Article Text

Abstract
Objective Receptor-interacting protein kinase 3 (RIPK3) is a key player in necroptosis execution and an emerging metabolic regulator, whose contribution to non-alcoholic fatty liver disease (NAFLD) is controversial. We aimed to clarify the impact of RIPK3 signalling in the pathogenesis of human and experimental NAFLD.
Design RIPK3 levels were evaluated in two large independent cohorts of patients with biopsy proven NAFLD diagnosis and correlated with clinical and biochemical parameters. Wild-type (WT) or Ripk3-deficient (Ripk3 −/−) mice were fed a choline-deficient L-amino acid-defined diet (CDAA) or an isocaloric control diet for 32 and 66 weeks.
Results RIPK3 increased in patients with non-alcoholic steatohepatitis (NASH) in both cohorts, correlating with hepatic inflammation and fibrosis. Accordingly, Ripk3 deficiency ameliorated CDAA-induced inflammation and fibrosis in mice at both 32 and 66 weeks. WT mice on the CDAA diet for 66 weeks developed preneoplastic nodules and displayed increased hepatocellular proliferation, which were reduced in Ripk3 −/− mice. Furthermore, Ripk3 deficiency hampered tumourigenesis. Intriguingly, Ripk3 −/− mice displayed increased body weight gain, while lipidomics showed that deletion of Ripk3 shifted hepatic lipid profiles. Peroxisome proliferator-activated receptor γ (PPARγ) was increased in Ripk3 −/− mice and negatively correlated with hepatic RIPK3 in patients with NAFLD. Mechanistic studies established a functional link between RIPK3 and PPARγ in controlling fat deposition and fibrosis.
Conclusion Hepatic RIPK3 correlates with NAFLD severity in humans and mice, playing a key role in managing liver metabolism, damage, inflammation, fibrosis and carcinogenesis. Targeting RIPK3 and its intricate signalling arises as a novel promising approach to treat NASH and arrest disease progression.
- cell death
- chronic liver disease
- lipid metabolism
- molecular carcinogenesis
- nonalcoholic steatohepatitis
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic steatohepatitis is already a relevant aetiology of cancer-related liver transplantation and the non-alcoholic fatty liver disease (NAFLD) mortality is alarmingly increasing.
Receptor-interacting protein kinase 3 (RIPK3)-dependent necroptosis is activated in the liver of patients with NAFLD, but its specific contribution to disease pathogenesis remains controversial.
Besides its well-established role in necroptosis execution, RIPK3-dependent signalling is arising as key player in metabolic reprogramming.
What are the new findings?
Hepatic RIPK3 levels positively correlate with NAFLD severity in humans, emphasising the role of RIPK3 signalling in regulating NAFLD progression.
RIPK3 deficiency ameliorates disease severity induced by mid-term and long-term choline-deficient L-amino acid-defined diet diet.
RIPK3 deficiency dampens signalling pathways involved in tumourigenesis.
RIPK3 differentially controls liver steatosis versus inflammation during hypercaloric diet feeding.
Deletion of RIPK3 impacts on hepatic lipidome and upregulates peroxisome proliferator-activated receptor γ.
How might it impact on clinical practice in the foreseeable future?
These results unveil a novel metabolic role of RIPK3, whose targeting might arrest NAFLD progression towards hepatocellular carcinoma.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in Western countries and its prevalence and incidence is increasing due to its strong association with obesity and metabolic syndrome, and the lack of pharmacological therapies.1 NAFLD encompasses a spectrum ranging from hepatic steatosis to non-alcoholic steatohepatitis (NASH), ultimately leading to liver cirrhosis and/or cancer.2 In turn, liver cancer displays a high mortality rate, ranking as the sixth most common cancer and the fourth most deadly worldwide.3 In the USA, NASH is already the second leading aetiology of cancer-related liver transplantation4 5 and the NAFLD mortality is predicted to increase by 178% between 2015 and 2030.6
Hepatocellular carcinoma (HCC), the most common primary liver cancer, arises almost exclusively in the setting of chronic hepatic inflammation and fibrosis. In turn, cell death represents a dominant trigger of inflammation and fibrosis, thus contributing to multiple hallmark features of cancer.7 In addition, chronic cell death in non-transformed hepatocytes increases cell turnover and selects for clones better adapted to survive and proliferate, thus driving tumour development.8 Still, knowledge on the association between liver cancer and chronic cell death and inflammation has not yet identified pharmacological targets that could alter the progression of chronic liver disease towards fibrosis and cancer.
Necroptosis represents a regulated necrotic cell death pathway activated downstream of death receptor stimulation and dependent on receptor-interacting protein kinase 3 (RIPK3) activity. On activation, RIPK3 phosphorylates mixed lineage kinase domain-like protein (MLKL), which plays a critical, non-redundant role in executing necroptosis.9 10 Necroptosis is highly immunogenic and, when sustained, may contribute to chronic inflammation and carcinogenesis.11 In addition, emerging evidence suggests that necroptosis-associated signalling could play a role in metabolic reprograming, including lipid metabolism and mitochondrial respiration,12–15 which are key for progression of chronic liver disease and carcinogenesis.
We and others have shown that RIPK3-dependent hepatocyte necroptosis is activated in patients with NAFLD and experimental NASH,16–18 but the specific contribution of RIPK3-dependent signalling in NAFLD pathogenesis remains controversial. For instance, activation of necroptosis was shown to contribute to liver damage, fibrosis, oxidative stress and inflammation in methionine-deficient and choline-deficient diet-induced NASH in mice,16 17 while in high-fat diet (HFD) RIPK3 deficiency exacerbates liver steatosis and inflammation, as well as circulating insulin/glucose levels induced.18 19 Similarly, RIPK3 was suggested to inhibit hepatocarcinogenesis in a genetic model of chronic hepatic inflammation,20 while necroptosis-associated hepatic cytokine microenvironment impinges on epigenetics triggering cholangiocarcinoma development from ontogenically transformed hepatocytes.11 In this study, we dissected the impact of RIPK3 signalling in lipid metabolism, steatosis, inflammation, fibrosis and carcinogenesis associated with NAFLD, by using human samples, mechanistic in vitro studies and an experimental model that recapitulates a broad spectrum of NAFLD and its sequel to HCC. We show that hepatic RIPK3 levels correlate with NAFLD severity in large independent human cohorts, emphasising the role of RIPK3-signalling in regulating NAFLD progression. Furthermore, RIPK3 differentially controls steatosis versus inflammation in choline-deficient L-amino acid-defined (CDAA)-fed mice, likely through peroxisome proliferator-activated receptor γ (PPARγ) modulation, suggesting that these events are dissociated in NAFLD progression to HCC.
Materials and methods
Additional materials and methods are included in online supplemental information.
