirm this, and data from Kaur and colleagues show that Cyp8b1–/– mice have higher levels of FFAs in the ileal lumen.11 These findings suggest two possibilities. The first is that triglyceride hydrolysis occurs normally in Cyp8b1–/– mice, but the altered bile acid composition is ineffective at ferrying hydrolysed fats across the unstirred water layer and permitting enterocyte uptake. The second possibility is that absorption per se of hydrolysed lipids is normal, but hydrolysis is slowed due to inefficiency, allowing fats to continue travelling down the alimentary tract. To distinguish between these, we tested the kinetics of absorption of radiolabelled versions of the products of triolein hydrolysis, namely [14C]-oleic acid and [3H]-2-oleoylglycerol. We found Cyp8b1–/– mice have normal absorption of oleic acid and 2-oleoylglycerol (figure 3A–B), supporting the second possibility described above. These findings suggest that Cyp8b1 deficiency in mice causes inefficient hydrolysis of intestinal triglyceride.

Absorption of hydrolysed lipids. Plasma disintegrations per minute (DPM) derived from (A) [3H]-2-oleoyl glycerol (2-OG) and (B) [14C]-oleic acid in WT and Cyp8b1–/– mice (n=5 for each group).
Dietary fat induces slow gastric emptying and low food intake in Cyp8b1–/– mice
Lipids in the lower small intestine suppress food intake by slowing gastric emptying and inducing gastric distention and satiation. Thus, we tested the hypothesis that the impaired lipid absorption of Cyp8b1 deficiency caused slow gastric emptying, thereby reducing food intake.
To evaluate gastric emptying, we used two methods. To measure solid-phase gastric emptying, we fasted mice for 16 hours, then allowed them to access chow for 1 hour, then removed food for 2 hours prior to tissue collection. We measured individual food intake and stomach contents to determine the percentage of food eaten that had emptied from the stomach. Of note, during these experiments, we determined that ‘refed’ food intake was not different between genotypes during the 1-hour feeding period after prolonged (16 hours) fasting (0.54±0.02 vs 0.45±0.05 g for control and Cyp8b1–/–, respectively, p=0.17). Liquid-phase gastric emptying was assessed by acetaminophen absorption23 after a liquid meal (Ensure:corn oil, 17:3). Both tests showed that Cyp8b1 deficiency impaired gastric emptying (figure 4A–B).
