Article Text

Abstract
Objective Hepatocellular carcinoma (HCC) is the fastest-growing cause of cancer-related mortality with chronic viral hepatitis and non-alcoholic steatohepatitis (NASH) as major aetiologies. Treatment options for HCC are unsatisfactory and chemopreventive approaches are absent. Chronic hepatitis C (CHC) results in epigenetic alterations driving HCC risk and persisting following cure. Here, we aimed to investigate epigenetic modifications as targets for liver cancer chemoprevention.
Design Liver tissues from patients with NASH and CHC were analysed by ChIP-Seq (H3K27ac) and RNA-Seq. The liver disease-specific epigenetic and transcriptional reprogramming in patients was modelled in a liver cell culture system. Perturbation studies combined with a targeted small molecule screen followed by in vivo and ex vivo validation were used to identify chromatin modifiers and readers for HCC chemoprevention.
Results In patients, CHC and NASH share similar epigenetic and transcriptomic modifications driving cancer risk. Using a cell-based system modelling epigenetic modifications in patients, we identified chromatin readers as targets to revert liver gene transcription driving clinical HCC risk. Proof-of-concept studies in a NASH-HCC mouse model showed that the pharmacological inhibition of chromatin reader bromodomain 4 inhibited liver disease progression and hepatocarcinogenesis by restoring transcriptional reprogramming of the genes that were epigenetically altered in patients.
Conclusion Our results unravel the functional relevance of metabolic and virus-induced epigenetic alterations for pathogenesis of HCC development and identify chromatin readers as targets for chemoprevention in patients with chronic liver diseases.
- hepatocellular carcinoma
- gene expression
- hepatitis C
- non-alcoholic steatohepatitis
- chemoprevention
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Chronic hepatitis C (CHC) and non-alcoholic steatohepatitis (NASH) are leading causes of hepatocellular carcinoma (HCC).
Chronic HCV infection induces genome-wide changes in histone modifications and associated gene transcription in the human liver persisting postcure.
Epidrugs are approved for the treatment of haematological malignancies and their efficacy in other diseases is under evaluation in clinical trials.
What are the new findings?
CHC and NASH result in similar epigenetic and transcriptional changes associated with HCC risk in patients.
Applying a human cell culture model that recapitulates the CHC-induced and NASH-induced genome-wide epigenetic changes associated with liver carcinogenesis, we identify compounds reverting gene expression associated with HCC risk in patients.
A bromodomain 4 inhibitor significantly reduces liver disease progression and tumour burden in a diet model of NASH and HCC.
How might it impact on clinical practice in the foreseeable future?
Inhibition of chromatin readers and modifiers provides a previously undiscovered approach for HCC prevention.
Introduction
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver with rising incidence.1 Chronic hepatitis C (CHC) is a major cause of HCC. The HCC risk still remains elevated postcure, especially in patients with advanced fibrosis.2 Patients with non-alcoholic steatohepatitis (NASH) are also at high risk of developing HCC. Given the change in lifestyle with increasing obesity and diabetes, NASH will replace viral hepatitis as major cause for HCC.3 Due to the high HCC mortality and unsatisfactory treatment options, the development of HCC chemopreventive strategies is a key unmet medical need.4
Epigenetic regulation is a major determinant of gene expression. Alteration of the epigenetic programme plays a key role for pathogenesis of human disease and cancer.5 Several studies have investigated the role of epigenetics in HCC, however the role of epigenome modifications for liver disease progression and hepatocarcinogenesis is only recently emerging. We and others have recently demonstrated that CHC results in genome-wide epigenetic modifications, which are associated with HCC risk and persist postcure with direct-acting antiviral agents (DAAs).6 7
The reversibility of epigenetic changes offers a therapeutic perspective to counteract the associated transcriptional changes and their functional consequences for disease biology. Small molecule inhibitors targeting chromatin modifiers or readers are currently explored as therapeutic approaches with a particular focus on cancer.8 9
Chromatin readers contain evolutionarily conserved motifs allowing their binding to modified histones to regulate gene expression. Bromodomain and extra terminal (BET) family display a bromodomain (BRD) motif to bind to acetylated histones and consists of BRD2, BRD3, BRD4 and BRDT proteins. Among them, BRD4 is a regulator of transcription elongation that can be targeted by small molecules.10
Materials and methods
Human subjects
We analysed adjacent non-tumourous liver tissue from: six control patients without known liver disease and without HCC, three patients without known liver disease (F0) and HCC (‘spontaneous’ HCC), three patients with CHC without HCC (F3-F4), three patients with CHC with HCC (F3-F4), six DAA-cured patients with CHC with HCC (F3-F4), three NASH and HCC (F1), three NASH and HCC (F4) and four NASH without HCC (F4) from patients undergoing surgical liver resection (see online supplementary table S1). Patients for isolation of tumour spheroids are described in online supplementary table S2. Tissues were obtained with informed consent from all patients for deidentified use. Some subjects have been described.6
Supplemental material
Research experiments on live vertebrates
C57BL/6 male mice (Charles River Laboratories) received a single intraperitoneal injection of 35 mg/kg diethylnitrosamine (DEN) on day 15. After the mice reached 6 weeks of age, they were fed a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) consisting of 60 kcal% fat and 0.1% methionine by weight (Research Diets A06071302). After the mice reached 12 weeks of age, they were randomised to receive intraperitoneal injections 5 days a week of either 50 mg/kg JQ1 in 10% (2-hydroxypropyl)-b-cyclodextrin or vehicle only. Livers were harvested and analysed at 30 weeks of age. The sample size estimate was based on a p value of 0.01 at 95% power assuming a 50% difference in means in tumour burden with 30% SD between control and drug-treated animals.