Supplemental material
Patient cohorts
NAFLD liver specimens were prospectively and sequentially collected from two independent cohorts of patients with biopsy-proven diagnosis of NAFLD (cohort A: n=71; cohort B: n=146). See online supplemental materials and methods for details.
Animals and diets
Male C57BL/6 mice aged 7–8 weeks wild-type (WT) or Ripk3-deficient mice (Ripk3−/ −)21 from the same genetic background were fed either a choline-deficient L-amino acid-defined (CDAA) diet (Envigo, Madison, USA) or a control choline-sufficient L-amino acid-defined (CSAA) diet for 32 or 66 weeks, and histopathology, serum analysis, glucose tolerance tests, liver lipidomics and gene expression analysis were performed as described in online supplemental materials and methods.
Quantitative RT-PCR
Total RNA was purified from frozen human liver tissue and extracted from animal liver and epididymal white adipose samples using the TRIzol reagent (ThermoFisher, Waltham, Massachusetts, USA). The mRNA level of human selected genes was normalised for the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). In turn, relative amounts of selected mouse genes were calculated based on the standard curve normalised to the level of hypoxanthine phosphoribosyltransferase (Hrpt) and expressed as fold change from CSAA WT mice at each respective time-point, except for the microarray validation where β-actin was used as reference gene and results expressed as fold change from non-tumorous (NT) tissue of CDAA WT mice. See online supplemental materials and methods for details.
Mouse liver cancer genes array
Differential gene expression in NT tissue from CDAA-fed WT mice versus NT from CDAA-fed Ripk3−/ − mice or tissue enriched in preneoplastic nodules from CDAA-fed WT or Ripk3−/ − mice was evaluated using a RT² Profiler Mouse Liver Cancer PCR Array (PAMM-133Z, Qiagen, Maryland, USA) according to the manufacturer’s instructions and as described in online supplemental materials and methods.
Cell culture and immunoblotting
Total protein extraction and immunoblotting were performed as previously described.22 Immunoblotting and cell culture and treatments are detailed in online supplemental materials and methods.
Statistical analysis
Regular statistical analysis was performed with GraphPad Prism 7 software (GraphPad Software, San Diego, California, USA). The Shapiro-Wilk test was used for testing the normality of data. Student’s t-test was applied when two groups were analysed. According to normality of values distribution, analysis of variance followed by Tukey’s multiple comparison or Krustal-Wallis followed by Dunn’s multiple comparison test were performed if three or more groups were compared. Association between two variables was assessed by Pearson’s or Spearman’s correlation coefficient for normally and non-normally distributed data, respectively. Values of p<0.05 were considered statistically significant.
Results
Hepatic RIPK3 levels are increased in patients with NASH and correlate with disease severity
RIPK3 levels were evaluated in two large independent cohorts of patients, with clinical and biopsy-proven diagnosis of NAFLD and correlated with clinical and biochemical parameters.
In cohort A, similar to data already reported for RIPK1 and MLKL,13 serum RIPK3 protein levels were significantly increased in patients with active disease (histological activity score ≥2 vs <2) (figure 1A), likely mirroring necro-inflammatory activity. Hepatic RIPK3 mRNA levels were also increased in patients with NAFLD, while the expression of other mediators of necroptosis and apoptosis were not altered between control, non-alcoholic fatty liver and NASH (figure 1B). Accordingly, hepatic RIPK3 mRNA levels positively correlated with the expression of several pro-inflammatory markers increased in the liver of patients with NASH (online supplemental figure S1A), namely tumour necrosis factor (TNF), interleukin-1β (IL-1β), NACHT, LRR and PYD domains-containing protein 3 (NLRP3), cluster of differentiation (CD)38 and CD68 (figure 1C, online supplemental table S1). In contrast, no correlation was found between RIPK3 expression and arginase-1 mRNA levels, a marker of anti-inflammatory macrophages (online supplemental table S1). Increased RIPK3 expression in patients with NAFLD further correlated with collagen1α1 (COL1α1) and transforming growth factor (TGF-β), as well as necroptosis-related factors MLKL and RIPK1 (figure 1C, online supplemental table S1). Noteworthy, RIPK3 expression was also positively correlated with caspase-1 and NLRP3 gene expression in the liver, but not with caspase-3 mRNA levels (online supplemental table S1), suggesting that RIPK3 is linked to inflammasome activation in NASH.
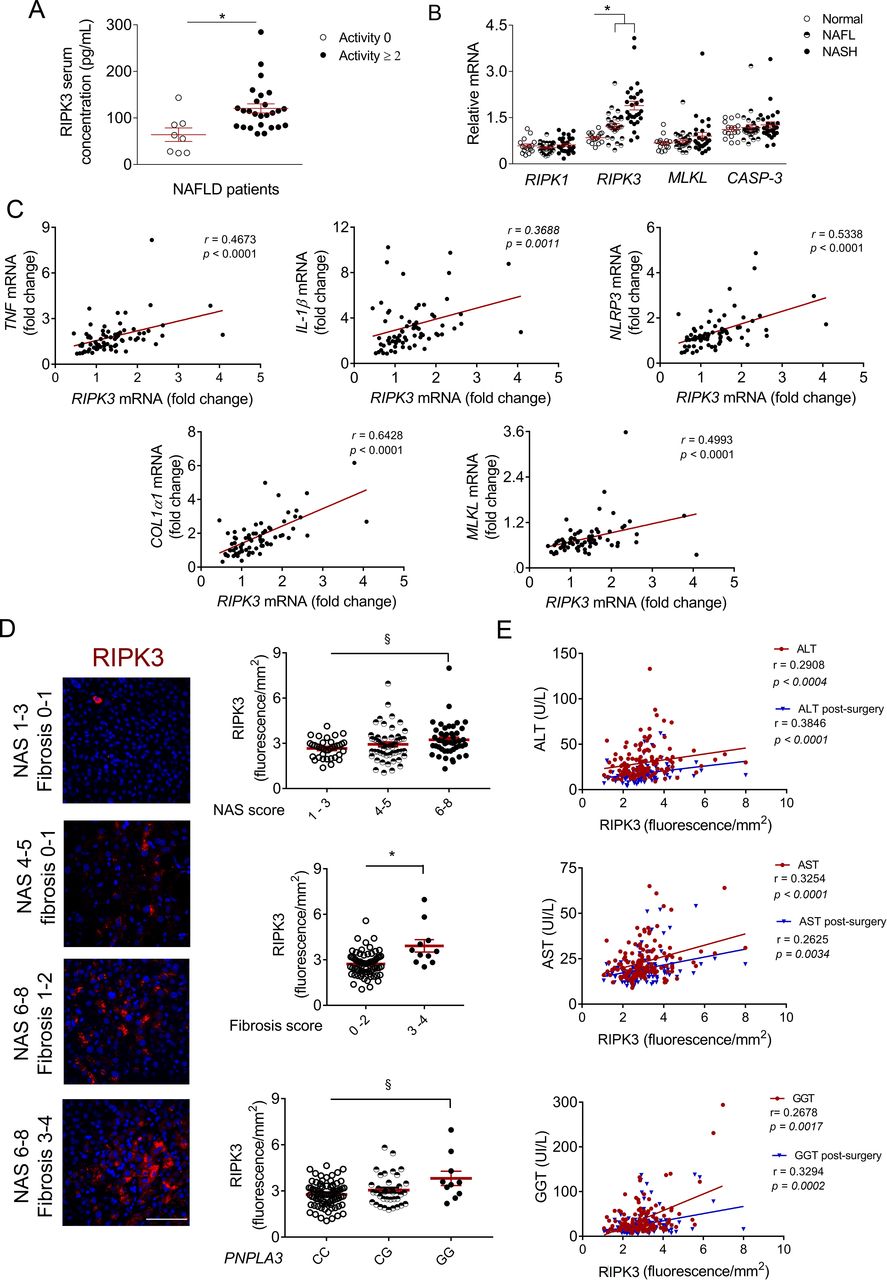
