Article Text

Abstract
Objective Impaired hepatic fatty acids oxidation results in lipid accumulation and redox imbalance, promoting the development of fatty liver diseases and insulin resistance. However, the underlying pathogenic mechanism is poorly understood. Krüppel-like factor 16 (KLF16) is a transcription factor that abounds in liver. We explored whether and by what mechanisms KLF16 affects hepatic lipid catabolism to improve hepatosteatosis and insulin resistance.
Design KLF16 expression was determined in patients with non-alcoholic fatty liver disease (NAFLD) and mice models. The role of KLF16 in the regulation of lipid metabolism was investigated using hepatocyte-specific KLF16-deficient mice fed a high-fat diet (HFD) or using an adenovirus/adeno-associated virus to alter KLF16 expression in mouse primary hepatocytes (MPHs) and in vivo livers. RNA-seq, luciferase reporter gene assay and ChIP analysis served to explore the molecular mechanisms involved.
Results KLF16 expression was decreased in patients with NAFLD, mice models and oleic acid and palmitic acid (OA and PA) cochallenged hepatocytes. Hepatic KLF16 knockout impaired fatty acid oxidation, aggravated mitochondrial stress, ROS burden, advancing hepatic steatosis and insulin resistance. Conversely, KLF16 overexpression reduced lipid deposition and improved insulin resistance via directly binding the promoter of peroxisome proliferator-activated receptor α (PPARα) to accelerate fatty acids oxidation and attenuate mitochondrial stress, oxidative stress in db/db and HFD mice. PPARα deficiency diminished the KLF16-evoked protective effects against lipid deposition in MPHs. Hepatic-specific PPARα overexpression effectively rescued KLF16 deficiency-induced hepatic steatosis, altered redox balance and insulin resistance.
Conclusions These findings prove that a direct KLF16–PPARα pathway closely links hepatic lipid homeostasis and redox balance, whose dysfunction promotes insulin resistance and hepatic steatosis.
- lipid metabolism
- fatty liver
- nonalcoholic steatohepatitis
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hepatic steatosis, which stems from a lipid metabolism disorder and is characterised by a dysfunction of hepatic fatty acids oxidation causing excessive ectopic lipid deposition and an altered intracellular redox balance, represents the greatest risk factor of non-alcoholic fatty liver disease (NAFLD) and insulin resistance in mammals.
However, the details of the underlying molecular mechanisms are still unclear, and consequently, the therapeutic approach to it remains inadequate.
In the liver, Krüppel-like factor 16 (KLF16) is an abundant transcription factor that is involved in antitumorigenicity and endocrine homeostasis. However, the function of KLF16 on hepatic lipid metabolism and insulin resistance is still undefined.
Significance of this study
What are the new findings?
Interestingly, hepatic KLF16 expression is significantly reduced in NAFLD patients and mice models, and the reduction of KLF16 is causatively linked to the suppression of genes involved in fatty acids oxidation and hepatic steatosis development.
The hepatocytes-specific KLF16 knockout mice promptly develops liver steatosis and insulin resistance, due to a dysfunctional lipid catabolism. KLF16 deficiency leads to severe mitochondrial stress, redox imbalance and inflammation.
Conversely, hepatic-specific KLF16 overexpression effectively attenuates hepatic steatosis and insulin resistance while improving hepatic mitochondrial function and mitigating both reactive oxygen species burden and inflammatory stress in HFD mice and db/db mice.
KLF16 impacts on cellular lipid catabolism by directly binding to the GGGGCCCG element mapped in the peroxisome proliferator-activated receptor α (PPARα) gene promoter (from −435bp to −426bp) and activating PPARα gene transcription. Restoration of PPARα expression does recover from hepatic steatosis and insulin resistance evoked by a KLF16 deficiency.
How might it impact on clinical practice in the foreseeable future?
Impaired fatty acid oxidation acts as a predominant risk factor of hepatic fat accumulation and insulin resistance. Our present findings prove the crucial importance of KLF16 in promoting hepatic lipid catabolism and insulin sensitivity in a PPARα-dependent manner. Targeting the KLF16–PPARα pathway supplies the molecular evidence of a novel therapeutic approach to the treatment of NAFLD.
Introduction
A well-coordinated lipid metabolism, including fatty acid oxidation, lipogenesis, and lipoprotein uptake and secretion, keeps mammals in healthy conditions.1 The ectopic overaccumulation of fatty acids drives the development of highly prevalent metabolic diseases, like hyperlipidaemia, diabetes and non-alcoholic fatty liver disease (NAFLD).2 Approximately 15%–80% of normal and obese individuals display early manifestations of hepatic steatosis due to a high-fat diet (HFD) or an excessive alcohol intake.3 Although a severely impaired energy homeostasis, inflammation and redox balance associate with liver steatosis progression, the underlying causative mechanisms of a progressively worsening NAFLD remain incompletely understood.
In patients with NAFLD, an impaired fatty acid oxidation is characterised by an inordinate deposition of triglycerides (TGs) inside the hepatocytes. The subsequent onset of insulin resistance, inflammation and oxidative stress aggravate NAFLD phenotype and drive the progress to non-alcoholic steatohepatitis, fibrosis, cirrhosis and even hepatocellular carcinoma.4 5 Thus, restoration of fatty acid oxidation was proposed as an effective strategy to protect against NAFLD.6 The transcriptional factor peroxisome proliferator-activated receptor α (PPARα) is a predominant regulator of hepatic lipid homeostasis. Under physiological conditions, PPARα pharmacological or genetic activation increased the expression of genes involved in mitochondrial and microsomal fatty acid oxidation, facilitating gluconeogenesis and acetyl-CoA synthesis to provide the needed ATP and nicotinamide adenine dinucleotide phosphate.7 Persistent PPARα activators could promote liver peroxisomal and mitochondrial fatty acid oxidation and have been used to treat NAFLD in clinical settings.8–10 Conversely, PPARα deficiency led to mitochondrial dysfunction and predisposed mice to hyperketonaemia and hyperglycaemic symptoms.11 PPARα knockout mice displayed a pronounced reactive oxygen species (ROS) generation when fed on HFD.12 Therefore, targeting PPARα may be an effective therapeutic strategy for NAFLD.
Krüppel-like factor 16 (KLF16), consisting of Cys2/His2 zinc-finger DNA-binding proteins, belongs to a family of transcription factors rich in GT-binding or GC-binding sites. KLF16 coordinates multiple biological processes including growth, cell metabolism and cell death.13 14 As a critical member of KLF family, KLF16 possesses a shared basic transcription element binding protein KLF group and associates with reproductive endocrinology by silencing specificity protein (Sp)/KLF sites.15 However, hitherto, the role of KLF16 in hepatic lipid metabolism and insulin response had not been investigated.