ChIPmentation-based ChIP-Seq and RNA next-generation sequencing
ChIPmentation-based ChIP-Seq was performed as described.6 11 RNA-Seq was performed as described.6 Mouse RNA-Seq data were processed as described for patient’s data but were instead mapped to the mouse genome mm10 and annotated using the Gencode vM15 gene annotation. Processing of ChIPmentation data was described,6 and data were partially derived from BioProject PRJNA506130. Since patients with late-stage fibrosis have the highest HCC risk, patient tissues from late-stage fibrosis (F3 and F4) samples were included for all H3K27ac analyses as well as from the external CHC RNA-Seq dataset (GSE84346).12 Transcriptomic data for patients with NASH were derived from external expression dataset (GSE115193)13 and data for DAA-cured patients were published in BioProject PRJNA506130.6 The RNA-Seq data from HCV-infected liver cells were published in GSE126831.14
Prognostic liver signature gene nCounter expression analyses
Profiling of the prognostic liver signature (PLS) was performed using Nanostring nCounter assay as described.15 Induction or suppression of the PLS in gene expression data was determined as previously reported using the Gene Set Enrichment Analysis (GSEA), implemented in GenePattern genomic analysis toolkits.16–18 False discovery rate<0.05 was regarded as statistically significant. Results are presented as heatmaps showing: (top) the classification of the PLS global status as poor (orange) or good (green) prognosis; (bottom) the significance of induction (red) or suppression (blue) of PLS poor-prognosis or good-prognosis genes. Global status corresponds to the difference between low-risk and high-risk gene enrichments.
Pathway enrichment and correlation analyses
Hallmark pathway enrichment and correlation analyses were performed as described.6
Statistical analyses
Statistical analyses of NGS data are based on DESeq (RNA-Seq) and MACS2/edgeR (ChIP-Seq) as described.6 The cell culture and tumour spheroids data are presented as the mean±SD except where mean±SEM is indicated and were analysed by the unpaired Student’s t-test or the two-tailed Mann-Whitney U test as indicated after determination of distribution by the Shapiro-Wilk normality test. The p values are indicated in the figure legends for each figure panel; p≤0.05 was considered to be statistically significant. Statistical analyses were performed with GraphPad Prism 6 software.
Organotypic ex vivo liver slice culture
Fresh liver tissue sections (300 µm thick) were made from surgically resected fibrotic livers from patients with HCC using Krumdieck Tissue Slicer MD6000 (Alabama Research and Development, Munford) as described.19 The tissues were cultured with JQ1 (0.1 or 1 µM) for 48 hours and harvested for gene expression analysis.
Tumour spheroids from liver tissues of patients with HCC
Patient-derived HCC spheroids were generated from fresh liver tissues from five HCC resections (online supplementary table S2) and cultured in spheroid culture medium in low attachment U-bottom plate similar as described.20 21 Spheroids were treated with 10 µM sorafenib or with 50 nM JQ1 for 6 days. Cell viability was assessed by ATP quantification using CellTiter-Glo 3D (Luminescent Cell Viability Assay).
HCC risk profiling
Transcriptome profiles of 72 NASH-affected liver tissues were obtained from NCBI Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo, accession number GSE49541). Transcriptomic molecular dysregulation was determined using Nearest Template Prediction algorithm as previously described16 and defined based on p≤0.05.
Supplementary materials
See online supplementary material for further details about reagents, protein expression, cell-based systems, CRISPR Cas9 and ChIP-qPCRs.
Results
Patients with NASH and CHC with advanced liver disease share similar epigenetic and transcriptional changes associated with HCC risk
To characterise epigenetic and transcriptional modifications in the liver driving HCC risk, we analysed NASH and CHC liver tissues with advanced liver fibrosis (F3/F4) using ChIPmentation-based ChIP-Seq profiling of the H3K27ac epigenetic marks of active promoters and enhancers combined with RNA-Seq (figure 1A and online supplementary figure S1). Within each aetiology, epigenetically modified genes significantly correlate with transcriptomic changes (figure 1B). Pathways analysis revealed that patients with NASH and CHC with advanced liver disease (F3 and F4) display increased H3K27ac levels on genes related to tumour necrosis factor α (TNFα) signalling via nuclear factor-kappa B (NF-κB), inflammatory response, epithelial-to-mesenchymal transition (EMT) and IL2-STAT5, and decreased levels of H3K27ac on genes related to xenobiotic, bile acid and fatty acid metabolism as well as adipogenesis, coagulation and oxidative phosphorylation (figure 1C). We then identified genes with H3K27ac modifications and concomitant alteration of corresponding transcript expression, and then intersected NASH (n=2721) and CHC (n=4017) groups to identify a gene set with modulated expression in both aetiologies present in the adjacent tissue of HCC. We uncovered a total of 1693 genes with common epigenetic and transcriptional changes (figure 1D,E and online supplementary table S3). Within the genes shared by CHC and NASH, we identified overexpressed oncogenes with increased level of H3K27ac and downregulated tumour suppressor genes (TSG) with decreased level of H3K27ac. Among the overexpressed oncogenes were FGFR1, a member of the fibroblast growth factor receptor (FGFR) family that plays a key role in the development and progression of HCC,22 the cyclin CCND2, reported to drive tumourigenesis and progression of various cancers including HCC, MLLT3, an oncogene associated with leukaemia, CDH11, a cadherin reported to be involved in liver fibrosis and in EMT23 as well as MAML2, a coactivator of the Notch signalling pathway known to mediate liver carcinogenesis.24 Downregulated TSG included FANCC, encoding a protein associated with the DNA damage response, and TSC2, a negative regulator of the mammalian target of rapamycin (mTOR) signalling pathway.25
Supplemental material

Patients with non-alcoholic steatohepatitis (NASH) and chronic hepatitis C (CHC) with advanced liver disease share similar epigenetic and transcriptional changes associated with hepatocellular carcinoma (HCC). (A) RNA-Seq (left panel) and ChIP-Seq (right panel) mapping of NASH-induced and CHC-induced transcriptomic and H3K27ac modifications from patient-derived liver biopsies and resections. Left panel: unsupervised clustering of significant 4790 differentially expressed genes in livers from NASH (n=3) and CHC (n=6) compared with control patients (n=3 and 5, respectively). Right panel: differential signals in H3K27ac ChIP-Seq peaks for corresponding genes in livers from NASH (n=7) and CHC (n=6) compared with control patients (n=6). (B) Significant H3K27ac modifications correlate (Spearman’s rank correlation coefficients and p values) with gene expression changes in both patients with NASH (left panel) and CHC (right panel). Prognostic association of gene expression was determined using Cox score for time to overall death in a cohort of patients as described in the ‘Materials and methods’ section. (C) Hallmark pathways significantly enriched for H3K27ac modifications in NASH (n=7) and CHC (n=6) compared with control (n=6) patient samples. (D) Significant H3K27ac changes of the 1693 genes with corresponding transcriptomic changes in patients with NASH and CHC derived from B. (E) Venn diagram showing the overlap of significant epigenetically modified genes (shown in D) with corresponding expression changes in patients with NASH and CHC with advanced liver disease derived from the ChIP-Seq and RNA-Seq experiments shown in B.