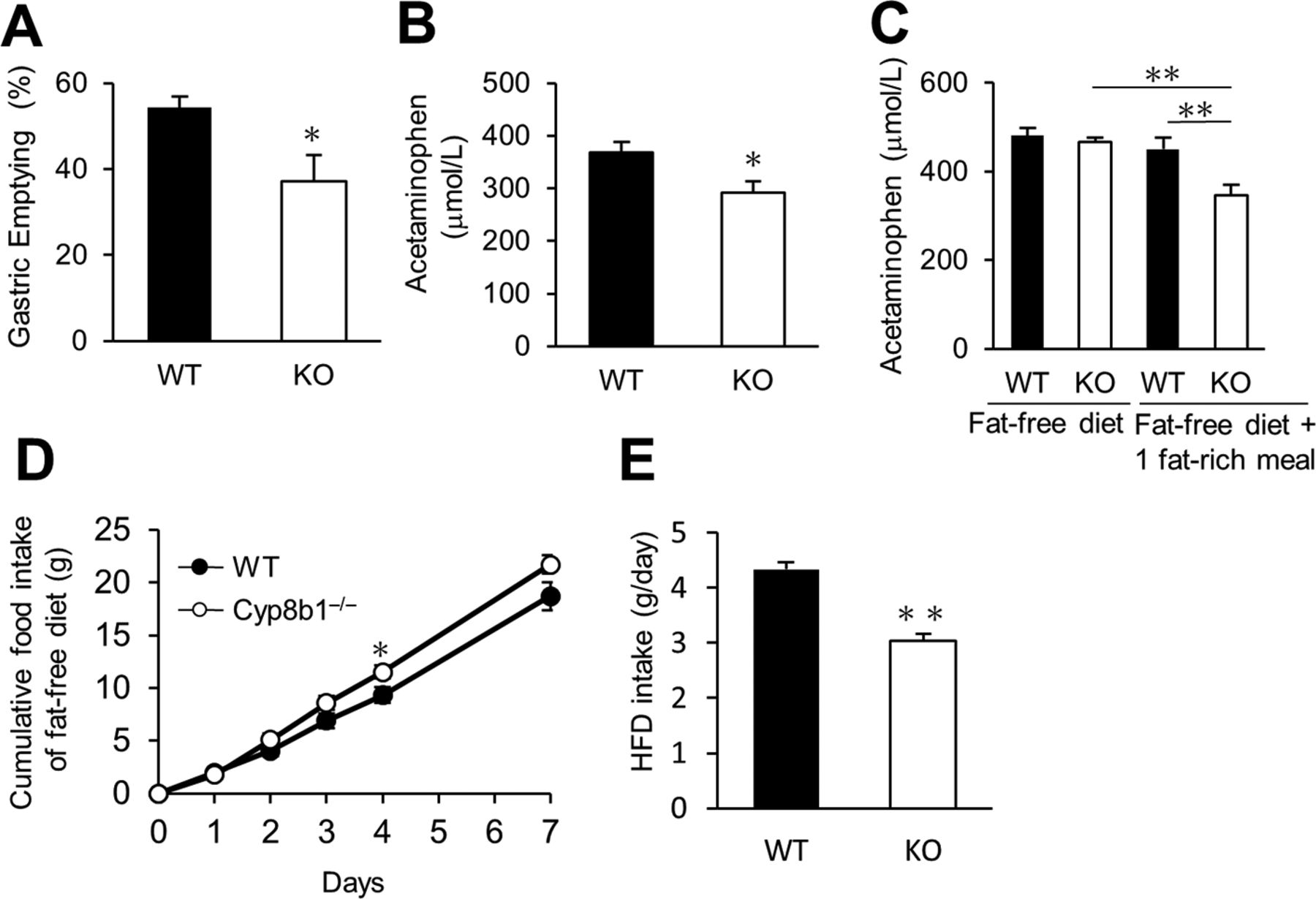
Effects of dietary lipid on gastric emptying and food intake in chow-fed WT and Cyp8b1–/– mice. (A) Solid gastric emptying (WT, n=3; and Cyp8b1–/–, n=4). *p<0.05 (Student’s t-test). (B) Liquid gastric emptying in chow-fed mice (n=8 for each group). *p<0.05 (Student’s t-test). (C) Liquid-phase gastric emptying in fat-free diet fed mice. Mice were given the fat-free diet. A fat-rich meal (Ensure:corn oil, 17:3) was given by oral gavage 17 hours before the gastric emptying test (n=8 for each group). (D) Cumulative fat-free diet intake for 7 days (n=8 for each group). *p<0.05 (Student’s t-test). (E) One-day food intake of high-fat diet (HFD) (WT, n=15; and Cyp8b1–/–, n=11). **p<0.01 (Student’s t-test).
Next, we directly evaluated the contribution of dietary fat to the impaired gastric emptying of Cyp8b1–/– mice. To test this, we gave mice a fat-free diet for 1 day, to eliminate lipids from the intestinal lumen. On this diet, there was no difference in gastric emptying between WT and Cyp8b1–/– mice (figure 4C). We also extended the fat-free diet feeding for 8 days, and we still observed no difference in gastric emptying between genotypes (480.6±17.1 vs 466.5±9.4 µmol/L acetaminophen in control and Cyp8b1–/–, respectively, p=0.48). However, when we stimulated mice with a single fat-rich meal the night before the test, we again observed decreased gastric emptying in Cyp8b1–/– mice (figure 4C). These results suggest that dietary lipid is necessary and sufficient to suppress gastric emptying in Cyp8b1–/– mice. We also measured jejunal epithelial levels of FAEs and endocannabinoids on the fat-free diet. In the fasted state, we observed no differences in jejunal AEA, DHEA, 2-AG or DHG between genotypes (online supplementary figure 4B–E). OEA was significantly reduced in Cyp8b1–/– mice (online supplementary figure 4A), although the physiological significance of this is unknown. Intragastric gavage with a liquefied preparation of the fat-free diet was completely unable to induce the levels of any of these specialised lipids, and there were no differences between genotypes after the meal (online supplementary figure 4A–E).