Materials and methods
Patient tissue preparation
Liver samples were taken from 66 patients with biopsy for cholecystitis and preoperative testing at the First Affiliated Hospital of Guangzhou University of Chinese Medicine. Written informed consent was obtained from each patient included in the study. Viral and alcoholic hepatitis were ruled out in this study. None of the patients were treated with a lipid/glucose-lowering therapy. Samples were divided into normal group and NAFLD group based on gold-standard histological classification (NAFLD activity score (NAS)) and liver pathology.
Tolerance test
Mice were intraperitoneal injection (i.p.) injected with D-glucose (1–2 g/kg) or pyruvate sodium (1–1.5 g/kg) after 16-hour fasting. For the insulin tolerance test, mice were i.p. injected with insulin (0.5–0.75 U/kg) after 6-hour fasting. Blood glucose levels were measured respectively from the tail vein using a glucose monitor (OneTouch Ultra; On Call EZII, China) at 0, 15, 30, 45, 60, 90 and 120 min.
Cell culture
Mouse primary hepatocytes (MPHs) were isolated from livers of male C57BL/6J mice, PPARα−/− mice or Klf16alb−/− mice (8 weeks old) as previously reported. Briefly, liver cells were isolated by injection of 0.5 mg/mL type collagenase (Sigma) through the inferior vena cava after anaesthesia in mice. Trypan blue dyes were used to detect cell activity (70% or more). Then, MPHs were cultured in RPMI-1640 Medium containing 10% fetal bovine serun (FBS), 100 units/mL penicillin and 0.1 mg/mL streptomycin. MPHs were infected with Ad-Gfp, Ad-Klf16, Ad-shLuci, Ad-shKlf16 or Ad-Pparα for 24 hours, and the medium was replaced with oleic acid and palmitic acid (OA and PA) containing fresh medium for another 24 hours. To inhibit PPARα, MPHs were treated with GW6471 (10 µM, MedChemExpress). Cells were then collected for further analysis.
Chromatin immunoprecipitation (ChIP) assay
Liver samples from indicated mice were lysised and collected. The ChIP assay were done using EZ ChIP Chromatin Immunoprecipitation Kit (Cell Signaling Technology, Danvers, USA), following the manufacturer’s instructions. The protein–DNA complexes were immunoprecipitated with mouse IgG antibody (control) or anti-KLF16 antibody. The promoter region of PPARα was amplified by PCR and qPCR, using primers: F:CACTGCCGTCCAGGGGGTGT; R:CCCTGCGGGGTCCCGCGGCG.
Fatty acid oxidation assays
Adenovirus-infected MPHs were incubated with 0.4 µCi/mL [9,10-3 H] oleic acid (PerkinElmer Life Science) and 100 µM unlabelled oleic acid (conjugated with bovine serum albumin (BSA)) in Krebs-Ringer buffer (119 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2.6 mM MgSO4, 24.6 mM NaHCO3, 2.6 mM KH2PO4, 10 mM HEPES, pH 7.4) for 1 hour at 37°C. Supernatants were collected and incubated with 1.3 M perchloric acid. All samples were next centrifuged at 16 000× g for 10 min. Supernatants were neutralised with 2 M KOH, 0.6M MOPS. Scintillation liquid (3 mL) was then added, and [3H] radioactivity was measured.
ROS detection
MPHs, seeded in six-well plates and treated as previously, were incubated with DCFH-DA (5 µM) at 37°C for 0.5 hour, avoiding light. Cells were rinsed thrice with PBS, and fluorescence emission was detected using fluorescence microscopy. For hepatic ROS analysis, mice were injected with DCFH-DA (10 mM) via tail vein 1 hour before sacrifice, and next their livers were collected. ROS levels were assessed using Berthold Technologies LB983 NC100.
Transmission electron microscopy
Fresh livers (1 mm3) or MPHs were collected and fixed for longer than 2 hours in 2% paraformaldehyde and 2.5% glutaraldehyde solution and next rinsed thrice with 0.1 M PBS. The specimens were fixed in 1% osmic acid, dehydrated first through an acetone gradient (50%, 70% and 90%), next thrice in 100% acetone and finally soaked and embedded in Epon 812 epoxy resin. Semithin sections were cut in a Reichert ultramicrotome, and next block laser positioning was performed. Ultrathin sections were stained with lead citrate and uranium acetate. The ultrastructural morphological features of hepatocytes and mitochondria were assessed under a transmission electron microscope.
Analytical procedures and chemicals
Serum concentrations of TGs, total cholesterol (TC), alanine aminotransferase (ALT) and aspartate aminotransaminase (AST) were determined using an automated Monarch device (Science and Technology Park, Guangzhou, China). Hepatic TGs and cholesterol were determined using a colorimetric diagnostic kit (Applygen Technologies, Inc, Beijing, China). Serum insulin concentrations were measured by ELISA (Mercodia, Uppsala, Sweden). Serum ketone body were determined by ELISA (R&D Systems, Minneapolis, USA). Penicillin, streptomycin, OA, PA and dimethyl sulfoxide (DMSO) were purchased from Sigma.
Statistical analysis
All results are given as means±SEMs. PRISM software (V.7.00, GraphPad Software, Inc) was used to assess statistical significance between two groups via Student’s t-test and one-way analysis of variance (ANOVA) followed by post hoc Tukey test for multiple groups comparisons. The curves of body weight and tolerance tests were analysed using a repeated measure two-way ANOVA. A p value <0.05 was considered as significant.
Additional methods
Other methods are detailed in online supplementary materials and methods.
Supplemental material
Results
Hepatic KLF16 expression is decreased in hepatic steatosis
To assess genes involved in hepatic steatosis, we performed mRNA assays on livers of wild type mice fed on chow diet and HFD, as well as db/m and db/db mice. Interestingly, KLF16 expression dramatically decreased in db/db and HFD mice (figure 1A and B), and a fasting and refeeding cycle, which stimulates lipid dynamics, significantly changed KLF16 expression (figure 1B and online supplemental figure 1A). Consistent with this finding, KLF16 protein levels were lower in patients with NAFLD than in controls, and an OA and PA cochallenge significantly reduced KLF16 expression in MPHs and HepG2 cells (figure 1C and online supplemental figure1B,C). Immunofluorescence analysis confirmed the decreased nuclear KLF16 protein in MPHs on OA and PA treatment (figure 1D). Moreover, forskolin treatment decreased KLF16 expression in MPHs and HepG2 cells, while the presence of insulin increased KLF16 levels, implying a key ability of KLF16 to sense these hormones (figure 1E,F).