Since our analysis included only a limited number of patients and the large majority having already established HCC without detailed longitudinal data available, we studied the association of the observed gene set modulation with HCC risk and overall mortality in a validation cohort of patients with HCV-related early stage cirrhosis and longitudinal analysis.26 Patients with dysregulation of the 1693 commonly changed genes exhibited a significantly shorter survival and a significantly earlier HCC development than those with transcriptionally intact liver (figure 2A). Furthermore, the dysregulation was more prevalent in patients with NASH-related advanced fibrosis defined as F3 or F4, whose HCC risk is higher,27 compared with those with mild fibrosis defined as F0 or F1 (p<0.001) (figure 2B). The identified gene set could be condensed to an intersected gene set of 25 genes with highest prediction of HCC risk. This gene set was termed ‘prognostic epigenetic signature’ (figure 2C,D and online supplementary table S4). Collectively, this validation analysis suggests that the identified gene expression changes are associated with HCC risk in advanced liver disease.

Risk for hepatocellular carcinoma (HCC) development and prognostic liver signature (PLS) expression in patients with advanced liver disease. (A) The probabilities of future hepatocarcinogenesis and overall survival according to the presence of the epigenetic dysregulation. The dysregulation was significantly associated with future HCC development and mortality in patients with HCV-related early stage cirrhosis. (B) The prevalence of the presence of the epigenetic dysregulation in patients with non-alcoholic steatohepatitis (NASH). The dysregulation was more frequently observed in patients with advanced fibrosis, one of the well-known HCC risks, compared with those with mild fibrosis. (C) The probabilities of future hepatocarcinogenesis and overall survival according to the presence of dysregulation of a gene subset termed the ‘prognostic epigenetic signature’ (PES). (D) The prevalence of the presence of the epigenetic dysregulation in patients with NASH. The PES, including 25 genes, showed better or similar capability to identify patients with higher HCC risk compared with the full signature. The PES was defined as commonly prognostic genes in both HCV and NASH (FDR<0.25). (E) Heatmap of the 186-gene PLS including modulation of the HCC high-risk (top) and low-risk (bottom) genes based on patient-liver transcriptome (left panel) and epigenome (right panel). Expression and H3K27ac changes of the gene members of the PLS in NASH, chronic hepatitis C (CHC) and DAA/HCC-cured compared with control patient livers measured using the RNA-Seq and ChIP-Seq experiments shown in figure 1A. (F) H3K27ac modifications correlate (Spearman’s rank correlation coefficients and p values) with transcriptomic changes of gene members of the PLS in patients with NASH (left panel) or CHC (right panel).
To assess transcriptional changes that were associated with the presence of advanced fibrosis (F3-F4), we intersected liver tissues with and without advanced fibrosis. Using this approach, we found that 43% of the epigenetically modified genes appeared to be linked to the presence of advanced fibrosis (742 of 1693). Interestingly, we found 27 genes with epigenetic modifications in the adjacent tissue of three patients with spontaneous HCC without fibrosis which were not altered in patients with HCC and fibrosis of any stage and not present in patients without liver disease. These findings suggest that epigenetic modifications associated with hepatocarcinogenesis can occur in the absence of fibrosis.
Next, we studied the role of epigenetic changes for the status of the prognostic liver signature (PLS)—a well-characterised stromal liver 186-gene expression signature that has been shown to predict survival and HCC risk in patients with advanced liver disease of all major HCC aetiologies.19 In line with previously published results,6 we observed a modulation of expression of genes predicting HCC high risk in both patients with NASH and CHC with advanced liver disease (figure 2E, left panel). Interestingly, H3K27ac levels on these 186 genes were correlated with their respective transcript expression (figure 2E, right panel and figure 2F), suggesting an association between disease-induced epigenetic modifications and the PLS gene expression. Interestingly, the poor-prognosis PLS status remained (figure 2E) after HCV cure in patients with advanced fibrosis and HCC.
Finally, we intersected the RNA-Seq and H3K27ac ChIP-Seq data among the three patient groups to uncover genes for which changes in their transcript were significantly correlated with H3K27ac modifications, assuming that these genes are most strongly associated with HCC risk. Seventy-six per cent (1286 out of 1693) of the genes identified in livers from both patients with NASH and CHC were similarly modulated in livers from DAA-cured patients who developed an HCC (online supplementary figure S2A–D and online supplementary table S5). H3K27ac changes and expression of the 186 genes from the poor-prognosis status of the PLS showed a high correlation in NASH (r=0.62; p<1.0×10−10), CHC (r=0.66; p<1.0×10−10) and DAA-cured patients with HCC (r=0.45; p<0.4×10−10), suggesting that the poor-prognosis/HCC high-risk status of the PLS provides a surrogate marker of epigenetic modifications of HCC-associated genes and of poor survival (figure 2F and online supplementary figure S2E).
Modelling of clinical epigenetic modifications in a liver cell-based model
We next aimed to model epigenetic changes observed in patients in a cell-based model that partially recapitulates transcriptomic and proteomic changes in patients with chronic liver disease.14 28 Dimethyl sulfoxide-differentiated Huh7.5.1 cells (Huh7.5.1dif cells) infected by HCV showed transcriptomic and proteomic changes found in liver tissue from HCV-infected patients.14 Simultaneously, we established a cell culture system aiming to model transcriptional changes in metabolic liver injury by using a co-culture of Huh7.5.1dif and LX2 stellate cells treated with free fatty acids (FFA). ChIPmentation-based ChIP-Seq profiling of the H3K27ac epigenetic marks and RNA-Seq analyses (figure 3A) revealed that both persistent HCV infection and FFA treatment led to epigenetic and transcriptomic changes that were also partially observed in patients with NASH and CHC (figure 3B–C, online supplementary figure S3A–B and online supplementary table S6). Comparative study of ChIP-Seq and RNA-Seq data from cells with viral and metabolic injury showed a significant positive correlation (epigenetic r=0.43 and transcriptomic r=0.47), suggesting shared epigenetic and transcriptomic changes by viral and metabolic stress in the model systems (online supplementary figure S3C). In concordance with the results found in patient-derived liver tissues, GSEA analyses revealed perturbation of pathways mediating TNFα signalling activation, E2F targets, G2M checkpoint and EMT signalling (figure 3D). Moreover, we observed an induction of the poor-prognosis status of the PLS on both epigenetic and transcriptomic levels and a positive correlation between H3K27ac and transcriptomic changes in both cell culture models (figure 3E and online supplementary figure S3C). Collectively, these analyses demonstrate that the cell-based models can partially recapitulate epigenetic and associated transcriptional alterations that are found in patients with CHC and NASH.