Supplemental material
Dietary fat was also necessary and sufficient for the hypophagia of Cyp8b1–/– mice. During fat-free diet feeding, cumulative food intake over 7 days in Cyp8b1–/– mice compared with WT was no longer reduced; if anything, it was slightly increased at some points (figure 4D). Cyp8b1–/– mice fed a high-fat diet for several weeks have been reported to have no differences in food intake10; however, the effects of intestinal lipid on food intake suppression are known to be blunted after chronic lipid stimulation.24–26 To test whether Cyp8b1 deletion affects high-fat diet intake in the short term, we gave mice high-fat diet (60% calories from fat) for 1 day. High-fat diet intake of Cyp8b1–/– mice was robustly decreased (approximately 30% reduction) compared with WT mice (figure 4E). These data support that Cyp8b1–/– mice have dietary fat–dependent decreases in gastric emptying, and this is associated with induced satiation.
Gpr119 is required for the gastric emptying and food intake phenotypes in Cyp8b1–/– mice
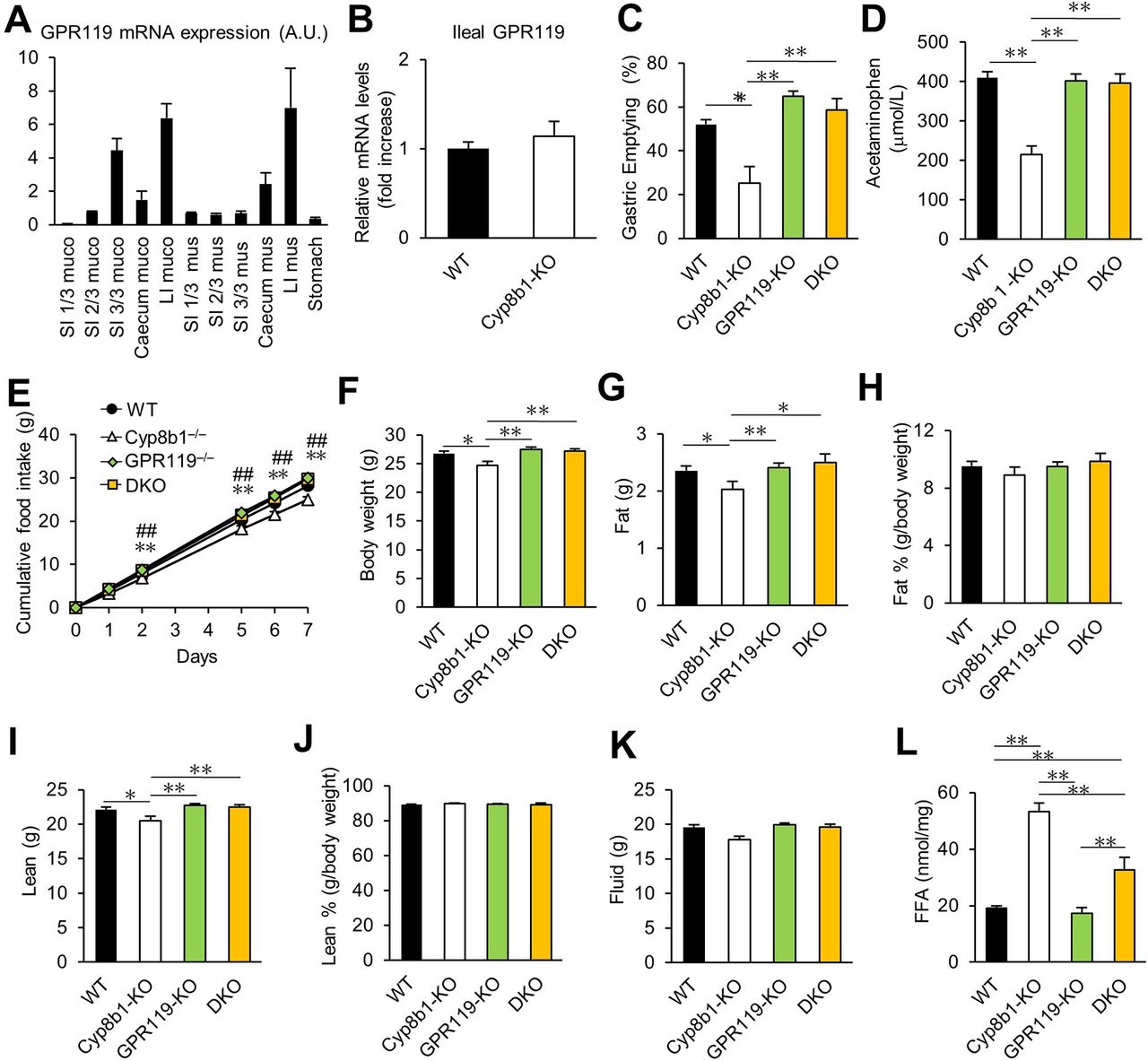
The fat sensor GPR119 is enriched in enteroendocrine cells of the ileum and colon,7 27 particularly L cells,28 and it is reported to mediate fat-induced GLP-1 secretion, based on nutritional experiments in humans.9 Activation of intestinal GPR119 slows gastric emptying via gut hormone–dependent and gut hormone–independent mechanisms.7 29 Consistent with prior data,8 28 we found GPR119 expressed primarily in distal small intestine and colon (figure 5A). However, there was no difference in the mRNA expression of Gpr119 between Cyp8b1–/– and control mice (figure 5B). We hypothesised that the Cyp8b1 deficiency induced slow gastric emptying through activation, not expression, of GPR119.