Hepatic KLF16 expression is downregulated in hepatic steatosis. (A) Heat map showing the expression of hepatic KLF16 and other genes regulating lipid metabolism in C57BL/6J, HFD, db/m and db/db mice (n=2/group). (B) Western blots of hepatic KLF16 in db/db (upper panel), HFD (middle panel) mice and C57BL/6J mice on fed and refeeding cycles (down panel) (n=6/group). (C) Western blots of KLF16 in liver samples from healthy volunteers and NAFLD patients, HepG2 cells and MPHs treated with OA and PA for 24 hours (n=6/group). (D) OA and PA treatment reduced nuclear KLF16 levels in MPHs. (E and F) FSK (E) and insulin (F) treatment changed KLF16 expression in HepG2 cells and MPHs (n=6/group). Comparable results obtained in three independent experiments. FSK, forskolin; HFD, high-fat diet; KLF16, Krüppel-like factor 16; MPH, mouse primary hepatocyte; NAFLD, non-alcoholic fatty liver disease; OA, oleic acid; PA, palmitic acid.
Hepatic KLF16 deficiency worsens hepatic steatosis and increases insulin resistance
To investigate the effects of KLF16 on hepatic steatosis and related metabolic disorders, we generated hepatic-specific KLF16 knockout mice (KLF16alb−/−) (online supplemental figure 2A). Hepatic KLF16 deletion did not alter the body weight of mice on chow diet, while the ratio value of liver weight/body weight was increased (figure 2A and online supplemental figure 2B). In keeping with this, serum and hepatic TGs were increased in KLF16alb−/− mice, while H&E and Oil red O staining also demonstrated the occurrence of a severe lipid accumulation in the same mice (figure 2B and online supplemental figure 2C). Conversely, TC content was not affected (online supplemental figure 2C). Moreover, we found that KLF16 deficit significantly increased the expression of genes involved in fatty acid synthesis and suppressed the genes involved in lipolysis and fatty acids oxidation, especially PPARα and its target genes (figure 2C,D).
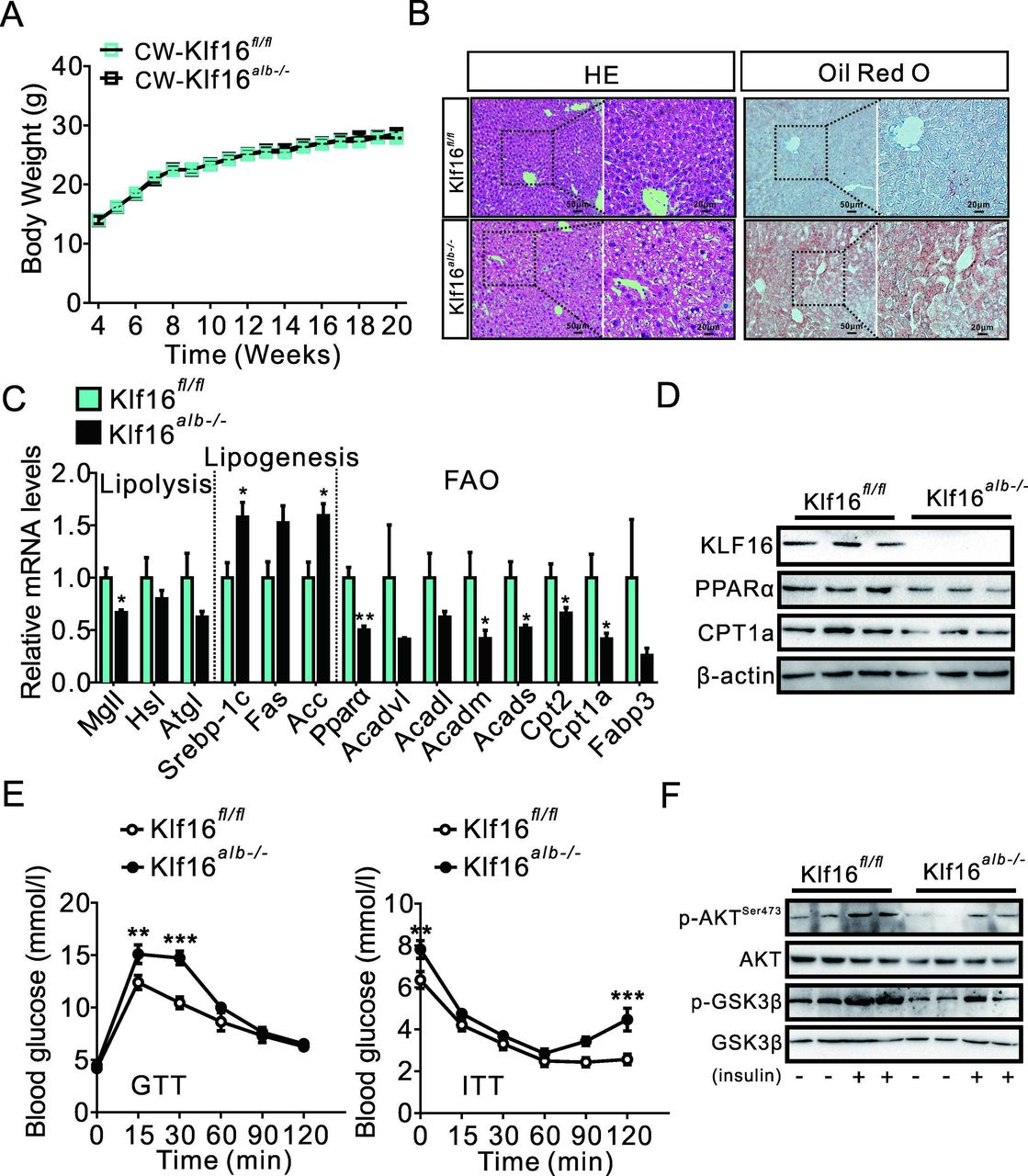
Hepatic KLF16 deficiency impairs lipid catabolism and insulin sensitivity. (A) Hepatic KLF16 deletion does not affect the body weight of mice on a chow diet; repeated measure two-way analysis of variance (ANOVA) was used to analyse the difference of two curves. (B) H&E and Oil red O staining increased lipid deposition in KLF16alb−/− mice. (C) KLF16 deletion changed the expression of genes involved in lipid metabolism. (D) KLF16 deletion reduced PPARα and CPT1A protein levels. (E) KLF16 deletion impaired glucose tolerance and insulin sensitivity; repeated measure two-way ANOVA was used to analyse the difference of two curves. (F) KLF16 deletion reduced phosphorylation of Akt/GSK-3β. Comparable results obtained in three independent experiments. Data are means±SEM; n=8 mice/group. *P<0.05, **p<0.01, ***p<0.001. KLF16,n Krüppel-like factor 16.
Interestingly, hepatic KLF16 deletion also increased fasting blood glucose and insulin levels (online supplemental figure 2D). KLF16 deficit impaired glucose tolerance, pyruvate tolerance and insulin sensitivity and went along with an elevated expression of gluconeogenesis-related genes (PEPCK and G6Pase) (figure 2E and online supplemental figure 2E,F). Moreover, western immunoblotting analyses revealed a decrease in the phosphorylation of AKT and GSK3β, suggesting that insulin signalling was deeply hampered in KLF16alb−/− mice (figure 2F).