Supplemental material
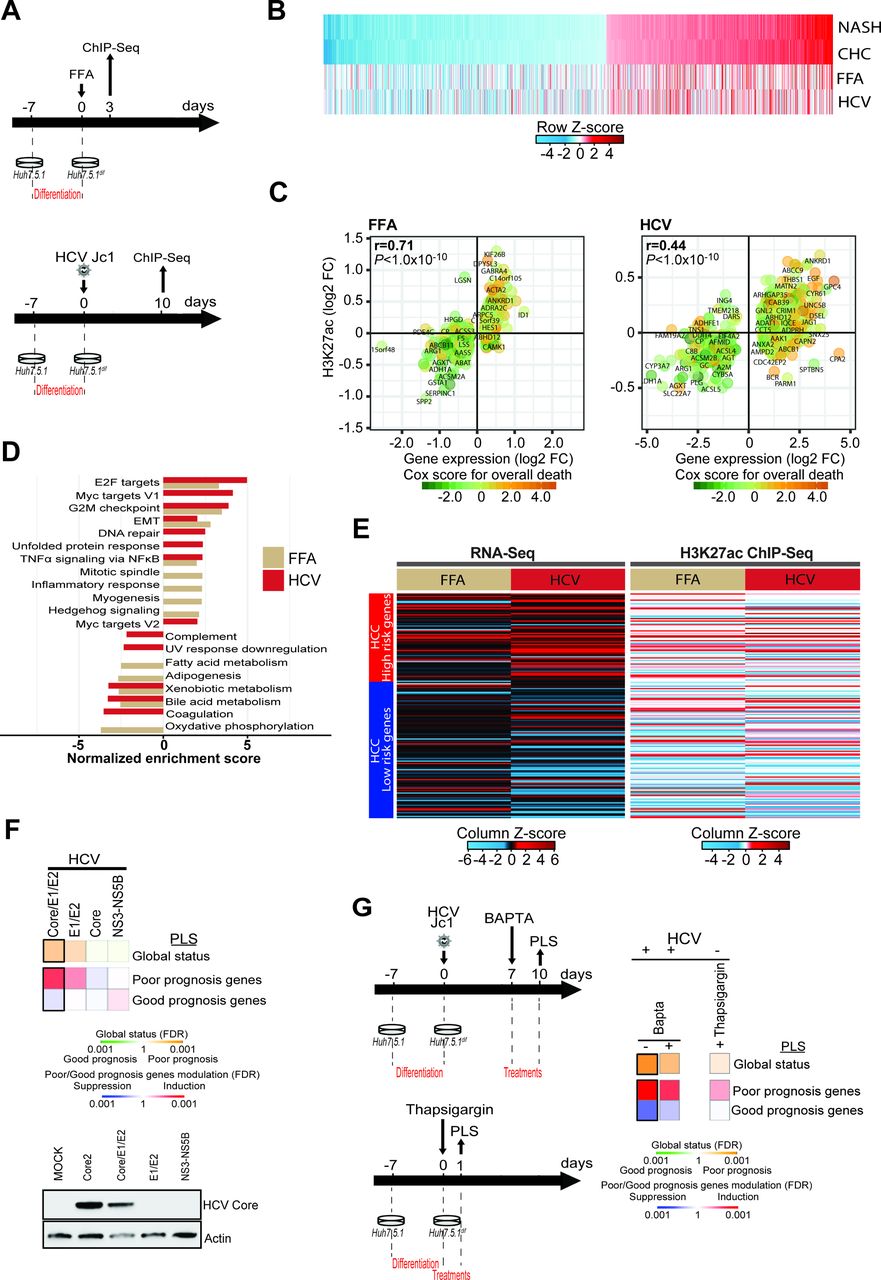
Modeling of HCV-induced and non-alcoholic steatohepatitis (NASH)-induced histone modifications associated with liver carcinogenesis in a cell culture model. (A) Schematic representation of the experimental setup. H3K27ac marks were profiled by ChIP-Seq following free fatty acid (FFA) treatment (top panel: day 3) or persistent HCV infection (bottom panel: day 10). (B) H3K27ac data of the 1693 genes with significant transcriptomic changes in patients with NASH and chronic hepatitis C (CHC) derived from figure 1B, and corresponding changes in FFA-treated or HCV-infected cells derived from the ChIP-Seq experiment shown in panels A and B. (C) Significant H3K27ac modifications correlate (Spearman’s rank correlation coefficients and p values) with gene expression changes in FFA-treated or HCV-infected cells. Prognostic association of gene expression was determined as described for figure 1B. (D) Gene Set Enrichment Analysis (GSEA) pathway analysis of genes associated with H3K27ac modifications in FFA-treated or HCV-infected compared with Mock or non-infected cells from the ChIP-Seq experiment shown in panels A and B. (E) Heatmap of the prognostic liver signature (PLS) based on the transcriptome (left panel) and epigenome (right panel) of FFA-treated or HCV-infected cells. (F, G) Effect of ectopic expression of HCV proteins, thapsigargin and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) treatment on the PLS status. Heatmaps show: (top) the classification of the PLS global status as poor (orange) or good (green) prognosis; (bottom) the significance of induction (red) or suppression (blue) of PLS poor-prognosis or good-prognosis genes. FDR, false discovery rate.
Next, we used the cell-based models to gain insights on the molecular mechanism of the induction of genes associated with liver disease progression and HCC risk. Expression of the HCV structural proteins resulted in the most robust induction of the PLS poor-prognosis status (figure 3F). Since HCV infection has been shown to induce an unfolded protein response (UPR),29 that is mediated by the viral envelope glycoproteins, we treated infected cells with BAPTA, a selective calcium chelator that mitigates UPR.30 Incubation of HCV-infected cells with BAPTA appeared to result in a partial reduction of HCC high-risk gene expression (figure 3G). To further corroborate the role of the UPR, we incubated naive cells with thapsigargin, a blocker of sarco/ER Ca2+-ATPase that disrupts cellular calcium homeostasis causing a sustained UPR.31 Thapsigargin resulted in a partial induction of HCC high-risk genes, suggesting that the UPR and cell stress contribute to the observed epigenetic and transcriptional changes associated with HCC risk.