Contribution of GPR119 to gastric emptying, body mass and food intake in Cyp8b1–/– mice. (A) GI expression of GPR119 in WT mice (n=3). Small intestines were cut into thirds. LI, large intestine; muco, mucosal layer; mus, muscle layer; SI, small intestine. (B) Ileal GPR119 expression in WT and Cyp8b1–/– mice (WT, n=4; and Cyp8b1–/–, n=5). (C) Solid gastric emptying. Mice were fasted overnight, then allowed access to chow diet for 1 hour, then food-deprived again for 2 hours. **p<0.01 (one-way ANOVA). (D) Liquid-phase gastric emptying. **p<0.01 (one-way ANOVA). (E) Cumulative food intake for 7 days. *p<0.05 WT vs Cyp8b1–/–, **p<0.01 WT vs Cyp8b1–/–, ##p<0.01 Cyp8b1–/– vs DKO (one-way ANOVA). (F) Body weight at 12 weeks, (G) fat mass, (H) fat %, (I) lean mass, (J) lean % and (K) fluid mass in chow-fed mice (WT, n=16; Cyp8b1–/–, n=10; GPR119–/–, n=19; DKO, n=9). (L) Faecal excretion of free fatty acids (FFAs) (WT, n=8; Cyp8b1–/–, n=6; GPR119–/–, n=12; DKO, n=6). *p<0.05, **p<0.01 (one-way ANOVA).
We investigated the role of GPR119 using both pharmacological and genetic approaches. We administered a GPR119 antagonist, Arvanil,30 and found that it normalised the gastric emptying of Cyp8b1–/– mice, suggesting that GPR119 activity is involved in reduced gastric emptying in Cyp8b1–/– mice (online supplementary figure 5). Because Arvanil may also act at non-GPR119 receptors, we also used a genetic model of GPR119 deficiency to evaluate its role in the fat-mediated slow gastric emptying in Cyp8b1–/– mice.30–32 We crossed Cyp8b1–/– mice and GPR119–/– mice to create double knockout mice (DKO). In DKO mice, the gastric emptying defect was completely normalised (figure 5C–D). Also normalised were food intake, body weight, fat mass and lean mass (figure 5E–K). Notably, DKO mice still displayed excess faecal fat excretion, compared with WT or GPR119-KO alone (figure 5L). We conclude that in the context of Cyp8b1 deficiency, concomitant GPR119 deletion does not eliminate the fat absorption phenotype, but rather it eliminates the ability to sense the excess lower-intestinal fat.
Supplemental material
Notably, we did not observe any differences in gastric emptying, food intake, body weight or body composition in mice with a single knockout of GPR119 compared with WT controls (figure 5C–K). This is consistent with published data,33 and with the concept that under normal conditions, GPR119 is dispensable for these functions. Instead, our data support the idea that GPR119 is activated in Cyp8b1–/– because of their impaired lipid absorption, providing ligands that would not normally access GPR119 in the distal intestine.
Ileal GLP-1 and PYY contribute to slow gastric emptying in Cyp8b1–/– mice
It is well established that GLP-1 decreases gastric emptying and body weight.34 GPR119 mediates GLP-1 and PYY secretion, and both of these hormones decrease gastric emptying.7 Furthermore, systemic plasma GLP-1 levels are increased after oral glucose gavage in an independently generated line of Cyp8b1–/– mice.11 Thus, one might predict GLP-1 and/or PYY to mediate the effects of Cyp8b1–/– on gastric emptying.