Additionally, the dysfunction of lipid metabolism impaired hepatic mitochondrial homeostasis in KLF16alb−/− mice, as revealed by decreased mitochondrial numbers and altered mitochondrial ultrastructure (online supplemental figure 2G,H). These results in a decrease in the expression of a cascade of genes involved in oxidative response and increases ROS production (online supplemental figure 2I,J). Moreover, the expression of hepatic inflammatory genes were upregulated due to KLF16 knockout, which promoted the development of hepatic steatosis and insulin resistance (online supplemental figure 2K). Collectively, these data showed that KLF16 deficiency impaired hepatic lipid and glucose metabolism, leading to hepatic steatosis and raised insulin resistance in mice.
KLF16 overexpression alleviates hepatic steatosis in db/db and HFD mice
We further set up hepatocyte-specific KLF16-overexpressing mice models through tail vein injections of AAV-Klf16 to confirm the function of hepatocyte KLF16 in hepatic steatosis and related metabolic disorders. AAV-Klf16 injection significantly increased hepatic-specific KLF16 overexpression in db/db mice, which coexisted with an upregulated expression of genes involved in fatty acid oxidation, while lipogenic genes expression did not change (figure 3A,B and online supplemental figure 3A). KLF16 overexpression did not alter the body weight but reduced the liver weight/body weight ratio value in db/db mice (figure 3C and online supplemental figure 3B). Moreover, AAV-Klf16-infected mice displayed a higher level of fatty acid oxidation, made plain by the elevated levels of circulating ketone bodies (figure 3D). This helped decrease hepatic lipid deposition in db/db mice and also reduced TGs levels, while TC level did not change (figure 3E and online supplemental figure 3C).
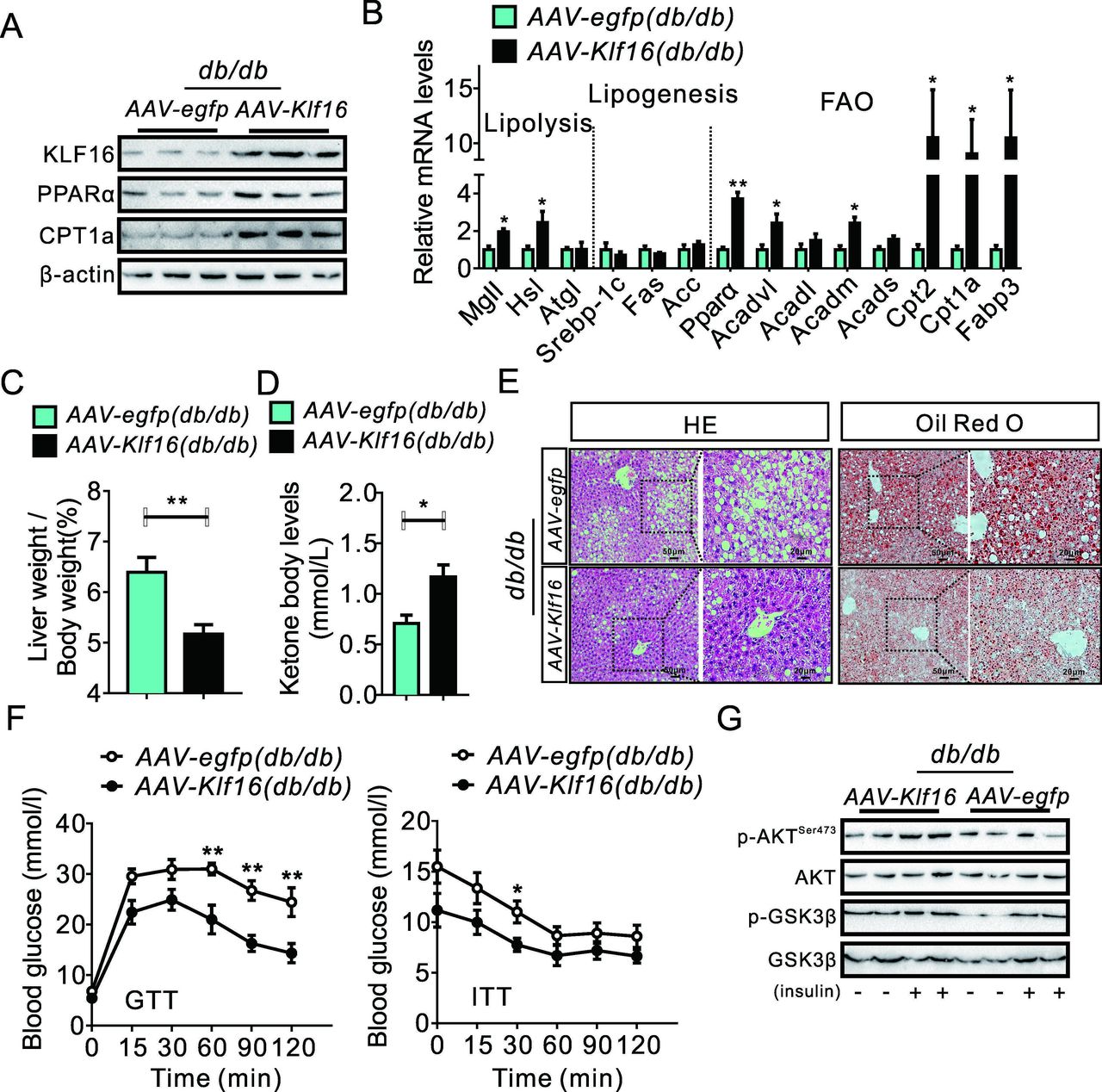
KLF16 overexpression alleviates hepatic steatosis in db/db mice. (A) KLF16 overexpression increased hepatic PPARα and CPT1A protein levels in db/db mice. (B) KLF16 overexpression changed the expression of genes involved in hepatic lipid metabolism in db/db mice. (C and D) KLF16 overexpression reduced the value of the ratio liver weight/body weight (C) and increased serum ketone body levels (D) in db/db mice. (E) H&E and Oil red O staining demonstrated a decreased hepatic lipid accumulation in AAV-Klf16-infected db/db mice. (F) KLF16 overexpression improved glucose tolerance and insulin sensitivity in db/db mice, repeated measure two-way analysis of variance was used to analyse the difference of two curves. (G) KLF16 overexpression increased the phosphorylation of Akt/GSK-3β in db/db mice. Comparable results obtained in three independent experiments. Data are means±SEM; n=8 mice/group. *P<0.05, **p<0.01. KLF16, Krüppel-like factor 16; PPARα, peroxisome proliferator-activated receptor α.
