Article Text

Abstract
Background & objectives Alcoholic hepatitis (AH) is a common but life-threatening disease with limited treatment options. It is thought to result from hepatocellular damage, but the presence of cholestasis worsens prognosis, so we examined whether bile ducts participate in the pathogenesis of this disease.
Design Cholangiocytes derived from human bile ducts were co-cultured with neutrophils from patients with AH or controls. Loss of type 3 inositol 1,4,5-trisphosphate receptor (ITPR3), an apical intracellular calcium channel necessary for cholangiocyte secretion, was used to reflect cholestatic changes. Neutrophils in contact with bile ducts were quantified in liver biopsies from patients with AH and controls and correlated with clinical and pathological findings.
Results Liver biopsies from patients with AH revealed neutrophils in contact with bile ducts, which correlated with biochemical and histological parameters of cholestasis. Cholangiocytes co-cultured with neutrophils lost ITPR3, and neutrophils from patients with AH were more potent than control neutrophils. Biochemical and histological findings were recapitulated in an AH animal model. Loss of ITPR3 was attenuated by neutrophils in which surface membrane proteins were removed. RNA-seq analysis implicated integrin β1 (ITGB1) in neutrophil-cholangiocyte interactions and interference with ITGB1 on cholangiocytes blocked the ability of neutrophils to reduce cholangiocyte ITPR3 expression. Cell adhesion molecules on neutrophils interacted with ITGB1 to trigger RAC1-induced JNK activation, causing a c-Jun-mediated decrease in ITPR3 in cholangiocytes.
Conclusions Neutrophils bind to ITGB1 on cholangiocytes to contribute to cholestasis in AH. This previously unrecognised role for cholangiocytes in this disease alters our understanding of its pathogenesis and identifies new therapeutic targets.
- alcoholic liver disease
- bilirubin
- chemokines
- cholestasis
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Alcoholic hepatitis is a life-threatening complication of alcoholic liver disease with limited effective therapies and is a leading cause of mortality worldwide.
The presence of cholestasis in alcoholic hepatitis is associated with disease severity and poor prognosis.
In other liver diseases, cholestasis occurs in part because of involvement of cholangiocytes and loss of expression of their primary intracellular calcium release channel, type 3 inositol 1,4,5-trisphosphate receptor (ITPR3), which is essential for bile secretion from cholangiocytes.
Although neutrophilia and hepatic neutrophil infiltration are hallmarks of alcoholic hepatitis, it is not known whether neutrophils associate with cholangiocytes or exert a pathological effect on them in this disease.
What are the new findings?
Endotoxin induces cholangiocytes to produce CXCL8, which attracts neutrophils.
Intercellular adhesion molecule and vascular cell adhesion molecule on neutrophils interact with integrin β1 on cholangiocytes to decrease ITPR3 expression through a RAC1/JNK/c-Jun pathway.
In patients with alcoholic hepatitis, cholangiocytes express increased amounts of integrin β1, and neutrophils associate with bile ducts in direct proportion to the degree of cholestasis.
How might it impact on clinical practice in the foreseeable future?
These findings reveal previously unrecognised roles for cholangiocytes and neutrophils in this disease and alter our understanding of the pathogenesis of alcoholic hepatitis.
Given currently limited therapeutic options for patients with alcoholic hepatitis, these findings suggest potentially druggable targets and pathways to ameliorate cholestasis and reduce disease severity and mortality.
Introduction
Excessive alcohol intake is the third leading cause of preventable death in the USA1 and is the leading risk factor for death globally among people aged 15–49 years.2 3 Moreover, cirrhosis-related mortality among people aged 25–34 years has been increasing by >10% per year in recent years, and this is driven entirely by alcohol-related liver disease.4 Alcoholic hepatitis is a severe and potentially life-threatening complication of alcoholic liver disease, with a short-term mortality ranging from 20% to 50%.4–6 Thus far, corticosteroids are the only pharmacological treatment that has demonstrated efficacy,6 and this has been in use for over 40 years.5 However, this therapy often is ineffective.7 Until now, alcoholic hepatitis (AH) has been attributed mostly to hepatocellular damage, resulting in varying degrees from ethanol and its metabolites, endotoxin or other bacterial toxins, oxidative damage and other inflammatory mediators.6–8 Indeed, animal models of AH mostly recapitulate infiltration of neutrophils into the hepatic lobule, steatosis, hepatocellular apoptosis and elevations in serum transaminases that are seen in patients with this disorder.9 10 However, emerging evidence suggests that AH also results in cholestatic liver injury and the presence of impaired bile secretion, or cholestasis, portends a worse prognosis.11–14 This is potentially a paradigm-shifting concept because cholestasis often is a separate process from hepatocellular injury, and so its presence may suggest previously unappreciated pathogenic mechanisms and therapeutic targets.
Although cholestasis may result from impaired secretory function of hepatocytes, it often instead reflects impaired secretion by cholangiocytes.15 16 A variety of liver diseases are largely due to cholangiocyte damage or dysfunction, such as primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), biliary atresia and biliary obstruction, and these cholangiopathies are characterised by cholestasis. Despite their widely different aetiologies, they all result in loss of expression of type 3 inositol 1,4,5-trisphosphate receptors (ITPR3) from cholangiocytes, which is sufficient to cause cholestasis.17 18 ITPR3 plays this important role because it is the primary intracellular calcium release channel in cholangiocytes,18 19 and its expression is needed to mediate biliary fluid and bicarbonate secretion.17 Cholangiocytes also can be involved in the cholestasis that occurs in AH13 and, as in other cholangiopathies, this is due to loss of ITPR3 as well.14 This in turn suggests that the clinically severe form of AH is due in part to a previously unrecognised mechanism (loss of ITPR3) in a cell type that previously has not thought to play a major role in this disease (cholangiocytes). Neutrophilia and the presence of neutrophils in the hepatic lobule both are part of the clinical picture of AH.6 12 20 Because neutrophilia is increased in patients with AH with cholestasis, relative to other patients with AH,14 we examined whether the cholestasis that occurs in AH is due to direct interactions between neutrophils and cholangiocytes.