Receptor-interacting protein kinase 3 (RIPK3) levels are increased in patients with non-alcoholic steatohepatitis (NASH) and correlate with disease severity. (A) RIPK3 protein levels in the serum of cohort A patients with non-alcoholic fatty liver disease (NAFLD), divided according to the histological activity score (<2, n=8 or ≥2 n=26), measured through ELISA. (B) Quantitative RT-PCR analysis of hepatic RIPK1, RIPK3, mixed lineage kinase domain-like protein (MLKL) and caspase-3 (CASP-3) in the liver of cohort A patients with NAFLD (n=71). (C) Correlations of hepatic mRNA levels of RIPK3 with tumour necrosis factor (TNF), interleukin-1β (IL-1β), NACHT, LRR and PYD domains-containing protein 3 (NLRP3), collagen1α1 (COL1α1) or MLKL in patients of cohort A. (D) Representative RIPK3 immunofluorescence (red) in liver sections from cohort B patients with NAFLD. Scale bar=100 µm. Histograms show the quantification of RIPK3 levels assessed by immunofluorescence according to NAFLD activity score (NAS) score, fibrosis score and PNPLA3 genotype (n=146). (E) Correlations of hepatic RIPK3 mRNA expression with serum levels of alanine transaminase (ALT), aspartate transaminase (AST) and γ-glutamyltranspeptidase (GGT), before (red) and 1 year after (blue) bariatric surgery in patients from cohort B. Each individual is represented by one dot. Data are expressed as mean±SEM arbitrary units or fold change. §P<0.05 and *p<0.01 compared with respective controls.
In cohort B, hepatic RIPK3 protein levels were increased in hepatocytes of patients with higher NAFLD activity score (NAS) and fibrosis scores (figure 1D, online supplemental figure S1B). Accordingly, RIPK3 positively and negatively correlated with circulating ferritin and platelet levels, respectively (online supplemental table S1), independent predictors of advanced fibrosis in NAFLD.23 24 In addition, hepatic RIPK3 positively correlated with circulating markers of liver injury before and 1 year after bariatric surgery (figure 1E). Interestingly, male patients with NAFLD (online supplemental figure S1C) and carriers of the rs738409 GG genotype in patatin-like phosphate domain-containing protein 3 (PNPLA3) presented significantly higher hepatic RIPK3 levels compared with controls (figure 1D). In turn, no association was found between RIPK3 and the transmembrane 6 superfamily member 2 nor the membrane bound O-acyltransferase domain-containing 7 (data not shown). Finally, although NAFLD is strongly linked with metabolic syndrome, RIPK3 levels were not associated with diabetes mellitus (online supplemental figure S1D), arterial hypertension or dyslipidaemia (data not shown), neither correlated with circulating levels of lipids or glucose (online supplemental table S1).
Overall, the analysis of a large number of patients with NAFLD shows that hepatic RIPK3 mRNA and protein levels are increased in advanced disease, correlating with NASH development, liver damage, fibrosis and inflammation.
RIPK3 deficiency ameliorates hepatic inflammation and fibrosis in CDAA diet-induced experimental NASH
Our data in human NAFLD cohorts strongly hinted that RIPK3-dependent signalling is involved in disease pathogenesis. To further dissect its contribution, we used a choline-deficient L-amino acid-defined (CDAA) diet model, mimicking both histological and pathophysiological features of NAFLD.25 26 An isocaloric choline-sufficient L-amino acid-defined (CSAA) diet was used as control.
Ripk3 −/− mice aged 7–8 weeks displayed increased body weight gain over time compared with WT mice on both diets (figure 2A). Total food intake was similar between experimental groups (online supplemental figure 2A). Although the relative increase in the liver-to-body weight ratio induced by the CDAA diet was similar between WT and Ripk3 −/− mice, CSAA-fed Ripk3 −/− mice exhibited hepatomegaly compared with CSAA-fed WT mice at 32 weeks (figure 2B). Furthermore, hepatic triglycerides (TG) and free fatty acids (FFA) were augmented in the liver of CDAA-fed animals, while FFA were also increased in CSAA-fed Ripk3 −/− mice (online supplemental figure S2B). Accordingly, the steatosis score was augmented in Ripk3 −/− mice compared with WT mice under the CSAA diet, without any histological evidence of inflammatory cell infiltration or hepatocyte injury (figure 2C, table 1). Intriguingly, serum FFA increased in CDAA-fed mice irrespective of genotype, while elevated TG were only found in Ripk3 −/− mice, irrespective of diet (online supplemental figure S2B). Despite the lack of impact of Ripk3 deletion on CDAA-induced circulating levels of liver enzymes (data not shown), Ripk3 deficiency decreased the grade of inflammatory cell infiltration induced by the CDAA diet at 32 weeks (figure 2C, table 1), as well as hepatic F4/80-positive cells and mRNA (figure 2D), and expression of pro-inflammatory markers Tnf-α, toll-like receptor 4 (Tlr4), Nlrp3 and cyclooxygenase 2 (Cox-2) (figure 2E). Furthermore, although hepatic fibrosis was mild at 32 weeks (figure 2F, table 1), hydroxyproline quantification showed less hepatic collagen content in CDAA Ripk3 −/− mice compared with WT mice (figure 2G).