Moreover, KLF16 overexpression effectively decreased fasting blood glucose and insulin levels (online supplemental figure 3D). Thus, KLF17 overexpression in hepatocytes improved glucose tolerance, pyruvate tolerance and insulin sensitivity in db/db mice and helped suppress gluconeogenesis-related genes and recovered of insulin signalling (figure 3F,G and online supplemental figure 3E,F).
In parallel, KLF16 overexpression improved hepatic function in db/db mice, as shown by the decreased levels of ALT and AST (online supplemental figure 3G). Also, electron microscopy pictures of hepatic tissue confirmed the improved mitochondrial biogenesis and ultrastructural features (online supplemental figure 3H,I), both of which may have helped reduce hepatic ROS and upregulate the expression of genes involved in oxidative stress (online supplemental figure 3JK). Finally, KLF16 overexpression effectively downregulated the inflammatory genes in db/db mice, which helped attenuate liver steatosis (online supplemental figure 3L).
Corresponding results obtained in HFD mice. AAV-Klf16 infection increased hepatic-specific KLF16 expression in HFD mice and upregulated the expression of genes involved in fatty acid oxidation (figure 4A and online supplemental figure 4A). KLF16 overexpression did not change the body weight but reduced the liver weight/body weight ratio value in HFD mice (online supplemental figure 4B,C). Likewise, AAV-Klf16-infected HFD mice displayed an improved NAFLD phenotype as attested by a reduced hepatic lipid deposition and TGs levels (figure 4B and online supplemental figure 4D). Moreover, KLF16 overexpression decreased serum ALT and AST levels, suggesting an improvement of hepatic function in HFD mice (online supplemental figure 4E). Of note, AAV-Klf16-infected HFD mice exhibited decreased fasting blood glucose and insulin levels (online supplemental figure 4F). Moreover, the KLF16 overexpression significantly improved the HFD-induced glucose intolerance, pyruvate intolerance and insulin resistance, which went along with a suppression of gluconeogenesis-related genes and an upregulated insulin signalling (figure 4C,D and online supplemental figure 4G). Additionally, KLF16 overexpression also increased the numbers and improved the ultrastructural features of liver mitochondria in HFD mice (figure 4E,F). This significantly mitigated the otherwise HFD-induced hepatic ROS overproduction and altered expression of genes involved in oxidative stress (figure 4G and H). Finally, KLF16 overexpression suppressed inflammatory genes in the livers of HFD mice, helping ameliorating liver steatosis (online supplemental figure 4H).

KLF16 overexpression improves hepatic steatosis in HFD mice. (A) KLF16 overexpression changed the expression of hepatic genes involved in lipid metabolism in HFD mice. (B) H&E and Oil red O staining demonstrated reduced lipid levels in AAV-KLF16-infected HFD mice. (C) KLF16 overexpression increased glucose tolerance and insulin sensitivity in HFD mice; repeated measure two-way analysis of variance was used to analyse the difference of two curves. (D) KLF16 overexpression increased phosphorylation of Akt/GSK-3β in HFD mice. (E and F) KLF16 overexpression promoted hepatic mitochondrial biogenesis (E) and improved the ultrastructural features (F) in HFD mice. (G and H) KLF16 overexpression both decreased HFD-induced ROS overproduction (G) and increased antioxidant genes expression (H). Equivalent results obtained in three independent experiments. Data are means±SEM; n=8 mice/group. *P<0.05, **p<0.01. HFD, high-fat diet; KLF16, Krüppel-like factor 16; ROS, reactive oxygen species.
KLF16 regulates hepatic lipid metabolism in a PPARα-dependent manner
To systemically explore the mechanism by which KLF16 affects hepatic metabolism, RNAseq data of liver samples from Ad-Klf16 infected C57BL/6J mice were analysed. They showed that 2545 genes were differently expressed with 1229 genes upregulated and 1313 gene downregulated (figure 5A). Kegg pathway analysis showed that KLF16 overexpression significantly altered hepatic fatty acid metabolism and PPAR signalling pathway (figure 5B), as it highly induced PPARα and its target genes, suggesting a potential mediation of KLF16-evoked effects by PPARα (figure 5C). We further conducted luciferase assays using reporter constructs (pPPARα−1299, pPPARα−866, pPPARα−577 and pPPARα−220) in HepG2 cells. Our results revealed that KLF16 increased the transcriptional activity of pPPARα−1299, pPPARα−866 and pPPARα−577, while the stimulatory effects disappeared when the promoter region was further truncated to −220 bp (figure 5D, left panel), suggesting a KLF16 binding site being located from −577bp to −221bp. Mutation of the −435/−426nt site significantly diminished the KLF16-mediated activation of PPARα (figure 5D, right panel). Additionally, the occupancy of KLF16 on PPARα promoter was confirmed using a ChIP assay, and our ChIP-qPCR data also revealed a decreased KLF16 enrichment of the PPARα promoter fragment in HFD mice (figure 5E,F). In MPHs, KLF16 overexpression increased fatty acids oxidation and reduced lipid deposition by upregulating PPARα and its target genes (figure 6A–C and online supplemental figure 5A). Conversely, silencing KLF16 elicited the opposite effects (figure 6D–F and online supplemental figure 5B), thereby implying a strong correlation between KLF16 function and PPARα-mediated fatty acid oxidation. To further validate the key role of PPARα to mediate the KLF16-induced protective effects, we treated wild-type MPHs with GW6471, followed by Ad-Klf16 infection and OA and PA treatment for another 24 hours. In keeping with our hypothesis, PPARα pharmacological inhibition significantly diminished the KLF16-evoked protective effects against lipid deposition, as shown by the unchanged Oil red O-stained lipid levels and cellular TGs contents (figure 6G). Very similarly, KLF16 overexpression did not alter the OA and PA induced lipid accumulation in PPARα-deficient MPHs isolated from PPARα−/− mice (figure 6H).

Via binding PPARα promoter region KLF16 affects fatty acid oxidation. (A and B) Scatter plot (A), KEGG analysis (B) and heatmap (C) of RNAseq data in Ad-Gfp-infected or Ad-Klf16-infected C57BL/6J mice showing that KLF16 affects fatty acid oxidation and the expression of PPARα and its downstream target genes. (D) Luciferase reporter gene assay demonstrated the existence of a potential KLF16 binding site from −220bp to −577bp (left) and that a mutation between −426bp and −435bp abrogates any KLF16-induced transcriptional activity (right). (E and F) ChIP (E) and ChIP-qPCR (F) assessed the endogenous KLF16 enrichment on PPARα’ promoter in the liver of mice on chow diet or HFD. Comparable results obtained in three independent experiments. Data are means±SEM; n=6/group. *P<0.05, **p<0.01, ***p<0.001. HFD, high-fat diet; KLF16, Krüppel-like factor 16; PPARα, peroxisome proliferator-activated receptor α.