Reverting epigenetic and transcriptional reprogramming associated with HCC risk using inhibitors of chromatin modifiers and readers
We next explored disease-induced epigenetic modifications as targets for HCC chemoprevention. To address this question, we treated liver cells expressing the PLS poor-prognosis status with inhibitors of chromatin modifiers/readers (figure 4A,B) at concentrations that did not induce major cell toxicity (figure 4C): C64632 and CTK7A inhibit p300 histone acetyltransferase,33 I-BET151 and JQ1 block the interaction of the bromodomain proteins BRD3/4 with acetylated lysines on histone tails; WDR5-0103 and MM-10234 inhibit the MLL/WDR5 complex required for the methylation of H3K4; SAHA blocks histone deacetyltransferases (HDAC) of class I and II activity and TSA and TMP15035 36 are inhibitors of histone deacetyltransferases. Transcriptomic analysis showed that I-BET151, JQ1 and MM-102 reversed the PLS from a HCC high-risk to low-risk status (figure 4D). Next, we analysed the effect of the compounds on EGF and NFkB expression as two well-characterised HCC risk drivers. CTK7A, I-BET151 and JQ1 significantly reduced the level of virus-induced EGF and NFkB2 transcript elevation confirming that epigenetic perturbations reverse transcriptional reprogramming driving HCC risk (figure 4E). None of these inhibitors significantly affect HCV infection, indicating that the observed effect was viral load independent (figure 4F). Several studies have reported that JQ1 is effective on HCC proliferation and had identified a modulation of c-Myc oncogene as a mode of action.37–40 We therefore quantified c-Myc protein expression in the cell-based model. JQ1 treatment partially decreased c-Myc levels but the decrease did not reach significance (p=0.1580) (figure 4G, online supplementary figure S4).

Effect of small molecules and knockdown targeting chromatin modifiers and readers on gene expression driving hepatocellular carcinoma (HCC) risk in cell culture. (A) Schematic representation of the experimental setup. (B) Mode of action of inhibitors of chromatin modifiers/readers targeting histone acetylation and methylation. TF, transcription factors. (C) Cytotoxicity of the inhibitors in Huh7.5.1dif cells depicted in panel B. Cell viability of non-infected cells (n=3) was measured 72 hours following incubation with inhibitors using MTT assay. Results (mean±SEM) show percentage of viable cells in compound-treated conditions relative to untreated cells. The concentrations used for functional assays shown in panel D are indicated by red asterisks. (D) Effect of compounds on the prognostic liver signature (PLS) gene expression. Heatmaps show: (top) the classification of the PLS global status as poor (orange) or good (green) prognosis; (bottom) the significance of induction (red) or suppression (blue) of PLS poor-prognosis or good-prognosis genes. FDR, false discovery rate. (E) mRNA expression of EGF and NFκB2 in cells treated with the indicated compounds for 72 hours (*p<0.05; **p<0.01; ***p<0.001, unpaired t-test). (F) Inhibitors of chromatin modifiers/readers do not modulate HCV viral load in the experiment shown in panel E. (G) Western blot analysis showing c-Myc protein expression in the cell-based system. One representative gel is shown (see online supplementary figure S4). Right panel graph shows the quantification of western blot analysis intensities in arbitrary units normalised to total protein level (n=3) (stain-free staining). Results show the mean±SEM of integrated blot densities of four independent experiments and are not significantly (‘n.s.’) modulated. (H) Gene silencing of BRD3/4 and HDAC9 reverses the HCC high-risk status of the PLS in Huh7.5.1dif-Cas9 cells. HCV-infected Huh7.5.1dif-Cas9 cells expressing the PLS poor-prognosis status were transduced with lentiviral vectors coding for sgRNA targeting p300, BRD3, BRD4 and HDAC9 genes or non-targeting sgRNA (sgCTRL) (means of two experiments). (I) Proliferation analyses: (left) JQ1-treated or untreated Mock and HCV-infected cells; (right) Mock or HCV-infected Huh7.5.1dif-Cas9 cells transduced with lentiviral vectors coding for sgCTRL or sgRNA targeting BRD3 and BRD4 genes. (J) Analysis of BRD3, BRD4, c-Myc and Actin protein expression levels in Huh7.5.1dif-Cas9 cells KO for BRD3 or BRD4 by western blot analysis. Effective sgRNA for BRD3 and BRD4 genes used in the screening experiment shown in (H) are labelled with a red asterisk.
We then sought to confirm the epigenetic drivers for the poor-prognosis status of the PLS by knocking out the targets of the compounds. BRD3, BRD4 and HDAC9 knockouts robustly prevented virus-induced induction of the PLS poor-prognosis status (figure 4H). These results confirm the functional impact of histone acetylation for the expression of HCC risk genes (figures 3E and 4E) and identify BRD3, BRD4 and HDAC9 as candidate genes for therapeutic intervention to decrease HCC risk. Of note, BRD3/4 knockout did not robustly alter c-Myc protein expression and cell proliferation (figure 4I–J).
As any cell culture model, the applied system only partially recapitulates the cell circuits in the diseased liver and represents a simplification of the liver transcriptome. However, the robust induction of the PLS suggests that the model is useful to assess pathways involved in the generation of the signature (as shown for chromatin drivers and readers in figure 3) or compounds reversing the signature (as shown in figure 4D).
BRD4 inhibition significantly reduces the tumour burden in a mouse model of NASH-induced hepatocarcinogenesis
Finally, to investigate whether inhibitors of chromatin modifiers/readers are an approach for HCC chemoprevention, we performed proof-of-concept studies choosing the BRD inhibitor JQ1 for further studies. Since no robust small animal model of HCV-induced hepatocarcinogenesis is available, we used a DEN/CDAHFD diet-induced mouse model of NASH-driven HCC to analyse the effect of JQ1 on liver carcinogenesis. Genetically, DEN-induced tumours resemble human tumours with poor prognosis41 and DEN administration changes the proportion of heterochromatin and euchromatin, suggesting an alteration of epigenetic marks. Mice were treated with JQ1 after 6 weeks of CDAHFD feeding when NASH was fully developed (figure 5A). Transcriptomic profiling of DEN/CDAHFD mouse livers (figure 5B and online supplementary figure S5, online supplementary table S7) unravelled gene expression changes on a large fraction of the 1693 genes that were similarly modulated in patients with CHC and NASH suggesting that the DEN/CDAHFD mouse model enables the study of epigenetic/transcriptomic changes associated with human liver carcinogenesis.