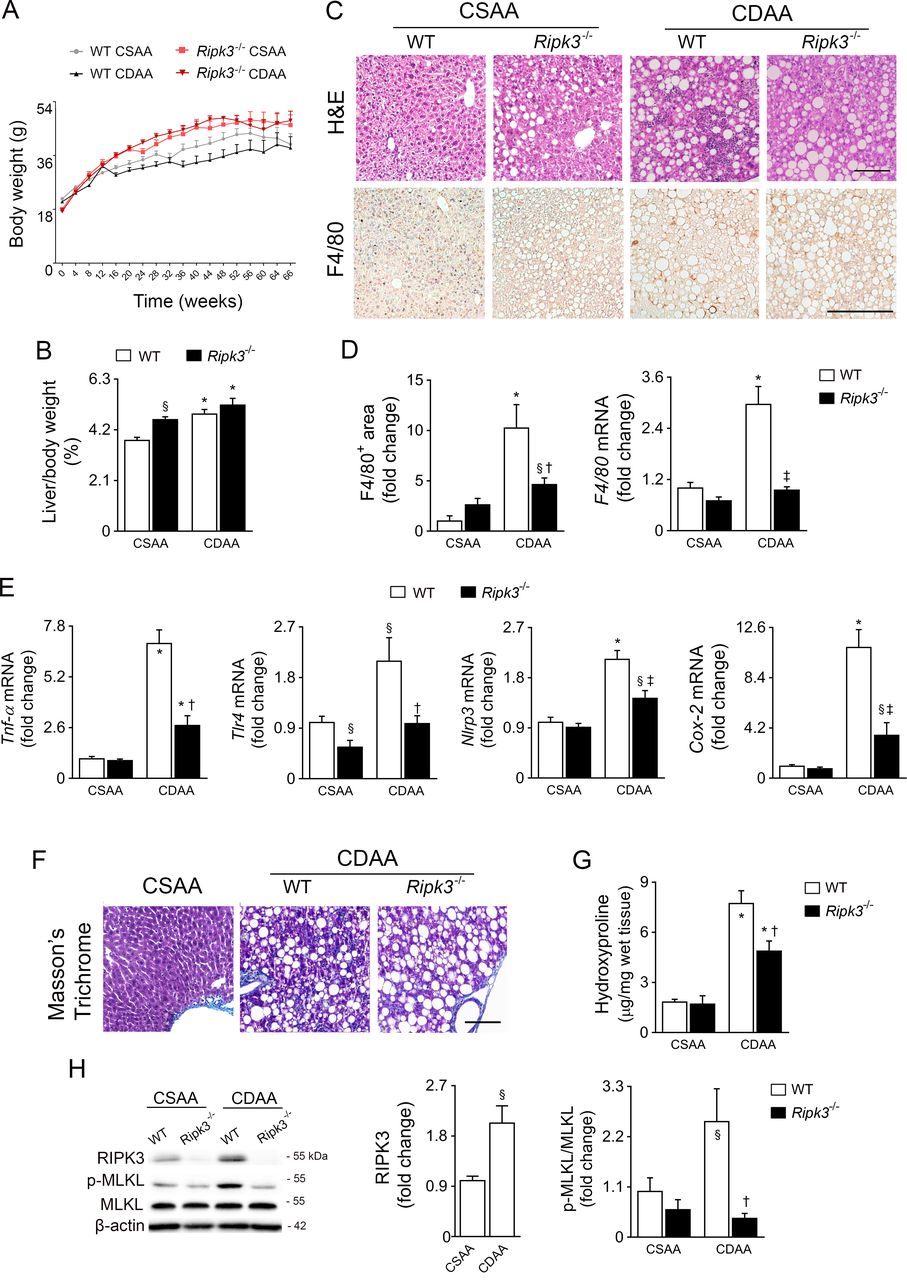
Absence of receptor-interacting protein kinase 3 (RIPK3) ameliorates hepatic inflammation and fibrosis during choline-deficient L-amino acid-defined diet (CDAA) diet-induced experimental non-alcoholic steatohepatitis (NASH). C57BL/6 wild-type (WT) or Ripk3−/− mice were fed a CDAA or an isocaloric control choline-sufficient L-amino acid-defined (CSAA) diet for 32 weeks. (A) Total body weight over time. (B) Liver-to-body weight ratio. (C) Representative images of H&E stained liver sections and immunoperoxidase staining for F4/80. Scale bar=100 µm. (D) Histograms show the quantification of F4/80-positive area (left) and F4/80 mRNA levels (right). (E) Quantitative RT-PCR analysis of Tnf-α, Tlr4, Nlrp3 and Cox-2 in mouse liver. (F) Representative images of Masson’s trichrome stained liver sections. (G) Liver hydroxyproline content. (H) Immunoblotting and densitometry of RIPK3, p-mixed lineage kinase domain-like protein (p-MLKL) and MLKL. Blots of RIPK3 were normalised to endogenous β-actin, whereas p-MLKL was normalised to MLKL. Representative immunoblots are shown. Results are expressed as mean±SEM arbitrary units or fold change of six to seven individual mice. Scale bar=250 µm. §P<0.05 and *p<0.01 from CSAA-fed WT mice; †p<0.05 and ‡p<0.01 from CDAA-fed WT mice.
Histological data of the CDAA murine model
Finally, similar to patients with NAFLD, liver RIPK3 levels were increased in WT mice fed the CDAA diet for 32 weeks, compared with the CSAA diet. MLKL phosphorylation was similarly increased but prevented by Ripk3 deletion (figure 2G). Altogether, targeting RIPK3 may halt NAFLD progression, at least in part, by preventing necro-inflammation associated with necroptotic cell death.
Absence of RIPK3 ameliorates disease severity induced by long-term CDAA feeding
Mice fed the CDAA diet for 66 weeks were used to evaluate the functional role of RIPK3-dependent signalling in NAFLD progression towards HCC. Much like at 32 weeks, Ripk3 deficiency ameliorated the grade of inflammation but not liver-to-body weight ratio nor steatosis at 66 weeks (figure 3A,B, table 1, online supplemental figure S2B). In agreement with the reduction of inflammatory cell infiltration, F4/80 protein and mRNA levels (figure 3C) and expression of pro-inflammatory mediators Tnf-α, Tlr4, Nlpr3 and Cox-2 were also decreased in CDAA-fed Ripk3 −/− mice, compared with WT mice (figure 3D). Furthermore, the NAS score was significantly lower in Ripk3 −/− than in WT mice, with Ripk3 −/− mice exhibiting only borderline NASH (NAS<5) (table 1). Histological examination also showed that CDAA diet promoted progressive pericellular and periportal hepatic fibrosis in WT mice, while Ripk3 deficiency significantly prevented the development of fibrosis in periportal spaces (figure 3E, table 1). Other histopathological abnormalities commonly observed in NASH, such as Mallory-Denk bodies and ceroid-laden macrophages, were also detected in liver sections of WT mice fed the CDAA-fed mice, and decreased in CDAA Ripk3 −/− mice (figure 3B, online supplemental figure S2C).