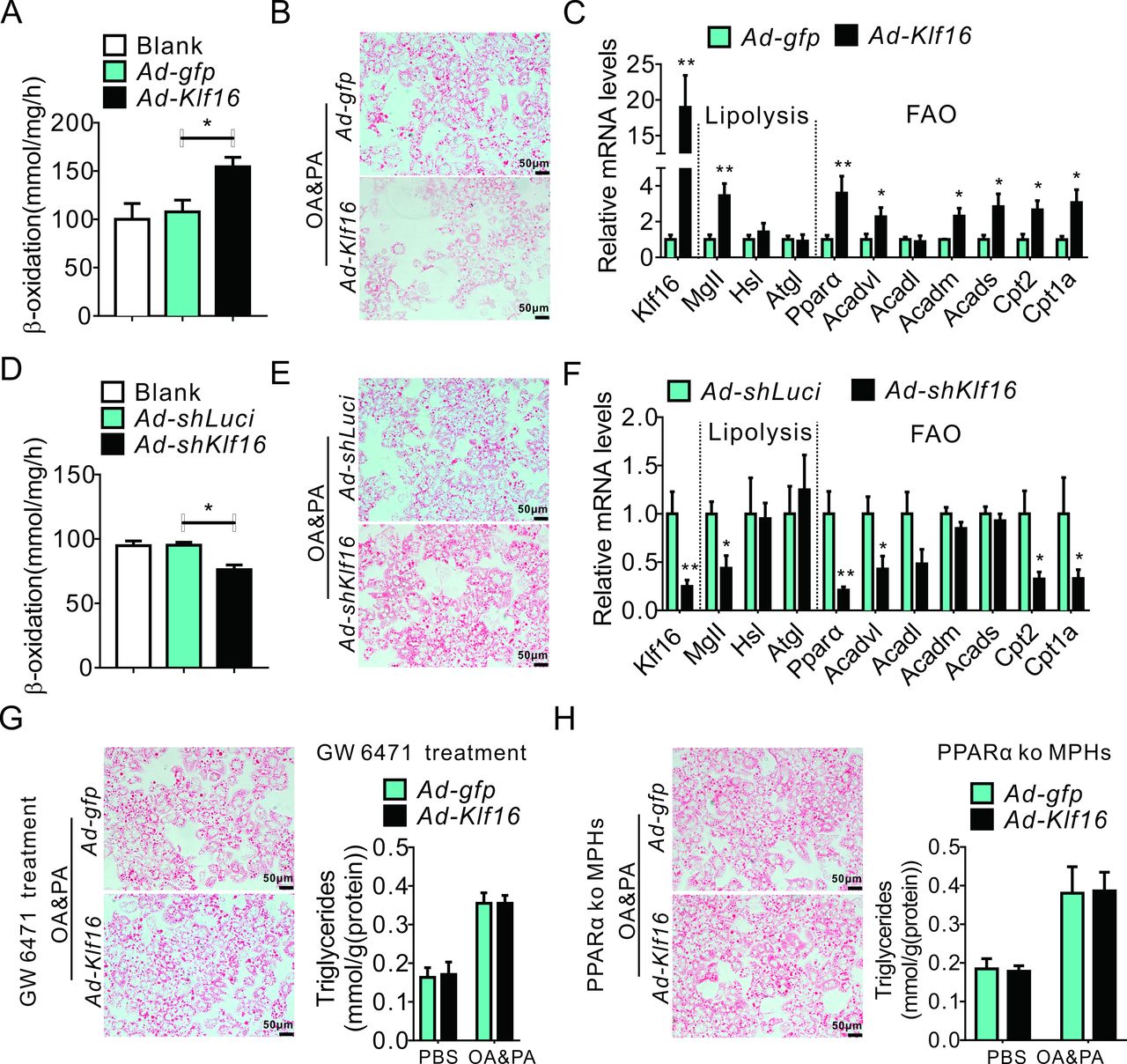
KLF16 regulates hepatic lipid metabolism in a PPARα-dependent manner (A–C) KLF16 overexpression increased fatty acid metabolism (A), decreased OA and PA induced lipid accumulation (B) and upregulated the genes involved in lipolysis and fatty acid oxidation (C). (D–F) KLF16 knockdown decreased fatty acid oxidation (D), worsened OA and PA induced lipid accumulation and suppressed the expression of genes involved in lipolysis and fatty acid oxidation (F). (G and H) Pharmacological inhibition or gene knockout of PPARα diminished the protective effects of KLF16 overexpression against lipid deposition. Equivalent results obtained in three independent experiments. Data are means±SEM; n=6/group. *P<0.05, **p<0.01. KLF16, Krüppel-like factor 16; MPHs, mouse primary hepatocytes; OA, oleic acid; PA, palmitic acid; PBS, phosphate buffered saline; PPARα, peroxisome proliferator-activated receptor α.
PPARα overexpression attenuated KLF16 deficiency-induced hepatic steatosis
To further confirm the importance role of PPARα in mediating KLF16-induced protective effects. MPHs isolated from KLF16alb−/− mice were infected with Ad-Pparα. As expected, Ad-Pparα infection induced a significant upregulation of PPARα in MPHs and livers (online supplemental figure 6A). Moreover, PPARα overexpression effectively rescued KLF16 deficit-induced lipid accumulation in MPHs (figure 7A,B).

PPPARα overexpression mitigated KLF16 deficiency-induced hepatic steatosis. (A and BB) PPARα overexpression reduced KLF16 deficiency-intensified lipid deposition after OA and PA treatment. (C) Hepatic PPARα overexpression changed the activity of genes that are involved in lipid metabolism. (D) PPARα overexpression reduced serum and hepatic TGs levels, and slightly decreased hepatic TCs content in KLF16alb−/− mice fed on HFD. (E) Ad-Pparα infection decreased hepatic lipid deposition in HFD KLF16alb−/− mice. (F) Ad-Pparα infection increased glucose tolerance, insulin sensitivity and pyruvate tolerance in HFD KLF16alb−/− mice; repeated measure two-way analysis of variance was used to analyse the difference of two curves. (G) PPARα overexpression increased the phosphorylation of Akt/GSK-3β in HFD KLF16alb−/− mice. Equivalent results were obtained in three independent experiments. Data are means±SEM, n=8 mice/group. *P<0.05, **p<0.01, ***p<0.001. HFD, high-fat diet; KLF16, Krüppel-like factor 16; OA, oleic acid; PA, palmitic acid; PPARα, peroxisome proliferator-activated receptor α; TC, total cholesterol; TGs, triglycerides.