Methods
Human liver biopsies, clinical data from patients with AH and cirrhosis and blood samples from healthy volunteers and from individual patients with AH and alcoholic cirrhosis were obtained from Yale-New Haven Hospital, Yale University.
Human blood samples and human neutrophil isolation
Blood samples were collected from healthy volunteers and from consecutive patients admitted to Yale-New Haven Hospital (YNHH) during 2019–2020 with the diagnosis of AH or alcoholic cirrhosis. The patients with AH included five men and three women, aged 43.5±10.0 years (mean±SD; range, 31–58), with a Model for End-Stage Liver Disease (MELD) score of 30±7 (range, 20–40) and a Maddrey’s Discriminant score of 56±20 (range, 22–82). The diagnosis of AH was confirmed by liver biopsy in five of the eight patients, and blood samples were collected before patients had begun corticosteroid treatment. All patients with alcoholic cirrhosis were abstinent for at least 3 months prior to admission. Peripheral venous blood samples were collected into K2 EDTA tubes (Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA) from healthy human volunteers, who were not taking any medication or alcohol, or from patients diagnosed with AH or alcoholic cirrhosis as above. Human neutrophils were isolated and purified by density gradient centrifugation using PolymorphPrep (Axis Shield, Oslo, Norway), according to the manufacturer’s instructions. Briefly, blood was layered over PolymorphPrep solution and centrifuged at 500× g for 35 min at 20°C without the brake. After centrifugation, the neutrophil layer was collected and washed with diluted N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-buffered saline to eliminate residues of PolymorphPrep. Neutrophils were harvested by centrifugation at 400× g for 10 min. To purify neutrophils and remove any residual erythrocyte contamination from the residual erythrocytes, cells were washed with ACK lysing buffer (Lonza, Bend, Oregon, USA). Neutrophils were harvested by centrifugation and resuspended in normal human cholangiocyte (NHC) medium. Cell density was quantified using a hemocytometer. Purity of neutrophil preparations was confirmed by a cytospin on the isolated neutrophils followed by Wright’s or May-Grünwald-Giemsa staining/Pappenheim staining. Only isolations with >95% neutrophils were used. For generation of ‘naked’ neutrophils, neutrophils were treated with phosphatidylinositol-specific phospholipase C (PI-PLC, 0.5 units/mL, Sigma-Aldrich, St. Louis, Missouri, USA) for 30 min at 37°C with gentle agitation.
Human liver specimens and histology
AH specimens were from patients with a clinical and morphological diagnosis of AH, who were treated at YNHH from 2012 to 2014. Clinical characteristics of patients with biopsy-proven AH at YNHH were analysed. Alcoholic cirrhosis specimens were obtained from patients with this diagnosis who underwent liver transplant at YNHH from 2014 to 2018, and so had been abstinent for at least the preceding 6 months. Histologically normal tissues were obtained from the peripheral area of liver resections performed for metastatic colon cancer, and cases were identified by a review of pathology reports from 2010 to 2017 at YNHH. Clinical, laboratory and histological characteristics of patients with non-alcoholic steatohepatitis (NASH), PBC and PSC whose liver biopsies were reviewed are summarised in online supplemental table S1. Liver biopsy or resection specimens were (previously) stained with H&E and were independently reviewed, and the histological diagnosis was confirmed by two experienced liver pathologists (MER, PTV). Cholestasis was classified histologically as either absent (no cholestasis), mild (with fine hepatocellular granules and scant canalicular bile), moderate (which additionally included coarse inspissated canalicular bile and occasional ductular bile plugs) or severe (which additionally included prominent ductular and canalicular bile plugs).
Supplemental material
To identify and quantify bile duct-neutrophil interactions, images from 10 different portal areas in each liver biopsy were captured using a 40× objective and the same camera settings on a digitalized Olympus BX51 microscope. For each image, the number of neutrophils in direct contact with the bile duct epithelium was counted. Both H&E stained slides and slides stained with the neutrophil-specific label chloroacetate esterase (CAE) were used, and these gave the same results (online supplemental figure S1). Slides were scored in a blinded fashion by an experienced liver pathologist (PTV) and results were represented as the number of neutrophils adjacent to bile ducts per 10 high-powered fields.
Supplemental material
Supplemental material
Naphthol AS-D chloroacetate esterase staining
Histochemical detection of a naphthol AS-D CAE enzyme, which is enzymatically hydrolysed by neutrophil-specific esterases, was chosen because formalin fixation destroys the activity of most enzymes in leucocytes, but CAE activity is retained. CAE staining is highly specific for granulocytes and is used to detect neutrophils in tissue sections that have been paraffin embedded.21 Liver tissue sections were warmed at 75°C for 15 min, deparaffinised in xylene for 15 min (5 min each) and then rehydrated in alcohol series (100%, 90%, 70% ethanol (5 min each) and tap water (15 min)). The slides were subsequently CAE-stained using the Naphthol AS-D Chloroacetate (Specific Esterase) kit (Catalogue # 91C, Sigma-Aldrich) in accordance with the manufacturer’s instructions. After that, liver sections were dehydrated with serial dilution of ethanol (70%, 90% and 100%) and xylene and then tissue sections were mounted with Cytoseal 60 (Thermo Scientific, Richard-Allan Scientific). Neutrophil counting was performed as previously described for H&E.