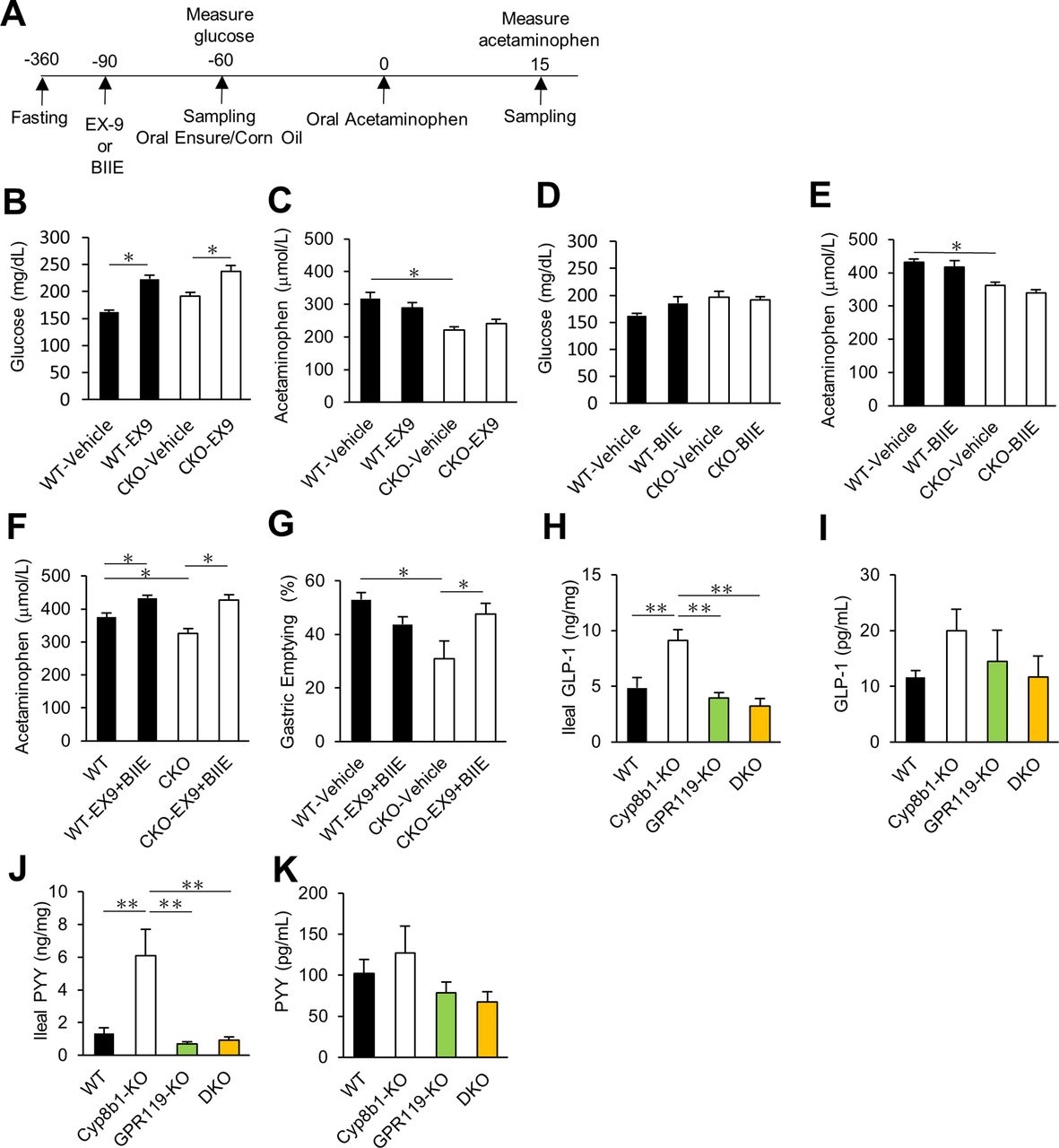
To investigate this, we used pharmacological receptor antagonists (experimental schedule is shown in figure 6A). Intraperitoneal injection of the GLP-1 receptor antagonist exendin-9 (0.3 mg/kg) successfully increased plasma glucose levels in all treated mice, thus validating the dose of exendin-9 (figure 6B). However, this treatment did not reverse the slow gastric emptying in Cyp8b1–/– mice (figure 6C). Intraperitoneal injection of the PYY Y2 receptor antagonist BIIE (2 mg/kg) did not change plasma glucose levels or normalise the slow gastric emptying in Cyp8b1–/– mice (figure 6D–E). To validate our dose of BIIE, we evaluated 5-hydoxytryptophan (5-HTP)–induced faecal pellet number because PYY suppresses 5-HTP induced defecation.35 The experimental schedule is shown in online supplementary figure 6A. In this experiment, 5-HTP treatment increased faecal pellet number, and this was suppressed by meal gavage (Ensure:corn oil, 17:3). This suppression was successfully reversed by BIIE (2 mg/kg) (online supplementary figure 6B), thus validating that this dose of BIIE blocked PYY signalling. Therefore, we successfully inhibited GLP-1 and PYY signalling, but individual blockade of these hormone signalling pathways did not reverse the gastric emptying phenotype of Cyp8b1–/– mice.