We also injected Ad-Pparα into KLF16alb−/− mice fed on HFD to confirm the PPARα capability to mediate the KLF16-induced effects. Ad-Pparα infection significantly stimulated the hepatic expression of PPARα and its target genes (figure 7C and online supplemental figure 6B). Ad-Pparα-infected KLF16alb−/− mice displayed reductions in body weight and liver weight/body weight ratio value (online supplemental figure 6C,D). Moreover, PPARα overexpression effectively decreased TGs levels and hepatic lipid accumulation in HFD KLF16alb−/− mice (figure 7D,E). However, hepatic TCs content only slight decreased (figure 7D). Notably, PPARα overexpression rescued HFD-induced mitochondrial dysfunction in KLF16alb−/− mice, as manifested by the increased numbers and improved ultrastructural features of the mitochondria (online supplemental figure 6E). These changes significantly reduced liver ROS overproduction and inflammatory genes elevation in HFD KLF16alb−/− mice (online supplemental figure 6F,G). PPARα overexpression effectively decreased both ALT and AST levels in KLF16alb−/− mice on HFD, indicating an improved hepatic function (online supplemental figure 6H). More importantly, PPARα overexpression significantly improved the glucose metabolism disorder in KLF16alb−/− mice fed on HFD, as shown by reductions in fasting blood glucose and insulin levels (online supplemental figure 6I). The PPARα overexpression-mediated recovery of insulin signalling improved glucose intolerance, pyruvate intolerance and decreased insulin resistance (figure 7F and G). Altogether, these data suggested that PPARα plays crucial roles in mediating the several KLF16-induced protective effects.
KLF16 maintains intracellular redox balance in a PPARα-dependent manner
Previous studies indicated that an immoderate fatty acids accumulation drives ROS overproduction and promotes NAFLD progression.16 17 Thus, we investigated KLF16 effects on the intracellular redox system. As expected, KLF16 overexpression effectively elevated mitochondrial biogenesis and promoted mitochondrial fission in MPHs under OA and PA treatment (figure 8A,B). Moreover, KLF16 overexpression also significantly upregulated genes playing antioxidative roles, for example, Nrf2, SOD2 and HO1 (figure 8C). This led to a remarkable decrease in OA and PA elicited ROS production, thereby improving the redox balance disorder in MPHs (online supplemental figure 7A). Conversely, KLF16 deletion decreased mitochondrial numbers while increasing MPHs sensitivity to OA and PA treatment, as shown by the mitochondrial deeply altered ultrastructural features (figure 8D,E). KLF16 deficiency after OA and PA treatment resulted in ROS overproduction due to antioxidant genes suppression (figure 8F and online supplemental figure 7B). Consistent with our above findings, PPARα plays a crucial role in mediating the protective effects of KLF16 on intracellular redox imbalance. PPARα overexpression effectively ameliorated OA and PA induced ROS overproduction in KLF16-deficient MPHs while also restoring the expression of genes involved in fatty acids oxidation (figure 8G). However, KLF16 overexpression failed to improve the OA and PA induced redox balance disorder after PPARα genetic deletion in MPHs, as revealed by the persistently heightened ROS levels (figure 8H). These data showed the existence of a KLF16–PPARα dependent cooperation keeping intracellular redox homeostasis, which affects hepatic lipid metabolism.

KLF16 maintains the intracellular redox balance in a PPARα-dependent manner. (A and B) KLF16 overexpression increased mitochondrial biogenesis (A) and improved mitochondrial ultrastructural features (B) in MPHs. (C) KLF16 overexpression increased the expression of antioxidant genes. (D and E) KLF16 deletion decreased mitochondrial biogenesis (D) and altered mitochondrial ultrastructural features (E) in MPHs. (F) KLF16 deficiency decreased the expression of antioxidant genes. (G) PPARα overexpression rescued OA and PA induced ROS overproduction (left panel) and the expression of the genes involved in fatty acid oxidation (right panel) in KLF16-deficient MPHs. (H) KLF16 overexpression failed to alter OA and PA induced ROS production in PPARαko MPHs. Comparable results obtained in three independent experiments. Data are means±SEM; n=6/group. *P<0.05. KLF16, Krüppel-like factor 16; MPHs, mouse primary hepatocytes; OA, oleic acid; PA, palmitic acid; PPARα, peroxisome proliferator-activated receptor α; ROS, reactive oxygen species.
Hepatic KLF16 deletion increases mice sensitivity to ethanol feeding
Previous studies revealed that an impaired hepatic fatty acids oxidation also contributes to the development of alcohol-induced fatty liver, alcoholic hepatitis and cirrhosis.18 19 Thus, we used these mice as further models to confirm the effects of the KLF16–PPARα axis on hepatic fatty acid oxidation. Interestingly, ethanol feeding significantly inhibited hepatic KLF16 expression and decreased KLF16 enrichment on the PPARα promoter (figure 9A,B and online supplemental figure 8A). KLF16alb−/− mice fed on ethanol displayed a severe hepatic lipid accumulation and a weightier liver (figure 9C,D). Moreover, hepatic TGs levels quickly rose in KLF16alb−/− mice fed on ethanol diet (figure 9E). KLF16 deletion also results in an increased of serum ALT and AST levels, along with an prompt upregulation of hepatic inflammatory genes after ethanol diet feeding (online supplemental figure 8B,C). In keeping with our previous findings, KLF16 deletion aggravated the alcohol-induced suppression of genes involved in fatty acid oxidation (figure 9F). Moreover, mitochondrial dysfunction and redox imbalance became irreversible, as shown by the grievous fusion of mitochondria and the overproduction of ROS in these mice (online supplemental figure 8D,E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hepatic KLF16 deletion renders mice more sensitive to alcohol feeding. (A and B) Ethanol feeding suppressed KLF16 expression (A) and reduced its enrichment on the PPARα promoter (B). (C) H&E and Oil red O staining demonstrated a severe hepatic lipid deposition in KLF16 alb −/− mice fed on an ethanol diet. (Dadn E) KLF16 deletion increased the liver weight/body weight ratio value (D) and hepatic TG content (E) during ethanol feeding. (F) KLF16 deletion suppressed the expression of genes involved in fatty acid oxidation in mice fed on an ethanol diet. Comparable results were obtained in three independent experiments. Data are means±SEM; n=8/group. *P<0.05, **p<0.01, ***p<0.001. KLF16, Krüppel-like factor 16; PPARα, peroxisome proliferator-activated receptor α; TG, triglyceride.
In summary, these data suggest that KLF16 directly binding PPARα promoter is essential to maintain a healthy hepatocytic intracellular environment and defend against NAFLD or alcohol-induced fatty liver disease via recovery of impaired hepatic fatty acid oxidation.