Supplemental material

Bromodomain (BRD)4 inhibitor JQ1 reduces liver tumour burden in a mouse model of non-alcoholic steatohepatitis (NASH)-hepatocellular carcinoma (HCC). (A) Schematic representation of the proof-of-concept study using a mouse model of DEN and choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD)-induced hepatocarcinogenesis. (B) Transcriptomic changes of genes with significant H3K27ac modifications from livers from patients with NASH and chronic hepatitis C (CHC) as explained in figure 1D (overlapping genes) and corresponding changes in vehicle or JQ1-treated DEN/CDAHFD mice. (C) JQ1 significantly reduces tumour burden in vivo. While body weights are stable, liver weights as well as the numbers of tumours are significantly (*p<0.05; ***p<0.001; ****p<0.0001, unpaired t-test) reduced in JQ1-treated (n=8) compared with untreated (n=8) mice. Results are expressed as means±SEM. (D) Representative macroscopic photographs of livers (x 1.5 magnified), H&E and Sirius red staining of liver sections from vehicle and JQ1-treated mice. Tumour nodules are indicated by an arrow head and are delimited by dashed lines. (E) JQ1 efficiently reduces liver fibrosis and inflammation. Fibrosis stage was evaluated through quantitative digital analysis of whole-scanned liver sections (collagen proportional area (CPA)) and fibrotic gene expression in JQ1-treated (n=3) compared with JQ1-untreated (n=3) mice. Results are expressed as means±SD. (D) Expression of inflammatory genes Ccl2 and Tnfα are shown as means±SD (*p<0.05; **p<0.01, unpaired t-test). Gene expression was assessed by quantitative reverse transcription (qRT)-PCR. (F) Gene Set Enrichment Analysis (GSEA) pathway analysis of transcriptional changes in JQ1-treated DEN/CDAHFD mouse livers. Normalised enrichment scores (NES) of significantly enriched hallmark pathways derived from RNA-Seq analysis of livers from vehicle (n=3) and JQ1-treated (n=3) mice compared with control/phosphate-buffered saline (PBS) (n=5) mice. JQ1 partially reverses the NASH-mediated and HCV-mediated induction (red labels) or repression (green labels) of pathways identified in patients with NASH and/or CHC. (G) PLS status and expression of HCC high-risk/low-risk genes from the RNA-Seq analysis shown in panel D. Heatmaps show: (top) the classification of the PLS global status as poor (orange) or good (green) prognosis; (bottom) the significance of induction (red) or suppression (blue) of PLS poor-prognosis or good-prognosis genes. FDR, false discovery rate. (H) Western blot analysis showing c-Myc protein expression in control/PBS (n=3), vehicle (n=3) or JQ1-treated (n=3) mouse livers. Bottom panel graph shows the quantification of western blot analysis from six different animals (n.s., non-significant; unpaired t-test). (I) Recruitment (ChIP-qPCR assays) to the promoter-enhancer regions of indicated genes, using liver of control, DEN/CDAHFD and JQ1-treated mice, using IgG and BRD4 antibodies (*p≤0.05; **p≤0.01; Mann-Whitney U test).
JQ1 treatment significantly reduced liver weights of DEN/CDAHFD mice without altering their body weight (figure 5C). Quantification of tumour nodules revealed an overall decrease in their number irrespective of their size and location (figure 5C,D). Moreover, JQ1 significantly (p<0.05) reduced liver fibrosis measured by collagen proportionate area (CPA) and expression of genes mediating fibrogenesis (Acta2, Tgfb-1 and Timp1) as well as liver inflammation (Ccl2 and Tnfα expression) (figure 5E). Comparative pathway analysis of patient and mouse livers revealed that JQ1 reverted the transcriptional liver reprogramming identified in patients with CHC and NASH, including the induced pathways related to EMT, inflammatory response, IL2/STAT5 signalling and allograft rejection, and the suppressed pathways related to oxidative phosphorylation, bile acid and fatty acid or xenobiotic metabolism (figure 5F). JQ1 reverted the PLS from a poor-prognosis to a good-prognosis status (figure 5G). Furthermore, our analyses revealed that expression of 59% (1007 out of 1693 genes) of H3K27ac-altered genes in patients were reverted by JQ1 treatment in DEN/CDAHFD mice (figure 5B and online supplementary figure S5 and online supplementary table S7). Of note, hepatic c-Myc protein expression (figure 5H and online supplementary figure S6) showed a partial and variable non-significant decrease (vehicle vs phosphate-buffered saline p=0.4196; JQ1 vs vehicle p=0.7233). We also addressed the relevance of the YAP/TAZ pro-tumourigenic activity causing the regression of neoplastic lesions by BRD4 inhibition.42 We did not find any alteration of YAP expression and phosphorylation (online supplementary figure S7), indicating that the effect of JQ1 is YAP-independent.
To assess whether BRD4 displacement from the promoter was the mode of action of JQ1 in DEN/CDAHFD mice, we performed a ChIP-qPCR on genes that were found epigenetically and transcriptionally changed in patients. We found that JQ1 treatment led to a significant reduced BRD4 occupancy on SOX9 (p<0.05), EPCAM (p<0.01), KRT7 (p<0.05) and NFκB2 (p<0.01) validating its mechanism of action (figure 5I). At the same time, the expression of chromatin readers remained unchanged (online supplementary figure S8). These data demonstrate that JQ1 inhibits liver disease progression and hepatocarcinogenesis by reverting the transcriptional liver reprogramming and identify chromatin readers as targets for HCC chemoprevention.