Immunohistochemistry staining for c-Jun
Immunohistochemical analysis of c-Jun was performed in liver biopsies from histologically normal livers and from patients with AH as previously described.22
Cell culture
The NHC cell line isolated from normal human liver was used for this study because it has been characterised at the transcriptomic,23 proteomic24 and functional14 25–29 level. These cells were cultured as previously described.14 26 27
Neutrophil extracellular trap formation
Neutrophils (1×106 cells) were initially stained with CellTracker Orange CMTMR Dye (5 µM, Invitrogen, Grand Island, New York, USA) for 30 min at 37°C, and then co-cultured with or without the NHC cell line. After co-culture and soon after the beginning of monitoring NET formation, the cell-impermeable DNA binding dye SYTOX Green (5 µM, Invitrogen) was immediately added into the co-culture system. The formation of neutrophil extracellular trap (NET) was monitored and visualised with fluorescence time-lapse imaging every 5 min for 15 hours using the Opterra II Swept-field confocal microscopy (Bruker, Middleton, Wisconsin, USA). Lipopolysaccharides (LPS) from Escherichia coli O111:B4 (5 µg/mL, Sigma-Aldrich) or phorbol 12-myristate 13-acetate (25 µM, Sigma-Aldrich) was added in the co-culture for 2 hours and used for positive controls of NET formation. For the quantification of NET formation, the baseline CellTracker (red fluorescence intensity) at the starting time was subtracted from the red fluorescence intensity at each subsequent time point and then divided by the SYTOX Green fluorescence intensity.
Cholangiocyte cultures in neutrophil-conditioned medium
The NHC cell line (2×105 cells/mL) was plated onto a 6-well plate and cultured overnight. Human neutrophils (1×106 cells) were then freshly isolated as above and incubated in NHC medium in the presence or absence of LPS (0.2 µg/mL) for 8 hours at 37°C. Neutrophils were removed by centrifugation (400× g×3 min) and the supernatant was used as the neutrophil-conditioned medium. The NHC cell line was incubated with the neutrophil-conditioned medium (2 mL) for an additional 18–24 hours. Cells were subsequently harvested and assessed for the expression of ITPR3.
Confocal fluorescence imaging
To identify NET formation, neutrophils (1×106 cells) and the NHC cell line (5×105 cells) were plated onto 6-well culture plates containing glass coverslips treated with collagen type I solution (Corning, New York, USA). After 2 hours, cells were fixed with 4% paraformaldehyde (Electron Microscopy Science, Hatfield, Pennsylvania, USA) and permeabilised with 0.5% Triton X-100 (Sigma-Aldrich). After washing in phosphate-buffered saline (PBS; GIBCO), non-specific binding was blocked using 1% bovine serum albumin (BSA) (Sigma-Aldrich), 0.05% Tween and 5% normal goat serum (Invitrogen) in PBS for 30 min at room temperature. For co-immunolabelling with primary antibodies, mouse anti-myeloperoxidase (MPO) antibody (Clone 2C7, ab25989, Abcam, 1:100 dilution) and rabbit anti-histone H3 (citrulline R2+R8+R17) antibody (ab5103, Abcam, 1:100 dilution) were incubated for 8 hours, at 4°C, followed by a 1-hour incubation at room temperature with goat anti-mouse Alexa 564 and goat anti-rabbit Alexa Fluor 488 secondary antibodies (1:500; Invitrogen), respectively. DNA was stained with DAPI (1:1000, Invitrogen). After labelling with fluorochromes, the glass coverslips (Thermo Fisher Scientific, Waltham, Massachusetts, USA) were mounted in an antifade fluorescent mounting medium (Invitrogen). Images were then obtained using a ZEISS LSM 710 or 880 confocal fluorescence microscope (Carl Zeiss, New York, USA).
For immunofluorescence staining of CD16, neutrophils were attached onto Superfrost Plus microscope slides (Thermo Scientific) by Shandon Cytospin (Thermo Scientific) and were fixed with 4% paraformaldehyde (Electron Microscopy Science) in PBS for 30 min. Slides were then washed with PBS and quenched in 50 mM NH4Cl (Sigma-Aldrich) in PBS for 20 min at room temperature to reduce autofluorescence. Slides were then washed 3 times with 0.05% tris-buffered saline (TBS)-Tween, blocked in 1% BSA and 5% normal goat serum in TBS for 1 hour at room temperature, and then incubated overnight at 4°C with rabbit anti-CD16 antibody (Clone 3G8, 1:200, Invitrogen) in blocking buffer. Cells were incubated with Alexa Fluor 488 conjugated goat anti-rabbit antibody (1:500 dilution, Invitrogen) for 1 hour at room temperature while protected from light. Cell nuclei were stained with Hoechst 33342 (1:1000 dilution, Invitrogen). Primary antibodies were omitted for background control specimens/images. Stained cells were washed in 0.05% TBS-Tween and cover-slipped with ProLong Gold Antifade Reagents (Invitrogen) and kept in the dark overnight at 4°C. Images were captured using a ZEISS LSM 880 Confocal Laser Scanning Microscope. All images were captured using the same settings, and images were processed using Zeiss Efficient Navigation software.