Supplemental material

GLP-1 and PYY levels and contribution of GLP-1 and Y2 receptors to gastric emptying. (A) Experimental schedule for GLP-1 receptor antagonist exendin-9 (EX-9) and Y2 receptor antagonist (BIIE). Exendin-9 (EX9, 0.3 mg/kg, i.p.) or BIIE (2 mg/kg, i.p.) were given to the mice 30 min before the fat-rich meal gavage, Ensure:corn oil (17:3), then 1 hour later, acetaminophen (100 mg/kg) was given by oral gavage. Blood samples were collected 15 min after acetaminophen gavage. (B–E) Effect of GLP-1 receptor antagonist and Y2 receptor antagonist on liquid-phase gastric emptying (WT, n=5–8; Cyp8b1–/–, n=4–6). *p<0.05 (two-way ANOVA). (F, G) Effect of GLP-1 receptor antagonist and Y2 receptor antagonist on liquid-phase and solid gastric emptying (WT, n=7–8; Cyp8b1–/–, n=6). *p<0.05 (two-way ANOVA). (H) IIeal GLP-1, (I) plasma GLP-1, (J) ileal PYY and (K) plasma PYY levels after solid gastric emptying (WT, n=5–17; Cyp8b1–/–, n=8–10; GPR119–/–, n=14–20; DKO, n=7–9). **p<0.01 (one-way ANOVA).
To test whether PYY and GLP-1 have overlapping functions on gastric emptying in Cyp8b1–/– mice, we administered both exendin-9 and BIIE to simultaneously block GLP-1 and Y2 receptors. Combined antagonist treatment normalised slow gastric emptying in Cyp8b1–/– mice (figure 6F,G). These data indicate that both GLP-1 and PYY play a crucial role in gastric emptying in Cyp8b1–/– mice.
Next, we examined the levels of GLP-1 and PYY. We collected the mucosal layer of ileal tissue in the same conditions as the solid gastric emptying test (16-hour fast, 1-hour chow feeding, 2-hour food deprivation, then tissue harvest). We observed a significant increase in GLP-1 and PYY in intestinal tissue of Cyp8b1–/– mice, and this was completely reversed in DKO mice (figure 6H,J). Unexpectedly, we did not observe significant differences between genotypes in systemic plasma GLP-1 or PYY (figure 6I,K). Also, there was no difference in plasma GLP-1 and PYY after a liquid meal (Ensure:corn oil, 17:3) or after olive oil infusion via chronic indwelling duodenal catheters (online supplementary figure 7A–F). This may be due to rapid degradation of these hormones in plasma. We found no differences in plasma levels of several other GI hormones including GIP, ghrelin and GDF15 (online supplementary figure 8). These data suggest that the levels of GLP-1 and PYY local to the intestine are elevated in Cyp8b1–/– mice, and this requires GPR119.