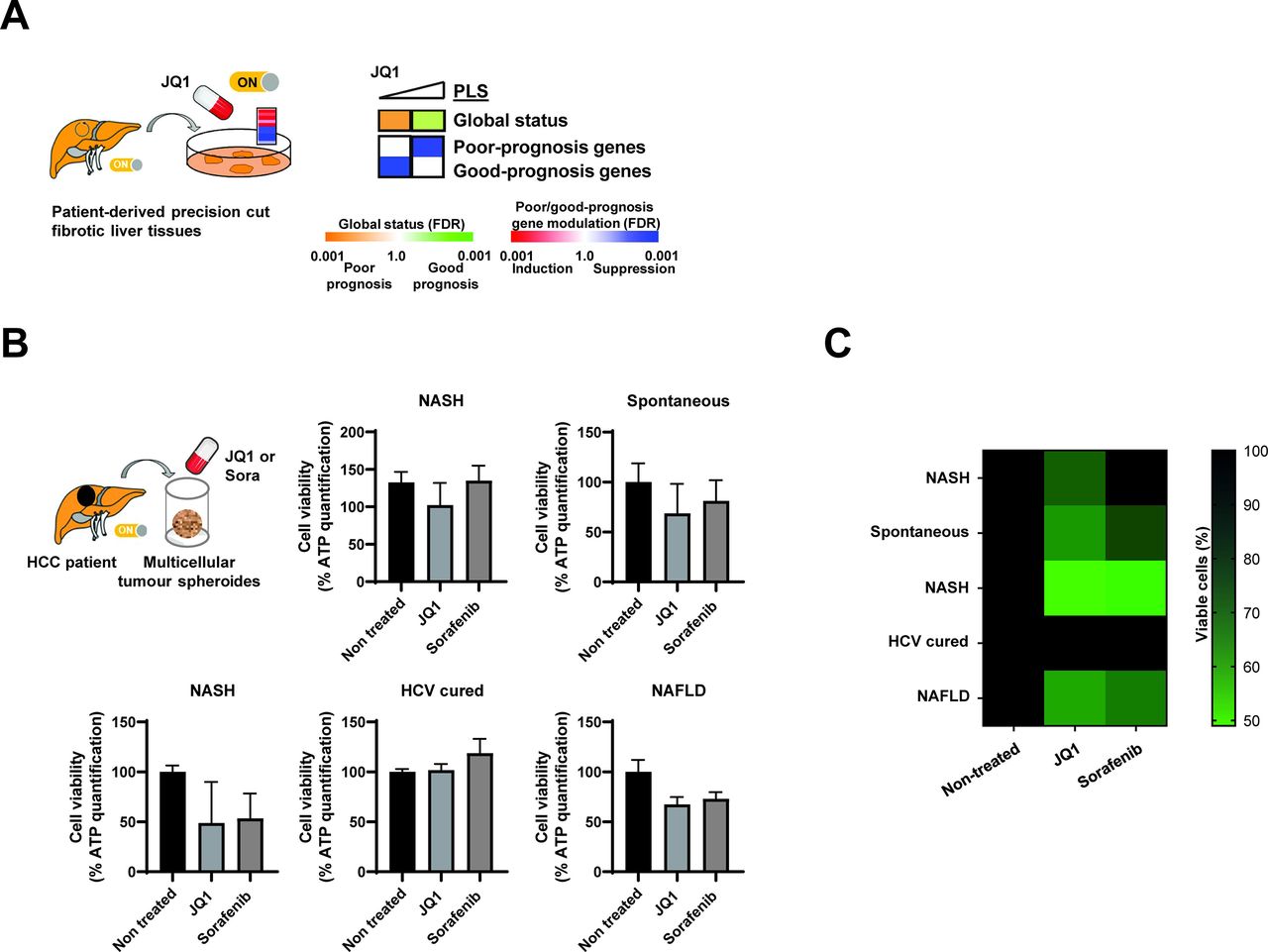
To obtain insights on whether JQ1 may have therapeutic efficacy in patients, we assessed the effects of JQ1 on the PLS in patient-derived precision cut fibrotic liver tissues. We found that JQ1 dose-dependently reverted the PLS poor-prognosis status with significant decreased expression of the PLS genes associated with high risk of HCC (figure 6A). Aiming to study whether JQ1 exerts a direct anticancer effect on established HCC, we used patient-derived HCC spheroids.20 21 JQ1 reduced HCC cell viability of patient-derived spheroids in four out of five patients similar to sorafenib (figure 6B,C), suggesting that JQ1 may also have a direct effect on HCC.

Proof-of-concept for therapeutic impact of JQ1 in patient-derived ex vivo in liver disease and hepatocellular carcinoma (HCC) models. (A) JQ1 reverts the poor-prognosis prognostic liver signature (PLS) in culture of patient-derived tissue that were surgically resected from patients diagnosed with chronic hepatitis C (CHC) (fibrosis stage=F3) and ex vivo treated with JQ1 0.1 and 1 µM. Heatmaps show: (top) the classification of the PLS status as poor (orange) or good (green) prognosis; (bottom) the significance of induction (red) or suppression (blue) of poor-prognosis or good-prognosis genes. FDR, false discovery rate. (B) JQ1 decreases HCC cell viability in a three-dimensional patient-derived tumour spheroid model. HCC spheroids were generated from HCC tissues with different aetiologies from five different patients and incubated with JQ1 (50 nM) or sorafenib (10 µM) as described in ‘Materials and methods’ section. Cell viability was assessed 6 days after treatment by measuring ATP levels. Each experiment shows mean±SEM in JQ1 and sorafenib-treated spheroids in percentage compared with dimethyl sulfoxide-treated tumour spheroids (n=4 for each condition). (C) Heatmap recapitulating the effect of JQ1 on the different patient-derived tumour spheroids shown in panel B. Colours indicate the percentage of viable cells (black=100%, light green=50%). NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Discussion
The key conceptual advancements of this study are the following: (1) CHC and NASH share epigenetic and transcriptional changes associated with HCC risk in patients; (2) integrative analyses in patients and model systems demonstrate that liver disease-induced epigenetic modifications are targets for HCC chemoprevention; (3) inhibition of chromatin reader BRD4 by a small molecule reduces liver cancer development in vivo by reverting the epigenetic and transcriptional reprogramming associated with cancer risk in patients. Therefore, the inhibition of chromatin readers or modifiers provides a previously undiscovered approach for HCC prevention. Here, we show that liver injury in both aetiologies results in similar epigenetic footprints and transcriptional changes associated with liver disease progression and HCC risk (figures 1–2). This finding is in line with the clinical and pathological observation that CHC and NASH share many phenotypes such as steatosis, insulin resistance, inflammation and fibrosis43 and that HCCs of CHC and NASH exhibit similar deregulated pathways and genetic footprints.44 45 These observations also indicate that HCV infection may serve as a model for understanding progression of liver disease progression and hepatocarcinogenesis in NASH.
Our data across models demonstrate that inhibition of disease-induced epigenetic changes robustly inhibits gene expression associated with HCC risk (figures 3–5) and markedly and significantly inhibits hepatocarcinogenesis in a state-of-the-art in vivo model for NASH-induced HCC (figure 5). Collectively, these data demonstrate that epigenetic modifications are a target for HCC prevention (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Targeting liver disease-induced epigenetic modifications and hepatocellular carcinoma (HCC) chemoprevention. Model: chronic hepatitis C (CHC) or non-alcoholic steatohepatitis (NASH) induce a transcriptional reprogramming of liver cells through genome-wide H3K27ac changes driving HCC risk and hepatocarcinogenesis. H3K27ac-mediated transcriptional reprogramming constitutes a target for HCC chemoprevention by bromodomain (BRD)4 inhibitors.