Absence of receptor-interacting protein kinase 3 (RIPK3) ameliorates disease severity induced by long-term choline-deficient L-amino acid (CDAA) feeding. C57BL/6 wild-type (WT) or Ripk3−/− mice were fed a CDAA or an isocaloric control choline-sufficient L-amino acid (CSAA) diet for 66 weeks. (A) Liver-to-body weight ratio. (B) Representative images of H&E stained liver sections and immunoperoxidase staining for F4/80. Scale bar=250 µm. (C) Histograms show the quantification of F4/80-positive area (left) and F4/80 mRNA levels (right). (D) Quantitative RT-PCR analysis of Tnf-α, Tlr4, Nlrp3 and Cox-2 in mouse liver. (E) Representative images of Masson’s trichrome stained liver sections. Scale bar=250 µm. (F) Representative macroscopic pictures of livers. The scale bar represents 0.5 cm; arrowhead depicts preneoplastic nodules. (G) Representative immunoperoxidase staining for Ki67. Histogram show the quantification of Ki67-positive cells in percentage. Scale bar=100 µm. (H) Immunoblotting and densitometry of RIPK3, mixed lineage kinase domain-like protein (p-MLKL) and MLKL. Blots of RIPK3 were normalised to endogenous β-actin, whereas p-MLKL was normalised to MLKL. Representative immunoblots are shown. §P<0.05 and *p<0.01 from CSAA-fed WT mice; †p<0.05 from CDAA-fed WT mice.
After 66 weeks of CDAA feeding, all mice developed macroscopic preneoplastic nodules (figure 3F), at a multiplicity of 5.2±2.1 in WT and 3.9±0.8 in Ripk3 −/− mice. In addition, one of seven mice in each WT and Ripk3 −/− CDAA diet groups developed HCC (online supplemental figure S3A). Histological analysis of HCC tumours revealed a trabecular growth pattern and cytoplasmic inclusions (online supplemental figure S3B). Livers from CSAA diet-fed mice lacked tumours. Of note, Ripk3 deficiency significantly reduced CDAA-induced hepatocyte proliferation in Ki67 immunohistochemistry (figure 3G). The reduction in the tumour burden provided by Ripk3 deletion was corroborated in established liver tumourigenesis induced by the chemical carcinogen diethylnitrosamine (DEN) (online supplemental figure S3C).
Importantly, in contrast with the 32 weeks time-point, CDAA feeding for 66 weeks reduced hepatic necroptosis, as seen by the significant downregulation of RIPK3 expression and MLKL phosphorylation in WT mice (figure 3H), suggesting that long-term CDAA feeding results in liver resistance to regulated cell death, contrasting with the active role of necroptosis in the pathogenesis of NAFLD.16 17
RIPK3 deficiency dampens signalling pathways involved in tumour development
For an unbiased analysis of liver carcinogenesis, we performed a comparative PCR array analysis of 84 genes associated with liver cancer progression in normal liver tissue (NT) or hepatic preneoplastic macroscopic nodules from WT and Ripk3 −/− mice fed a CDAA diet for 66 weeks. The heatmap of differentially expressed genes showed a distinct transcriptional profile between WT liver nodules and WT or Ripk3 −/− NT or Ripk3 −/− nodules (figure 4A,B, online supplemental table S4), corroborated by independent quantitative RT-PCR validation on randomly selected genes (Akt, Cdkn1a, Rac1 and Rhoa) (online supplemental figure S3D). Pathway enrichment analysis indicated that genes associated with cancer, Wnt signalling pathways and oncogenes were upregulated in nodules of CDAA-fed WT mice, comparing with Ripk3 −/− nodules (figure 4C). Indeed, the protein levels of the oncogene β-catenin (Ctnnb1) were only upregulated in WT nodules (figure 4D).

Receptor-interacting protein kinase 3 (RIPK3) deficiency impacts on transcriptional profiles associated with tumour development during long-term choline-deficient L-amino acid (CDAA) feeding. C57BL/6 wild-type (WT) or Ripk3−/− mice were fed a CDAA or an isocaloric control choline-sufficient L-amino acid (CSAA) diet for 66 weeks. (A) Heatmap of differentially expressed genes in NT from CDAA Ripk3−/− mice, nodules from CDAA WT and Ripk3−/− mice compared with NT from CDAA WT mice. The colour code represents the transformed ratios between means of the groups (log2(fold change)), where red indicates overexpressed transcripts and green represents underexpressed transcripts. (B) Venn diagrams showing the number of upregulated and downregulated transcripts. (C) Gene Ontology (GO) process, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and oncogenes enrichment analysis for the differentially expressed genes in WT and Ripk3−/− nodules. The false discovery rate (FDR)-adjusted P value was indicated. (D) Immunoblotting and densitometry of β-catenin, p53, cyclin D1 and p21 in mouse liver. Blots were normalised to endogenous β-actin. Representative immunoblots are shown. (E) Caspase-3/Caspase-7 activity assay. Results are expressed as mean±SEM arbitrary units or fold change of six to seven individual mice. NT, non-tumorous tissue (tissue without macroscopic discernible nodules). §P<0.05 and *p<0.01 from CSAA-fed WT mice; †p<0.05 from and ‡p<0.01 from CDAA-fed WT mice. φP<0.01 from NT tissue of CDAA WT mice.
Gene Ontology process enrichment suggested that the absence of RIPK3 decreased cell proliferation and increased apoptosis in preneoplastic nodules (figure 4C). Indeed, caspase-3/caspase-7 activity was decreased in preneoplastic nodules-enriched samples from WT mice compared with nodules from Ripk3 −/− mice; caspase-3/caspase-7 activity was unaffected by CDAA feeding in Ripk3 −/− mice (figure 4E). Furthermore, in CDAA-fed WT mice, cyclin D1 (Ccnd1) expression increased in nodules comparing with NT, while p53 (Tp53) and p21 (Cdkn1a) expressions were decreased. Strikingly, p21 was strongly increased in the liver of Ripk3 −/− mice irrespective of the diet, while p53 and cyclin D1 expression in preneoplastic nodules was restored to basal levels by Ripk3 deficiency (figure 4D). Still, cellular senescence hallmarks such as β-galactosidase-positive cells and p16 expression were not differentially modulated in Ripk3 −/− livers compared with WT mice (online supplemental figure 3E,F). α-Fetoprotein (AFP) was also increased in both NT and preneoplastic nodules of CDAA-fed WT mice, but not in liver of Ripk3 −/− mice (figure 5A). Protein levels of additional stemness-associated markers, including Nanog and aldehyde dehydrogenase 1/2 (ALDH), were only upregulated in preneoplastic lesions of WT mice. Importantly, DNA damage sensor γH2AX expression was uniquely increased in preneoplastic lesions of WT nodules (figure 5A), whereas Ripk3 deficiency also abrogated CDAA-induced reactive oxygen species in the liver of mice fed a CDAA diet for 66 weeks (figure 5B). We have previously shown that absence of Ripk3 attenuates oxidative stress in experimental NASH.16 Strikingly, the hepatic expression of α-smooth muscle actin, macrophage inflammatory protein 2 and vimentin, markers of epithelial-mesenchymal transition implicated in liver tumourigenesis and angiogenesis,27 were markedly increased in WT preneoplastic nodules, while the absence of Ripk3 prevented its upregulation in nodular lesions (figure 5C–D). Finally, CDAA-induced p38 phosphorylation in preneoplastic nodules was abrogated by Ripk3 deficiency (figure 5D) in agreement with reports that RIPK3 promotes vascular permeability through necroptotic-independent activation of p38.28 Taken together, these findings suggest that the absence of Ripk3 dampens signalling pathways implicated in transformation of dysplastic hepatocytes and aggressiveness of tumours.