Neutrophil Ca2+ signalling
Cytosolic Ca2+ signalling in neutrophils in response to N-Formyl-Met-Leu-Phe (fMLP) was monitored with a Zeiss LSM 710 confocal microscope. Briefly, human neutrophils attached to a glass coverslip were incubated with HEPES buffer (pH 7.4), then loaded with the calcium-sensitive fluorescent dye Fluo-4/AM (5 µM; Molecular Probe) for 30 min at 37°C. Coverslips containing the labelled neutrophils were transferred to a custom-built perfusion chamber on the stage of the confocal microscope, and the cells were perfused with the HEPES buffer while stimulated with 10 µM fMLP (Sigma-Aldrich), a potent neutrophil agonist of the G protein-coupled N-formylpeptide receptor. Relative Fluo-4 fluorescence intensity after fMLP stimulation was analysed in selected regions of interest using ImageJ (National Institutes of Health) and compared with baseline Fluo-4 fluorescence intensity as described.27
Fluorescence-activated cell sorting
For the assessment of viability and surface expression of CD16, 1×106 neutrophils were aliquoted into FACS tubes. Neutrophils were stained with anti-CD16 monoclonal antibody (Invitrogen) followed by FITC-labelled anti-IgG (Invitrogen), APC-labelled Annexin V (BioLegend, San Diego, California, USA) and propidium iodide (BD Biosciences). Cells were analysed using a FACSCalibur flow cytometer (BD Biosciences). FACS data were analysed with FlowJo software (FlowJo, Ashland, Oregon, USA).
Transwell migration assays
The NHC cell line (1×105 cells) was seeded on the underside of 24-well plates for 18 hours and incubated with LPS (5 µg/mL) for 6 hours at 37 °C. The supernatant was removed and rinsed with PBS then maintained in NHC medium. Meanwhile, human neutrophils were isolated and preincubated with CellTracker Orange CMTMR Dye (5 µM) for 30 min at 37°C. Transwell inserts with 3.0 µm pore-size transparent PET membranes (FALCON-353096) were placed in a 24-well plate. A C-X-C motif chemokine ligand-8 (CXCL8) antagonist (1 µg/mL) was added to the bottom compartment, and neutrophils (5 ×105 cells) in NHC medium were added to the top of each Transwell. As a positive control, recombinant CXCL8 (1 µg/mL) was added to the bottom side. All neutrophils were allowed to migrate for 1 hour. The migration of neutrophils into the bottom compartment was monitored using a Zeiss Axio Observer epifluorescence microscope with a 20× objective, and the number of migrating neutrophils was quantified using ImageJ.
Connexin 43, integrin β1, nuclear factor kappa B-p65 or c-Jun silencing
Connexin 43 (CX43), integrin β1 (ITGB1), nuclear factor kappa B (NFκB)-p65, or c-Jun were suppressed by the human CX43-specific small interfering RNA (siRNA) (Ref.# L-011042-00-0005, Horizon Discovery, formerly Dharmacon, Lafayette, Colorado, USA), the human ITGB1-specific siRNA (SO-2826724G, Horizon Discovery), the human NFκB-p65-specific siRNA (Ambion-Thermo Fischer Scientific) or the human c-Jun-specific siRNA (Ambion-Thermo Fischer Scientific), respectively. A non-targeting siRNA (scrambled-siRNA) was also obtained from Ambion (Thermo Fischer Scientific) for negative control transfections. In brief, the NHC cell line was seeded overnight, then transfected with 50 nM of CX43-siRNA, ITGB1-siRNA, NFκB-p65-siRNA, c-Jun-siRNA or scrambled-siRNA using Lipofectamine RNAi MAX Transfection Reagent (Invitrogen). After 24 hours post-transfection, the magnitude of CX43, ITGB1, NFκB-p65 or c-Jun silencing was assessed at the messenger RNA level by TaqMan real-time PCR or at the protein level by western blot analysis.
RNA isolation and quantitative real-time PCR. Total RNA was extracted and purified by a RNAqueous Total RNA Isolation kit (Invitrogen) according to the manufacturer’s protocol. One microgram of total RNA was reverse-transcribed into cDNA using an iScript cDNA Synthesis kit (Bio-Rad, Hercules, California, USA). All TaqMan primers and probes were obtained from Applied Biosystems (Foster City, California, USA): CX43 (Hs00748445_s1), ITGA5 (Hs01547673_m1), ITGB1 (Hs01127536_m1), ITGB3 (Hs01001469_m1), NFκB-p65 (RELA) (Hs01042014_m1), CXCL1 (Hs00236937_m1), CXCL8 (Hs00174103_m1), GAPDH (Hs02758991_g1), ACTB (Hs01060665_g1). Real-time PCR reactions were performed in triplicate using an ABI 7500 Sequence Detection System (Applied Biosystems). The expression of target genes was normalised to GAPDH or ATCB and quantification of relative expression was determined as previously described.22
Immunoblotting analysis
Western blot analyses of NHC lysates were performed as previously described.22 The following primary antibodies were used: ITPR3 (Clone 2/IP3R-3, 610312, BD Transduction Laboratories), NFκB-p65 (Clone E379, ab32536, Abcam), RAC1 (PA1-091, Invitrogen), ITGB1 (Clone 12G10, ab30394, Abcam), JNK (9252, Cell Signaling Technology), phospho-JNK (pJNK, 9251, Cell Signaling Technology) and GAPDH (Clone 6C5, AM4300, Invitrogen). The band intensities of proteins of interest were quantified and analysed using FOTODYNE Imaging and TL-100 image analysis or ImageJ software.