HCC prevention in patients with advanced liver fibrosis is likely the most effective strategy to improve patient survival, because tumour recurrence after surgical treatment is frequent, and therapeutic approaches for advanced disease remain unsatisfactory.46 47 Approved therapies for NASH are absent and late-stage clinical trials show only moderate success. In CHC, DAA-cured patients with advanced fibrosis remain at risk for HCC.2 Addressing this key unmet medical need, we identify BRD4 as a candidate target for HCC chemoprevention.
Mechanistically, our findings revealed a partial decrease in c-Myc expression following JQ1 treatment indicating that the HCC chemopreventive effect may be partially mediated by c-Myc, a well-known pathway for BRD4-mediated downstream signalling in lymphoma cells.38 In this regard, it is of interest to know that the pro-oncogene c-Myc is frequently overexpressed in HCC and it has been described that JQ1 inhibits c-Myc-positive tumour cell growth.37 39 40 However, the absence of a robust and significant reduction of c-Myc levels suggests that additional mechanisms are likely involved in the chemopreventive effect of JQ1. Our analyses in patients and models (figures 1–5) rather suggest that altered inflammatory gene expression such as NF-κB expression or antifibrotic responses may play a mechanistic role. This is in line with observations showing that BRD4 inhibition results in amelioration of liver inflammation and fibrosis (figure 5E)48 49 and that fibrosis is one of the most important risk factors for HCC.46 The inhibition of cell viability of patient-derived tumour spheroids (figure 6) suggests that JQ1 may also have a direct effect on HCC in addition to indirect chemopreventive effects via reducing fibrosis and inflammation (figure 5E 48 49).
Epidrugs are entering the clinic with the first licensed compounds. BRD inhibitors are currently in clinical trials for treatment of various malignancies with acceptable safety profiles.50 In summary, our study uncovers epigenetic regulation of transcriptional reprogramming as a potential target for HCC chemoprevention—a key unmet medical need for patients with advanced fibrosis.
Supplemental material
Acknowledgments
The authors would like to thank R. Bartenschlager (University of Heidelberg, Germany) and C. Rice (Rockefeller University, New York, New York, USA) for providing plasmids for HCVcc production, T. Wakita (National Institute of Infectious Diseases, Tokyo) for HCV JFH1 protein expression constructs and F. Chisari (The Scripps Research Institute, La Jolla, California, USA) for the gift of Huh7.5.1 cells. psPAX2 (Addgene plasmid: #12260). pMD2.G (Addgene plasmid: #12259) was a gift from D. Trono (EPFL, Lausanne, Switzerland) and LentiGuide-Puro (Addgene plasmid # 52963) was a gift from F. Zhang (Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA). pXPR_BRD111 (Addgene plasmid: #78166) was a gift from W. Hahn (Harvard Medical School, Boston, Massachusetts, USA). The authors would like to thank the Centre de Ressources Biologiques-Biological Resource Centre, Strasbourg, France, for the management of patient-derived liver tissues. The authors would also like to thank the IGBMC high-throughput sequencing facility, a member of the 'France Génomique' consortium (ANR10-INBS-09-08) for NGS sequencing.
Footnotes
FJ, NH, EC and SL contributed equally.
BCF and TFB contributed equally.
Contributors TFB initiated and coordinated the study. TFB, BCF, FJ, NH, EC, SL, KG and YH designed experiments and analysed data. NH, EC, SL, MAO, CT, AARS, KG, AO, MS and GA performed experiments. FJ performed the computational analyses of NGS data. HES performed PLS drug screen analyses. KKT contributed to the concept and approach of the study. CS and CB performed sequencing of ChIPmentation experiments. ID, AO, HES, SJP, MBZ, NF, AS, AM, KG, AARS and YH analysed data. PT, MS, KC, HA and PP provided clinical liver tissue samples. RM and AS analysed the histopathology of mouse liver tissues. NH, FJ, EC, FHTD, BCF, MBZ, SJP, AM, CS and TFB wrote the manuscript.
Funding This work was supported by the European Union (ERC-AdG2014 HEPCIR #671231 (TFB and YH) and H2020 HEPCAR #667273, ERC PoC-HEPCAN #862551 to TFB). ARC, Paris and Institut Hospitalo-Universitaire, Strasbourg (TheraHCC and TheraHCC2.0 IHUARC IHU201301187 and IHUC201901299 TFB), the Foundation of the University of Strasbourg and Roche Institute (HEPKIN), the Agence Nationale de Recherches sur le Sida et les Hépatites Virales (2017/1633), the US Department of Defense (W81XWH-16-1-0363 to TFB and YH), the Cancéropôle du Grand-Est, the National Institutes of Health (DK099558 and T32CA073145-20 to SL, R01CA233794 to YH and TFB, NCI 1R21CA209940-01A1 to TFB), AMED (19fk0210020h0003 to KC), AASLD Foundation (Pinnacle Research Award) and the Massachusetts General Hospital Department of Surgery (KKT) and the Cancer Prevention & Research Institute of Texas RR180016 to YH. This work has been published under the framework of the LABEX ANR-10-LABX-0028_HEPSYS and PLAN CANCER 2014-2019 HCCMICTAR and benefits from a funding from the state managed by the French National Research Agency as part of the Investments for the Future Program, INCa (National Institute for Cancer) and INSERM.
Competing interests The University of Strasbourg, INSERM, the IHU Strasbourg and Mount Sinai Hospital have filed a patent application on the clinical gene signature-based human cell culture model and uses thereof with YH and TFB as co-inventors (WO 2016174130 A1), which has been licensed to Alentis Therapeutics, Basel, Switzerland.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval The protocols were approved by the Ethics Committee of the Strasbourg University Hospitals (DC-2016–2616), Mount Sinai Hospital, New York (HS13-00159), Basel University Hospital (EKNZ 2014–362) and Hiroshima University Hospitals (E-1049–1).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data, i.e. raw reads from ChIP-seq and RNA-seq experiments in fastq files, are available in a public, open access repository without restrictions on the use or distribution of the data. The Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) project accession number for accessing the data produced for this study is PRJNA606244.