Receptor-interacting protein kinase 3 (RIPK3) deficiency impacts on oxidative damage and signalling pathways interactions associated with cancer phenotypes. C57BL/6 wild-type (WT) or Ripk3−/ − mice were fed a choline-deficient L-amino acid (CDAA) or an isocaloric control choline-sufficient L-amino acid (CSAA) diet for 66 weeks. (A) Immunoblotting and densitometry of α-fetoprotein (AFP), Nanog, aldehyde dehydrogenase 1/2 (ALDH) and γH2AX in mouse liver. Blots were normalised to endogenous β-actin. Representative immunoblots are shown. (B) Fluorescence intensity from whole cells lysates stained with the fluorescence probe H2DCFDA. Values were normalised with total protein levels. (C) Quantitative RT-PCR analysis of α-smooth muscle actin (α-Sma) and macrophage inflammatory protein 2 (Mip-2) in mouse liver. (D) Immunoblotting and densitometry of vimentin, p-p38 and p38. Blots of vimentin were normalised to endogenous β-actin, whereas p-p38 was normalised to p38. Representative immunoblots are shown. Results are expressed as mean±SEM fold change of six to seven individual mice. NT, non-tumorous tissue (tissue without macroscopic discernible nodules). §P<0.05 and *p<0.01 from CSAA-fed WT mice; †p<0.05 from and ‡p<0.01 from CDAA-fed WT mice. φP<0.01 from non-tumour tissue of CDAA WT mice. ROS, reactive oxygen species.
Deletion of RIPK3 impacts on hepatic lipidome and upregulates PPARγ
Because alterations in liver fat accumulation and body weight were observed in Ripk3 −/− mice, we next analysed the mouse hepatic lipidome at 32 weeks, the time-point where increased RIPK3 levels were observed in CDAA-fed WT mice. Univariate analysis and non-supervised principal components analysis of the lipidomics data evidenced the existence of metabolic clusters associated with diet and mice genotype (figure 6A, online supplemental figure S4A). In particular, Ripk3 deficiency decreased levels of diglycerides (DG) and TG with longer acyl chains and high number of double bonds (figure 6B,C, online supplemental figure S4B). Furthermore, monounsaturated FA (MUFA) and polyunsaturated (PUFA)/saturated FA (SFA) or MUFA/PUFA ratios were decreased in CDAA-fed WT mice, but increased in CDAA Ripk3 −/− versus CDAA WT mice (table 2). Ripk3 deletion associated with increased FA 16:1n-7/16:0 and FA 18:1n-9/18:0 ratios, irrespective of the diet (table 2), indicating increased stearoyl-CoA desaturase (SCD) activity in Ripk3 −/− mice. Similarly, fatty acid desaturases appear to be modulated in Ripk3 −/− mice fed the CDAA diet, as evidenced by the change in FA 18:3n-6/18:2n-6, 20:4n-3/20:3n-3 and 20:5n-3/20:4n-3 ratios (table 2). In line with increased liver fat accumulation in CSAA Ripk3 −/− mice, the FA 16:0/18:2n-6 ratio, surrogate of lipogenic index, and fatty acid synthase (Fasn) and acetyl-CoA carboxylase mRNA levels were also increased (table 2, online supplemental figure S4C). Conversely, FA 22:5n-3/20:5n-3 and FA 18:0/16:0 ratios, indicative of elongation of very long chain fatty acid protein (ELOVL2/5) and ELOVL6 activities, respectively, were not affected by Ripk3−/ − deficiency. Of note, very long chain fatty acids have been functionally implicated in necroptosis14 15; here, we show that Ripk3 deficiency could increase the expression of the first committed enzymes in the de novo lipogenesis pathway, but species with long acyl chains were even decreased in Ripk3−/ − mice. In addition, very long-chain specific acyl-CoA dehydrogenase and medium-chain-specific acyl-CoA dehydrogenase that catalyse the first step of the mitochondrial fatty acid β-oxidation pathway were increased in CDAA-fed Ripk3−/ − mice compared with WT counterparts. This is in agreement with our previous observations showing that Ripk3 −/− deficiency improves mitochondrial respiratory chain enzymatic complexes activity during CDAA feeding, while inhibition of the downstream target of RIPK3 increased β-oxidation on steatotic stimuli.13 Finally, TG/DG and omega-6/omega-3 ratios, which are increased in human NAFLD,29 were augmented in CDAA-fed WT mice, a phenomenon that was prevented by Ripk3 deletion (table 2). Overall, Ripk3 deletion induced global changes in the hepatic lipidome of mice fed both the CSAA and CDAA diets at 32 weeks, towards a better lipid profile in the context of NASH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Deletion of receptor-interacting protein kinase 3 (RIPK3) impacts on hepatic lipidome and upregulates peroxisome proliferator-activated receptor γ (PPARγ) in vivo. C57BL/6 wild-type (WT) or Ripk3−/ − mice were fed a choline-deficient L-amino acid (CDAA) or an isocaloric control choline-sufficient L-amino acid (CSAA) diet for 32 weeks. (A) Score scatter plot corresponding to a principal components analysis (PCA) of the lipidomic data from hepatic tissue samples. Each individual is represented by one dot. (B) Relative intensity of two selected triglycerides (TG). (C) Heatmap representation of the TG number of carbons and double bond content in Ripk3−/ − mice versus WT mice. The colour code represents the transformed ratios between means of the groups (log2(fold change)). *P<0.05, **p<0.01, p<0.001. (D) Serum fasting insulin and glucose levels from mice. (E) Glucose tolerance test of CSAA, CDAA or standard diet (SD)-fed mice. (F) Liver glycogen content. (G) Serum fibroblast growth factor-21 (FGF-21) from mouse. (F) Immunoblotting and densitometry of PPARγ in mouse liver. Blots were normalised to endogenous β-actin. Representative immunoblots are shown. Results are expressed as mean±SEM arbitrary units or fold change of six to seven individual mice. §P<0.05 and *p<0.01 from CSAA-fed WT mice; †p<0.05 from CDAA-fed WT mice. (G) Representative immunoblots of RIPK3 and PPARγ in the liver of patients from cohort A (n=22) and their Spearman’s correlation scatter plot. Blots were normalised to endogenous β-actin. NAFL, non-alcoholic fatty liver; NASH, non-alcoholic steatohepatitis.