RNA sequencing and enrichment analysis
RNA was isolated from the NHC cell line co-cultured with neutrophils (n=4) and without neutrophils (n=3). The RNeasy kit (QIAGEN) was used and RNA integrity number was determined using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, California, USA). On average, 40 million sequencing reads (100 bp paired end) of each sample were obtained using an Illumina 6000 Sequencer. The sequencing data were analysed using the public server at usegalaxy.org. Quality control was performed by FastQC (Galaxy V.0.2.6+galaxy0) and Trimmomatic (Galaxy V.0.2.6+galaxy0). To remove Illumina adaptor sequences, the TruSeq3-paired-end file was used in Trimmomatic. The reads were mapped to the University of California Santa Cruz hg19 human genome by STAR (Galaxy V.2.6.0b-1), 78.62% of which were uniquely aligned to the reference. The raw read counts of genes were counted by feature Counts (Galaxy V.1.6.4+galaxy1) and the differential expression analysis was conducted by DESeq2 (Galaxy V.2.11.40.6). A Volcano plot was generated using the Enhanced Volcano package (V.3.10) in R (V.3.6.1). The cut-off value was set at 0.01 for p values and at 1.5 for fold-change. Pathway enrichment analysis of significantly differentially expressed genes (p≤0.01) in different gene subsets was determined by IPA (Ingenuity Pathway Analysis, QIAGEN). The RNA-seq data have been deposited in the Gene Expression Omnibus under the accession number GSE146899.
Functional blocking interventions
To inhibit the function of ITGB1 in cholangiocytes, an anti-β1-integrin function blocking antibody (AIIB1, Developmental Studies Hybridoma Bank) was used. In brief, the NHC cell line (5×105 cells/well) was seeded overnight and preincubated with ITGB1 antibody (2.2 µg/mL) for 2 hours at 37°C. The ITGB1 antibody was removed and cells were rinsed with PBS and co-cultured with or without neutrophils for an additional 18 hours. Cells were then harvested and assessed by western blot analysis. For adhesion molecule blockade on the surface of neutrophils, the human vascular cell adhesion molecule-1 (VCAM-1)/CD106 antibody (Clone BBIG-V1, BBA5, R&D Systems) or the human intercellular adhesion molecule-1 (ICAM-1)/CD54 antibody (Clone BBIG-I1 (11C81), BBA3, R&D Systems) were employed. Briefly, isolated neutrophils (1×106 cells) were preincubated with or without 25 µg/mL of VCAM-1 and/or 25 µg/mL of ICAM-1 antibodies for 2 hours at 37°C. After that, neutrophils were washed with diluted HBSS without Ca2+/Mg2+, and co-cultured with NHCs for an additional 18 hours. NHCs were then harvested and assessed by western blot analysis.
Transient transfection and luciferase reporter gene assay
The NHC cell line (2×105 cells/well) was plated onto 24-well plates 24 hours prior to transfection. Luciferase reporters for the human ITPR3 and the expression plasmid for c-Jun (100 ng) were transiently transfected into the cells, along with a renilla luciferase (Promega) as a control for transfection efficiency using FuGENE HD Transfection Reagent (Promega). Cell lysates were harvested 48 hours after transfection to measure firefly and renilla luciferase activity using a dual-luciferase reporter assay system (Promega) as previously described.22
Mouse model of alcoholic hepatitis
AH was induced in male C57BL/6N mice aged 9–10 weeks (Charles River Laboratories) using a modification of the National Institute on Alcohol Abuse and Alcoholism (NIAAA) model of chronic plus binge ethanol feeding.30 Lieber-DeCarli control and ethanol liquid diets were obtained from BioServ (Flemington, New Jersey, USA). After acclimatisation to tube feeding with the control diet ad libitum for 5 days, mice received either the control liquid diet (paired-fed control group) or the ethanol liquid diet (ethanol-fed group) for 10 days. However, in the ethanol-fed group, mice received the ethanol liquid diet plus 30% fructose (Sigma-Aldrich) along with intermittent injection of LPS (Escherichia coli O111:B4, 0.05 mg/kg body weight, Sigma-Aldrich) on the 4th day, 7th day and 10th day of the ethanol liquid diet regimen. The control group was fed with the same amount of food consumed by ethanol-fed mice and with a non-isocaloric control diet. On day 11, mice were gavaged with 5 g ethanol/kg body weight and sacrificed 9 hours later. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were determined by the YNHH Chemistry Laboratory. All animals received humane care in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Statistical analysis
Data are expressed as mean±SEM of multiple independent experiments, unless stated otherwise. Statistical analyses were performed using the paired, unpaired Student’s t-test, or one-way analysis of variance with Tukey’s multiple comparison test where appropriate. Differences with p<0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism 7 Software (GraphPad, La Jolla, California, USA).
Results
Neutrophils are in contact with bile ducts in alcoholic hepatitis and this correlates with degree of cholestasis
To investigate the association between neutrophils and cholangiocytes, liver biopsy specimens from patients with AH, patients with alcoholic cirrhosis who were abstinent and normal controls were reviewed. Neutrophils in contact with bile ducts were identified in a blinded fashion by a liver pathologist (figure 1A,B) and their frequency was quantified. The frequency of neutrophils in contact with bile ducts was the same regardless of whether the slides were stained with H&E or with the neutrophil-specific stain CAE (online supplemental figure S1A). Neutrophils were adjacent to bile ducts in nearly all AH specimens but rarely in alcoholic cirrhosis or normal control specimens (figure 1). Moreover, the frequency of neutrophil-bile duct contacts correlated significantly with serum levels of bilirubin and alkaline phosphatase (figure 1C,D), which are the two serum markers most commonly used to asses for the presence of cholestasis. Furthermore, histological evidence for cholestasis in the hepatic lobule was only seen when neutrophils were in contact with bile ducts (figure 1E). The frequency of neutrophil-bile duct contacts was not related to serum levels of ALT and AST, which are serum markers of hepatocellular injury (online supplemental figure S1B,D). The frequency of neutrophil-bile duct contacts was slightly but significantly correlated with MELD score (R2=0.25, p<0.05; online supplemental figure S1E), but was not correlated with serum C reactive protein or ferritin (online supplemental figure S1F,G). To understand the specificity of this histological finding for AH, we also investigated whether it was present in patients with NASH, PBC and PSC (online supplemental table S1). The frequency of neutrophil-bile duct contacts was very low in patients with NASH or PBC but was frequently seen in PSC (figure 1F). Moreover, in PSC the frequency of neutrophil-bile duct contacts correlated significantly with serum alkaline phosphatase (R2=0.65, p<0.02; online supplemental figure S2), similar to what was observed in AH (figure 1D).