Discussion
NAFLD has become the most prevalent aetiological factor of chronic liver diseases.20 The earlier pathogenetic view about the development of NAFLD was the ‘two-hit hypothesis’, envisaging that concurring imbalances of hepatic lipid synthesis and oxidation cause a local surplus accumulation of fatty acids, promoting hepatic steatosis development.21 22 Recent studies proposed the ‘multiple hit hypothesis’, which envisions several insults acting together on genetically predisposed subjects to induce NAFLD, thus more accurately explaining the latter’s pathogenesis.23–26 Such hits include an increased insulin resistance, adipose tissue-secreted hormones, nutritional factors, altered gut microbiota and genetic and epigenetic factors. Despite the ‘two-hit’ or ‘multiple hit’ hypotheses, one of the key consequences of NAFLD is the inordinate lipids accumulation, such as free fatty acids, in the liver. This fact critically points to a potential therapeutic strategy for the treatment of NAFLD.
Emerging evidence have revealed lipogenesis (de novo fatty acid synthesis), fatty acid oxidation, and lipoprotein uptake and secretion tightly control hepatic lipid homeostasis.8 27 Deregulations of lipogenesis or fatty acid oxidation cause NAFLD development. Therefore, any strategy aimed at decreasing liver’s lipid load by effectively repressing the de novo fatty acid synthesis and by increasing fatty acid oxidation might represent a beneficial treatment of NAFLD. Sterol regulatory element binding protein-1 (SREBP-1c), a key transcription regulator of lipogenesis and TGs storage, is synthesised as an inactive precursor that is initially attached to the nuclear envelope and the endoplasmic reticulum membranes.28 The activation of SREBP-1c plays a crucial role in metabolic diseases, especially NAFLD.29 However, in this study, we found that KLF16 did not alter the protein levels of SREBP-1c in MPHs, both in cytoplasm and nucleus (online supplemental figure 9). At variance with SREBP-1c role in lipogenesis, PPARα is an important nuclear receptor involved in the regulation of fatty acid oxidation.30–32 In current study, we reported that liver-specific KLF16 overexpression did remarkably enhance the expression of PPARα and its target genes including Acadvl and Cpt2, which intensified lipolysis and reduced hepatic lipid load, suggesting that the KLF16 overexpression-mediated reduction in hepatic lipids levels relates to the PPARα-mediated elevated hepatic lipolytic effect, rather than the SREBP-1c-mediated lipogenesis.
KLF16 is a member of Krüppel-like family of transcriptional factors (KLFs), which regulates cellular homeostasis playing roles in growth, development and programmed cell death.33–35 We and others previously showed that KLFs strongly associated with the regulation of metabolic syndromes.13 14 36–41 For instance, we previously proved that mouse KLF9, KLF10 and KLF14 partake in hepatic glucose homeostasis by modulating the expression of peroxisome proliferator-activated receptor-γ coactivator-1 alpha.13 14 36 KLF15 was identified as a pivotal component of transcriptional circuitry coordinating the physiological fluxes of three basic kinds of cellular nutrients: that is, glucose, amino acids and lipids.37–39 We also showed that mouse KLF11 regulates hepatic lipid metabolism by targeting the PPARα signalling pathway via an unknown mechanism.40 Recently, Bechmann et al 41 revealed that KLF6 is a novel regulator of glucose and lipid metabolism by mediating the PPARα post-transcriptional activation. Differently from previous reports, in this study, we offer the direct evidence that, being a transcription factors, KLF16 directly binds PPARα’s promoter domain at a location placed from −577bp to −221bp endowed with GT-rich or GC-rich elements. PPARα deletion could remarkably abrogate KLF16 overexpression-mediated alleviation of OA and PA induced lipid deposition in MPHs. Conversely, liver-specific PPARα overexpression could reverse the KLF16 deficiency-induced hepatic steatosis, demonstrating that PPARα is a bona fide target gene of KLF16. In keeping with our results, KLF1 demonstrated a similar transcriptional regulatory mode.42
Accumulating evidence suggest that an excessive lipid deposition stimulates the intracellular production of ROS by impairing the lysosomal–mitochondrial axis.43 Besides, chronic HFD exposure activated JNK and drove its migration into the mitochondria triggering a vicious ROS-JNK feed-forward loop resulting in oxidative stress and eventually causing hepatocytes’ death. Healthy hepatocytes are endowed with systems comprising antioxidant enzymes and antioxidant genes such as PPARα, Nrf2 and Ho1, which eliminate ROS surpluses and maintain the redox balance. PPARα activation effectively protected mice against cholestasis-induced hepatic injury by inhibiting JNK signalling. A PPARα antagonist effectively inhibited liver glycogen synthase kinase-3β (GSK-3β) and p(Thr183/Tyr185)-JNK. We proved that KLF16 overexpression efficaciously improved HFD-induced or alcohol-induced hepatic mitochondrial stress and ROS overproduction and eventually benefiting the hepatotoxicity and fatty liver phenotype. Conversely, KLF16-deficient mice showed a fast and severe lipid deposition during HFD or alcohol exposure. Moreover, ROS surpluses induce DAMP molecules release and trigger an irreversible secondary inflammatory injury through an abnormal innate immune system activation.44 KLF16 was previously reported to suppress human glioma and glioblastoma cells proliferation by targeting TFAM, implying its potential anti-inflammatoty role. Here, we show that hepatic KLF16 acts as a negative regulator of proinflammatory genes in the liver. Moreover, we also prove that KLF16 exerts a protective effect against hepatocytes apoptosis in vivo and in vitro (online supplemental figure 10).
In summary, our results reveal that KLF16 targets the PPARα gene as it directly binds to the KLF16 promoter region, thus contributing to upregulate its expression at the transcriptional level, which crucially promotes fatty acids oxidation and acts as a new target for the treatment of fatty liver and metabolic syndrome.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the ethics committees of the First Affiliated Hospital of Guangzhou University of Chinese Medicine (NO. K [2019]082).
Acknowledgments
We would like to thank Professor Hongbing Zhang and Yongsheng Chang for providing alb-cre mice and Ad-Pparα adenovirus.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
CL, YC and YG are joint senior authors.
NS, CS, LZ, XW, YY, XY and CY are joint first authors.
Contributors YG and YC conceived the idea and designed the experiment. NS, XW, CS, LZ and CS performed experiments. CZ, CL, ZG, WM, ZY, WG, XY, YY, LH and KW analysed the data and improved the manuscript.
Funding This work was supported by the National Natural Science Foundation of China (grant nos. 81800738, 81773969, 81102883, 81800718 and 82070891), the first-class discipline construction major project of Guangzhou University of Chinese Medicine (Guangzhou University of Chinese Medicine Planning (2018, No. 6), Guangzhou University of Chinese Medicine Planning (2019, No. 5)), the Guangdong Science and Technology Collaborative Innovation Center for Sport Science (2019B110210004), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 18KJB310015), the Jiangsu Shuangchuang Programme and the Starting Foundation for Talents of Xuzhou Medical University (No. D2018006). Shandong key research and development program (2019GSF108270).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.