Relative hepatic lipid content of the study groups from the CDAA murine model at 32 weeks
Since lipid metabolism, including FA desaturation, and insulin/glucose metabolism are closely interrelated,30 we also found that Ripk3 −/− mice displayed increased fasting insulin irrespective of diet (figure 6D), without impacting on glucose tolerance (figure 6E) and even improving hepatic glycogen levels in CDAA-fed mice (figure 6F). Furthermore, serum levels of fibroblast growth factor-21 (FGF-21), a metabolic regulator of glucose and lipid metabolism, increased in Ripk3 −/− mice irrespective of diet (figure 6G).
The phenotypic and metabolic changes found in Ripk3−/ − mice fed either a CSAA or CDAA diet, namely reduced inflammation, less cell proliferation and augmented body weight, hepatic lipid storage, SCD activity and insulin, prompted us to investigate the regulation of PPARγ by RIPK3. We found increased PPARγ protein and mRNA levels in the liver of Ripk3 −/− mice (figure 6H, online supplemental figure S4E). Moreover, in the liver of patients from NAFLD cohort A, PPARγ negatively correlated with RIPK3 (figure 6I). In agreement, on treatment with palmitic acid (PA), Ripk3−/ − primary hepatocytes displayed increased Pparγ promoter activity and mRNA levels compared with WT hepatocytes (online supplemental figure S5A and B). Furthermore, in Ripk3−/ − primary hepatocytes, PA-induced Fasn expression and intracellular fat accumulation as assessed by Nile red fluorometric quantification were, at least in part, PPARγ-dependent (online supplemental figure S5B and C). Knockdown of RIPK3 was also associated with decreased levels of collagen and vimentin in immortalised human hepatic stellate cells LX-2 after TGF-β treatment, while co-silencing of PPARγ abrogated this phenotype (online supplemental figure S5D and E), suggesting that increased PPARγ levels in Ripk3 cells also contributes to impaired fibrosis development.
Also, in line with increased FGF-21 levels, Pparγ mRNA was upregulated in white adipose tissue of CDAA-fed Ripk3 −/− mice, accompanied by increased expression of uncoupling protein 1, a target of PPARγ involved in thermogenesis (online supplemental figure S3E). Circulating levels of leptin, an adipokine that reflects adipose tissue mass, regulates energy homeostasis and whose expression is controlled by PPARγ, diminished on CDAA feeding in WT mice, whereas deletion of Ripk3 −/− raised leptin levels compared with their WT CSAA or CDAA-fed counterparts (online supplemental figure S4G). Conversely, the diet or genotype did not impact on mouse serum adiponectin levels. The white adipose tissue preserved the normal architecture with well-differentiated adipocytes. However, the infiltration of mononuclear inflammatory cells, such as macrophages and occasional lymphocytes, was increased in CSAA-fed Ripk3 −/− mice (online supplemental figure S4H), possibly as a side effect of augmented leptin levels from the baseline.
These results suggest that RIPK3 plays an intricate role in regulating metabolism, likely through PPARγ targeting, in parallel with its role in necroptosis.
Discussion
Despite several ongoing clinical trials, no effective pharmacological approach against NASH-induced fibrosis and HCC has yet been approved, pointing the need for a better understanding of pathogenic mechanisms underpinning NAFLD. The present study provides evidence that RIPK3-dependent necroptosis plays a determinant role in the pathogenesis and progression of NAFLD, facilitating fibrosis development and cancer initiation. We show increased RIPK3 levels in serum and liver of patients with NAFLD that correlate with disease severity, including markers of liver damage, inflammation and fibrosis. Furthermore, liver RIPK3 levels prior to bariatric surgery also positively correlated with increased circulating markers of liver injury 1 year after surgery. Interestingly, hepatic levels of RIPK3 were increased in male patients with NAFLD and carriers of the rs738409 C>G variant in the PNPLA3 gene, well-known risk factors of NAFLD, advanced fibrosis and HCC.3 31 In sum, augmented hepatic RIPK3 levels associate with key genetic and phenotypic determinants of NAFLD progression, emphasising the role of RIPK3-dependent necroptosis in regulating NAFLD pathogenesis.
We show that the association between RIPK3 and inflammation/fibrosis in human NAFLD is back translated in the CDAA murine model. Indeed, our results demonstrate that RIPK3 deficiency prevents hepatic inflammation, while reducing liver fibrosis, in response to CDAA feeding for both 32 and 66 weeks. Most cancers likely result from a cooperation between (micro)environmental factors and genetic somatic mutations, with the link between cancer and inflammation/fibrosis being well established in HCC. Indeed, pro-inflammatory TNF-α has been implicated in tumour growth and poor prognosis of HCC, at least in part, by activating tumour progenitor cells.32 Similarly, COX-2 overexpression in hepatocytes has been shown to induce spontaneous HCC development in mice,33 likely through modulation of progenitor/stem Wnt/β-catenin signalling pathways.34 In this regard, we show that CDAA-fed Ripk3−/ − mice display reduced hepatic expression of Tnf-α and Cox-2, accompanied by decreased protein levels of β-catenin, AFP and other markers of stemness. Our results further demonstrate that long-term CDAA feeding dampens p53-dependent signalling pathway in WT preneoplastic nodules, alongside increased compensatory hepatocyte proliferation, which were restored by Ripk3 deficiency. Taken together, our results suggest that deletion of Ripk3 associates with a better prognosis, at least in part, by reducing the inflammatory milieu that favours survival of dysplastic hepatocytes. Still, the inflammatory role of RIPK3-dependent signalling could play an intricate, cell/context-dependent role in hepatocarcinogenesis in vivo. On one hand, it was recently reported that RIPK3 deficiency exacerbates DEN plus CDAA-induced HCC in mice by promoting an immunosuppressive microenvironment,35 while here we showed that Ripk3−/ − mice were protected from tumour burden induced by both CDAA or DEN. On the other, it was recently shown that the necroptosis-associated hepatic cytokine microenvironment could govern cholangiocarcinoma development from ontogenically transformed hepatocytes.11 However, further studies are needed to corroborate this hypothesis in the context of NAFLD progression towards liver cancer.