Supplemental material
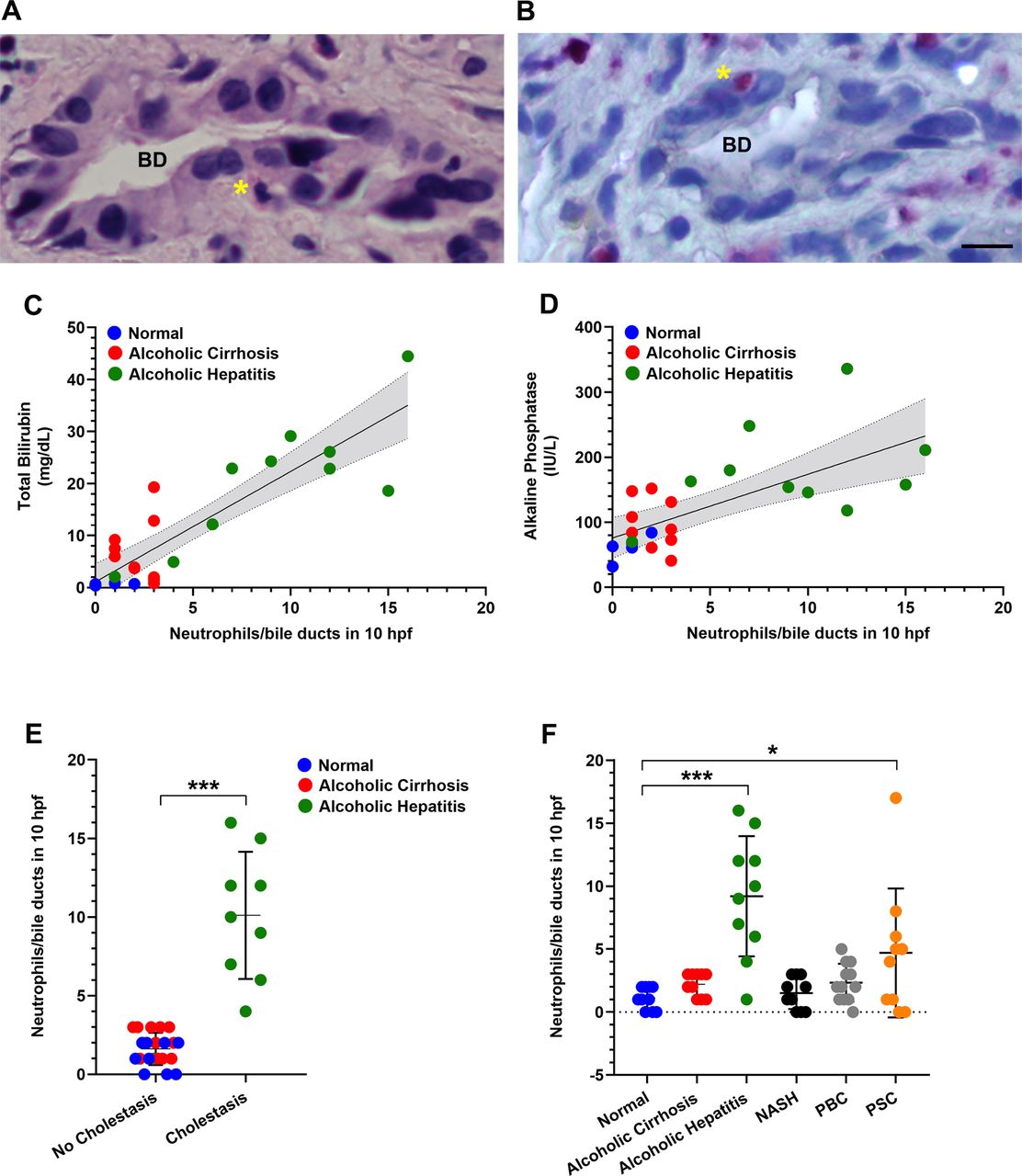
Neutrophils are in contact with bile ducts in alcoholic hepatitis and this correlates with disease severity. (A) Representative micrograph of a liver biopsy specimen from a patient with alcoholic hepatitis stained with H&E shows a neutrophil (asterisk) adjacent to a bile duct (BD). Scale bar: 20 μm. (B) Representative image of a chloroacetate esterase (CAE)-stained neutrophil (asterisk) adjacent to a bile duct in a liver biopsy from a patient with alcoholic hepatitis. Scale bar: 20 μm. (C) Degree of hyperbilirubinaemia correlates with the number of neutrophils adjacent to bile ducts. There is a positive correlation between serum total bilirubin levels and the frequency of neutrophil-bile duct contacts (R2=0.753, p<0.0001). (D) The cholestatic marker serum alkaline phosphatase correlates with the numbers of neutrophils adjacent to bile ducts (R2=0.4477, p<0.001). Data were analysed using the Pearson’s correlation coefficient. (E) Histological evidence for cholestasis in the hepatic lobule (including bile pigments in hepatocytes and canalicular bile plugs) was only observed when neutrophils were adjacent to bile ducts. Data reflect blinded observations from liver biopsies of patients with alcoholic hepatitis (n=10), abstinent patients with alcoholic cirrhosis (n=10) and histologically normal control subjects (n=10). Data represent mean±SEM, ***p<0.0001 compared with non-cholestatic biopsies. (F) Analysis of the number of neutrophils adjacent to bile ducts among patients with a spectrum of liver diseases. Neutrophils adjacent to bile ducts were rarely observed in alcoholic cirrhosis (n=10), non-alcoholic steatohepatitis (NASH; n=10) and primary biliary cholangitis (PBC; n=10) but were common in alcoholic hepatitis (n=10) and primary sclerosing cholangitis (PSC; n=10). Data represent mean±SEM, *p<0.05, ***p<0.001 compared with histologically normal liver biopsies.