Supplemental material
Supplemental material
Non-12-OH BA CDCA impairs lipid absorption and slows gastric emptying in WT mice
We predicted that the impaired gastric emptying of Cyp8b1–/– mice was secondary to the altered BA composition and the consequential impairment in upper intestinal lipid absorption. To test this, we carried out oral gavage of CA (the major BA missing from Cyp8b1–/– mice) for 4 days. This was sufficient to normalise the lipid absorption and slow gastric emptying in Cyp8b1–/– mice (figure 7A,B). This demonstrates that the altered BA composition is the key aspect of Cyp8b1–/– mice that is responsible for the impaired gastric emptying.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Influence of exogenous BAs on gastric emptying and lipid absorption. (A–B) Effect of CA on faecal free fatty acid and gastric emptying in WT and Cyp8b1–/– mice. (C–D) Effect of CA, CDCA and α-MCA on faecal free fatty acid and gastric emptying in WT mice. Each BA was given daily for 4 days (50 mg/kg) before overnight fasting (n=5 for each group). Faeces were collected 24 hours before gastric emptying experiment (n=5 for each group). *p<0.05, **p<0.01 (one-way ANOVA). (E) Effects of 12-OH BAs on food intake. Lowering BA functionality by altering BA composition causes inefficient hydrolysis from triglyceride (TG) to free fatty acid (FFA) and 2-monoacylglycerol (2-MAG). This results in insufficient lipid absorption in the upper intestine. Entry of unabsorbed lipid such as 2-MAG into the lower intestine activates GPR119 and induces the secretion of gut hormones GLP-1 and PYY. This causes slow gastric emptying, gastric distention and satiation.
We next tested whether we could induce slow gastric emptying in WT mice by manipulating the BA pool composition with exogenous BAs. There are two major changes to the BA pool composition when Cyp8b1 is ablated: (1) reduction in 12-OH BAs—CA, DCA and their conjugates; and (2) compensatory increases in non-12-OH BAs, such as CDCA, MCAs and their conjugates.12 13 MCAs are the most abundant BAs in Cyp8b1–/– mice, and one might predict the MCAs cause the poor triglyceride absorption and consequent slow gastric emptying. This prediction is supported by the finding that feeding mice MCAs strongly impairs cholesterol absorption.36 However, cholesterol and triglyceride absorption are carried out by different mechanisms, and the effects of MCAs on triglyceride absorption have not been directly evaluated. We gave CA, CDCA or α-MCA to WT mice by oral gavage (50 mg/kg) daily for 4 days, and then measured faecal fat excretion and solid gastric emptying. CA treatment increased intestinal fat absorption, whereas CDCA decreased fat absorption (figure 7A). CDCA caused slow gastric emptying compared with CA in WT mice (figure 7C). These data show that the effect size of bile acid–induced variations in fat absorption are sufficient to alter gastric emptying. Unexpectedly, α-MCA had no effect on fat absorption or gastric emptying. These data suggest that providing mice with exogenous CA promotes lipid absorption and gastric emptying, whereas exogenous CDCA is inefficient in these functions.
Discussion
In this study, we showed that altering BA composition by Cyp8b1 ablation induces changes in two intestinal lipid sensing pathways: (1) impaired OEA production after refeeding in jejunum and (2) slow gastric emptying and increased satiation, which are reversed after concomitant GPR119 knockout. These findings indicate that (3) enhanced GPR119 signalling can override decreases in jejunal OEA, and that (4) BAs impact GPR119-dependent intestinal fat sensing and play a role as regulators of food intake.
Typically, feeding increases OEA levels in jejunum, by providing dietary oleic acid as a substrate for the OEA synthesis pathway, the last step of which is carried out by the enzyme NAPE-PLD.4 17 Magotti and colleagues showed that BAs—especially DCA, with its hydroxyl groups at carbon 3α and 12α—tightly bind the substrate-binding pocket of the NAPE-PLD dimer, which reinforces the enzyme’s stability and activity.14 Kinetic experiments demonstrated that DCA (3α, 12α) increases NAPE-PLD activity better than CDCA, another dihydroxy BA (3α, 7α).15 Consistent with these reports, our data showed that jejunal OEA levels were decreased in Cyp8b1–/– mice, which lack DCA, compared with littermate control mice. Thus, deficiency of 12-OH BAs may reduce OEA production due to impaired NAPE-PLD activity, with a potential additional contribution of impaired oleic acid uptake in jejunum. We noted that other FAEs and endocannabinoids were produced at normal levels in jejunum of Cyp8b1–/– mice, suggesting the possibility that DCA (or 12-OH BAs more generally) impacts stability and also substrate selectivity of NAPE-PLD. We did not find any differences between genotypes in OEA or other NAPE-PLD products in the hypothalamus. This may reflect differences in neuronal NAPE-PLD structure or substrate availability. But furthermore, there are only modest data available on whether—and which—BA species exist in brain.37 38
The presence of dietary fat in the lower small intestine induces slow gastric emptying and satiation.39 40 This effect is mediated by GPCR-dependent enteroendocrine cell activation, which promotes gut hormone secretion. We propose that the inefficient intraluminal triglyceride hydrolysis in Cyp8b1–/– mice allows entry of unabsorbed lipid in the lower intestine and colon, as demonstrated by the fact that a small amount of unabsorbed lipid reaches all the way into faeces. This accesses and activates GPR119 to induce GLP-1 and PYY, which slow gastric emptying and reduce food intake (figure 7E).