Emerging evidence has shown that RIPK3-dependent signalling could also play a pivotal role in metabolic reprograming in vitro,12 14 15 but its contribution to metabolic liver disease remains to be elucidated. Inflammation may sometimes precede steatosis in human NAFLD, while only 10%–20% of patients with simple steatosis advance towards inflammatory and fibrotic stages,36 suggesting that liver steatosis and inflammation constitute independent events. Indeed, although expression of PPARγ in the normal liver is very low, Pparγ deletion in hepatocytes was shown to ameliorate liver steatosis but increase inflammation in mice on HFD‐plus‐binge ethanol.37 Noteworthy, both the CSAA and CDAA diets (~4.4 kcal/g) contain higher calories than a standard chow (~3 kcal/g).38 Thus, our results show that Ripk3−/ − mice under the hypercaloric CSAA diet display increased hepatic lipogenesis and fat accumulation in the absence of liver inflammation, together with increased hepatic expression of PPARγ. Noteworthy, PPARγ agonists, which induce moderate weight gain, are used off-label in patients with NASH39 and may also impact on the hepatic lipidome by inducing SCD1,40 a desaturase implicated in the conversion of SFA to MUFA. Accordingly, Ripk3−/ − mice fed either a CSAA or a CDAA diet were more obese than their WT counterparts, but display augmented MUFA+PUFA/SFA. Thus, our results support that, by upregulating PPARγ, Ripk3 deletion shifts the hepatic lipid profile to buffer the accumulation of detrimental lipid species and could trigger lipoexpediency, a phenomenon in which lipid signals generated by de novo lipogenesis can be directed towards benefit.41 Besides its well-known metabolic and anti-inflammatory effects, PPARγ also displays antiproliferative properties. For instance, PPARγ ligands have been shown to upregulate p21 and reduce cyclin D1 gene expression, inhibiting human lung carcinoma cell growth.42 Of note, in our study, Ripk3−/ − mice at 66 weeks of CDAA feeding show a similar pattern. PPARγ has been suggested to act as a tumour suppressor in HCC,43 whereby its activation could contribute to arrest carcinogenesis in Ripk3−/ − mice-fed a CDAA diet. Finally, in line with the reported antifibrogenic role of PPARγ activation,44 our results suggest that the upregulation of PPARγ in Ripk3-deficient hepatic stellate cells prevented collagen production and, thus, could contribute to impaired fibrosis development in CDAA-fed Ripk3−/ − mice.
In agreement with our results, it was shown that HFD-fed Ripk3−/ − mice also become obese and display increased liver steatosis compared with WT counterparts, and display augmented fasting insulin and glucose levels compared with chow diet-fed Ripk3−/ − mice. In addition, PPARγ and SCD were also upregulated in HFD-fed Ripk3−/ − mice.18 However, in contrast with our results, inflammation and fibrosis were aggravated, likely due to increased apoptosis in the liver and adipose tissue.18 19 The different outcomes of Ripk3 deletion in liver inflammation and fibrosis on different NAFLD models may rely on the specific features of each experimental model; the methionine-deficient and choline-deficient and CDAA diets cause substantial steatosis, inflammation, fibrosis and liver damage, whereas the HFD results solely in mild fibrosis and no liver damage, despite inducing steatosis.45 As such, although liver necroptosis is also activated in response to a HFD,18 its extent and relevance in these conditions is likely minor.
Whether PPARγ-related effects are solely a consequence of the genetic deletion of Ripk3 in mice under a hypercaloric diet, or whether they could also be achieved with the pharmacological inhibition of core components of the necroptotic machinery, including RIPK3, remains unknown. Further insights on possible metabolic events resulting from the pharmacological inhibition of necroptosis should also be provided by the first clinical trials using a small molecule inhibitor of RIPK1 kinase for autoimmune diseases.46
In conclusion, our study provides a comprehensive characterisation of the role of RIPK3-dependent signalling in a stepwise progression of NASH towards HCC, unravelling a paradoxical new role. While RIPK3 deficiency increases obesity, liver fat accumulation and insulin/glucose levels, it also impairs long-term inflammation and signalling pathways implicated in transformation of dysplastic hepatocytes. Thus, RIPK3 plays an intricate role in the interplay between cellular metabolism, cell death and inflammation, whose modulation could offer new venues to arrest NASH progression to HCC.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Human sample collection was performed after patient informed consent and Institutional Review Board approval, in accordance with the Declaration of Helsinki.All animal experiments were carried out with the permission of the local Animal Welfare Organ in accordance with the EU Directive (2010/63/EU), Portuguese laws (DL 113/2013, 2880/2015, 260/2016, and 1/2019) for the use and care of animals in research and all relevant legislations. The experimental protocol was approved by the competent national authority, Direcção Geral de Alimentação e Veterinária, Portugal.
Acknowledgments
We thank Dr Vishva Dixit, Molecular Oncology Department, Genentech, Inc. (San Fransisco, CA, USA) and Dr Miguel Soares, Instituto Gulbenkian de Ciência (Oeiras, Portugal) for kindly providing WT and Ripk3 − / − from the same genetic background. We also thank Dr Madalena Monteiro, Laboratório de Patologia, Instituto Nacional de Investigação Agrária e Veterinária (Oeiras, Portugal) and Dr Pedro Ruivo, Unidade de Patologia Comparada, Instituto de Medicina Molecular (Lisbon, Portugal) for technical assistance. The authors thank Mediagnost, GmbH (Reutlingen, Germany) for providing leptin and adiponectin ELISA kits and Nina Bierig for technical assistance. Finally, we thank all members of the laboratory for insightful discussions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it published Online First. The names for authors Amine Majdi, Jesus M Banales and Alvaro Santos-Laso have been corrected.
Contributors MBA performed most of the experiments, analysed and interpreted results, and wrote the manuscript. PMR, ALS and MMG. helped in animal experiments and MM-P in data analysis. EA and CA performed lipidomics. AS, RJ-A, EG, LB, MJP, VR, JG. and JB. provided human samples and clinical data. AM performed experiments with samples of cohort A. JB, VR and JG. provided important intellectual input. REC was involved in planning and organisation of the study. CMPR was the principal investigator of the study responsible for study concept and design. All authors critically revised the manuscript.
Funding Main funding is provided by FEDER funds through the COMPETE programme and by national funds through Fundação para a Ciência e a Tecnologia to CMPR (grants SAICTPAC/0019/2015 - LISBOA-01–0145-FEDER-016405 and PTDC/MED-FAR/29097/2017 - LISBOA-01-0145-FEDER-029097). Additional funding comes from research grant APEF (Portuguese Association for the Study of Liver)/BAYER 2020 to MBA. JG is funded by the Fondation pour la Recherche Médicale (ARF20170938613), the Institute of Cardiometabolism and Nutrition (PAP17NECJG), the Société Francophone du Diabète (R19114DD) and the Mairie de Paris (Emergences – R18139DD). MBA, PMR, MMP and ALS were investigators or students funded by Fundação para a Ciência e a Tecnologia.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.