Neutrophils inhibit ITPR3 expression in cholangiocytes
To determine if neutrophils affect cholangiocytes, freshly isolated human neutrophils were examined in co-culture with the cholangiocyte-derived NHC cell line.14 23–29 Loss of the subapical intracellular calcium release channel ITPR317 is part of a final common pathway in cholestatic disorders of the bile ducts,14 18 26 27 so this was used to monitor whether neutrophils exerted a cholestatic effect. Neutrophils inhibited ITPR3 expression in the NHC cell line in a concentration-dependent fashion that followed first order kinetics (figure 2A). Furthermore, neutrophils isolated from patients with clinically severe AH more potently reduced ITPR3 expression than neutrophils from normal control subjects or from abstinent patients with alcoholic cirrhosis (figure 2B). However, ITPR3 expression was not reduced in the NHC cell line treated with neutrophil-conditioned medium (figure 2C) or when the cells were separated from neutrophils by a semi-permeable membrane (figure 2D).

Neutrophils inhibit ITPR3 expression when they are co-cultured with cholangiocytes. (A) Human neutrophils decrease ITPR3 expression in the normal human cholangiocyte (NHC) cell line in a concentration-dependent fashion. Cells were co-cultured with freshly isolated human neutrophils at given concentrations for 18 hours. Following co-culture, cells were harvested and assessed for ITPR3 expression by immunoblotting. Representative blot of ITPR3 expression in the cells in co-culture with human neutrophils from healthy controls at given neutrophil concentrations (left panel) and quantification (n=5, right panel). The relationship between neutrophil concentration (x-axis) and ITPR3 expression in the NHC cell line (y-axis) is closely described by a mono-exponential decay curve (correlation coefficient R2=0.991 in this example and ranged from 0.988 to 0.992). (B) Neutrophils from alcoholic hepatitis patients are more potent than neutrophils from normal controls or from abstinent patients with alcoholic cirrhosis in inhibiting ITPR3 expression in the NHC cell line. Logarithmic scale of the neutrophil concentration versus ITPR3 expression in NHCs illustrates a steeper slope for neutrophils from alcoholic hepatitis patients, reflecting more potent inhibition (left panel). The individual value for the rate constant k for each patient from the three groups is shown (n=5–8, right panel), *p<0.05. (C) Conditioned medium (CM) from human neutrophils does not decrease ITPR3 expression in the NHC cell line. Neutrophils were stimulated either with or without lipopolysaccharides (LPS) (0.2 µg/mL) for 8 hours prior to obtaining CM that was used for the cells. After 18–24 hours of co-culture, cells were harvested and assessed for ITPR3 expression by immunoblotting. Representative blot (top panel), and quantitative analysis (bottom panel) reflect no effect of conditioned medium on ITPR3 expression in the NHC cell line (n=6). (D) Neutrophils do not inhibit ITPR3 expression in the NHC cell line when they are separated by a semipermeable membrane. Neutrophils were placed in the upper compartment of 3 μM pore Transwell system and co-cultured with NHCs cultured in the lower compartment. After 18–24 hours of co-culture, NHCs were harvested and assessed for ITPR3 expression by immunoblotting. Representative blot (top panel) and quantitative analysis (bottom panel) reflect no significant change in NHC ITPR3 expression in the Transwell co-culture system (n=4).
Mouse model of alcoholic hepatitis with cholestasis
To investigate whether cholestatic features of AH can be recapitulated in a mouse model, the NIAAA model of chronic plus binge ethanol feeding30 was modified to include a high fructose diet plus intermittent injections of LPS. These mice developed histological evidence of steatohepatitis (online supplemental figure 3A), an increase in body weight and liver weight (online supplemental figure 3B,C) and elevated serum transaminases (online supplemental figure 3D,E), as would be expected in a mouse model of AH. However, they also developed an increase in serum alkaline phosphatase (online supplemental figure 3F) and confocal immunofluorescence imaging revealed neutrophils in contact with bile ducts (online supplemental figure S4A) and loss of ITPR3 from the apical region of individual cholangiocytes within the bile ducts (online supplemental Figure S4B). Therefore, this mouse model recapitulated biliary findings in patients with AH with cholestasis.