Our work suggests that when dietary lipid absorption is chronically impaired, the GPR119 pathway is chronically activated by physiological ligands. Which lipid species are responsible for this? Although OEA can activate GPR119 with an EC50 of 0.2–4.4 µM,7 this is unlikely to explain the phenotype of Cyp8b1–/– mice because intestinal OEA levels are lower in Cyp8b1–/– mice. However, multiple other lipid species can also activate GPR119, with EC50s in the same order of magnitude, including 2-MAG such as 2-OG.7 2-MAG is abundant after a meal because it is a primary product of dietary triglyceride hydrolysis. Moreover, our previous lipidomics study identified higher levels of MAG in the faeces of Cyp8b1–/– mice, demonstrating that excess was accessible to their lower intestines. Thus, we predict that 2-MAG is the activator of GPR119 in Cyp8b1–/– mice.
Satiation mechanisms in the GI tract are intricately regulated by gut hormones. Gut hormones are released from cells throughout the GI tract, but GLP-1 and PYY secretion in response to GPR119 is enriched in the lower intestine, where GPR119 expression is highest. Flock et al showed that the effect of a pharmacological GPR119 agonist to slow gastric emptying could not be reversed with either Y2 receptor antagonist or GLP-1 receptor ablation.29 After Roux-en-Y gastric bypass surgery, single blockade of GLP-1 or PYY signalling had no effect on food intake, but inhibition of both signalling pathways increased food intake.41 This suggests that multiple gut hormones regulate gastric emptying and food intake. In agreement with these reports, either of exendin-9 or BIIE treatment did not normalise the slow gastric emptying of Cyp8b1–/– mice, but combined treatment reversed this phenotype. Thus, our data indicate that lowering 12-OH BAs enhances GLP-1 and PYY signalling via GPR119 activation in lower intestine to slow the rate of gastric emptying.
We used Cyp8b1–/– mice as a tool to understand the contributions of BA-dependent intestinal lipid metabolism on gastric emptying and satiation. This study reveals that the effect of BAs to promote efficient hydrolysis and absorption of dietary fat is an endogenous mechanism that prevents enteroendocrine cell–induced satiation. Would this particular change in BA composition—decrease of 12-OH BAs and increase in non-12-OH BAs—cause similar changes in humans? Our data suggest that abundance of CDCA, the primary non-12-OH BA in humans, is sufficient to impair lipid absorption and slow gastric emptying in WT mice. Further experiments would be required to fully test this. Nonetheless, it is tempting to speculate that the preferential synthesis of 12-OH BAs during insulin resistance in humans42–44 could diminish intestinal lipid sensing. Conversely, it is possible that therapeutic alterations in BA composition could sustain chronic, low-level activation of endogenous intestinal lipid sensing pathways for obesity and diabetes treatment.
Acknowledgments
The authors would like to acknowledge Rudolph Leibel, Enrico Bertaggia and Anna Chen for helpful discussions and Thomas Kolar for technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it published Online First. The author names have been amended and figure 5 updated.
Contributors SH and RH conceived and designed research; SH, TRA, DAA, PAP and CZ performed experiments; GJS taught the method for implanting duodenal catheters and provided valuable feedback; SH, ND and RH analysed data; SH and RH drafted manuscript; all authors edited the manuscript.
Funding This work was supported by the Russell Berrie Foundation, NIH/NIDDK (R01DK115825), and the American Diabetes Association (1-18-IBS-275) to RH, Kanae foreign study grant to SH, and NIH grants T32DK007328 to TRA, DK119498, DA034009 and DK114978 to ND.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experiments were approved and conducted according to the guidance of the Columbia University Institutional Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.