Supplemental material
Supplemental material
Direct contact with neutrophils is required to decrease ITPR3 expression in cholangiocytes
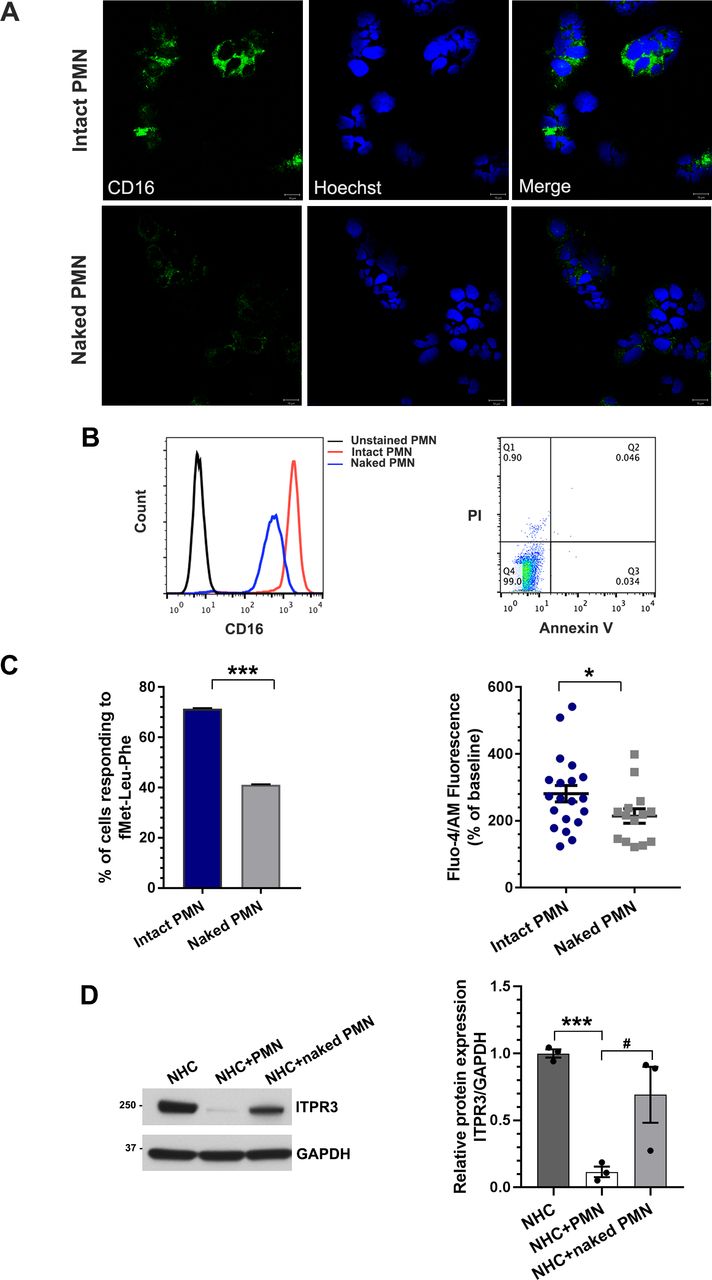
To determine the mechanism of interaction between neutrophils and cholangiocytes, three possibilities were investigated. First, we investigated whether there were direct receptor-ligand interactions between neutrophils and cholangiocytes, by generating ‘naked’ neutrophils in which CD16 surface membrane proteins were removed by PI-PLC31 32 (figure 3A). Flow cytometry analysis showed that >98% of PI-PLC-treated neutrophils were viable but there was a~50% reduction in surface expression of CD16 (figure 3B). PI-PLC treatment also significantly reduced fMet-Leu-Phe-induced cytosolic Ca2+ signals in the neutrophils (figure 3C), which is mediated by the G protein-coupled formylpeptide receptor 1 in the plasma membrane.33 34 These data collectively provide both structural (loss of CD16) and functional (loss of fMet-Leu-Phe-induced Ca2+ signals) evidence that the naked neutrophils remain viable but have lost their surface receptors. ITPR3 expression was only minimally reduced in the NHC cell line co-cultured with naked neutrophils (figure 3D), consistent with the idea that neutrophil surface receptors are necessary to reduce ITPR3 in cholangiocytes. Second, NETs can form in tissues in various inflammatory conditions as a way for neutrophils to signal to nearby parenchymal cells,35 and NET formation among hepatocytes in AH has been described.36 Neutrophils use this mechanism to bring factors such as extracellular DNA, citrullinated histone, high mobility group box 1 protein (HMGB1), neutrophil elastase and myeloperoxidase into proximity with target cells.37–39 Immunofluorescence staining illustrated the local accumulation of co-localised citrullinated histone-3 and myeloperoxidase (figure 4A). To verify NET formation, swept-field confocal microscopy was used to investigate NET formation with cholangiocytes, and time lapse measurements of the cell-impermeant DNA dye SYTOX Green demonstrated that neutrophils undergo NET formation upon contact with the NHC cell line (online supplemental figure S5A). However, addition of neutrophil elastase, HMGB1, or exogenous DNA to NHCs failed to decrease ITPR3 expression (figure 4B,C and online supplemental figure S5B). Therefore, although neutrophils are capable of forming NETs in proximity to cholangiocytes, this does not contribute to loss of ITPR3. Third, neutrophils can transiently form gap junctions with some cell types. Neutrophils express connexin 43 (CX43) hemichannels, and expression increases significantly in conditions of endotoxaemia,40 such as AH. Marginating neutrophils form functional gap junctions with endothelial cells by this mechanism,40 and cholangiocytes also express CX43.41 To determine whether neutrophils affect ITPR3 expression in cholangiocytes via gap junctions, NHCs were pretreated with either the gap junction blocker, 18α-glycyrrhetinic acid (18α-GA) or CX43 siRNA, then co-cultured with neutrophils for 18 hours. Neither 18α-GA (figure 4D) nor CX43 (figure 4E and online supplemental figure S5C) altered the neutrophil-induced downregulation of ITPR3 in the NHC cell line, suggesting that the effect of neutrophils on cholangiocytes is not mediated by gap junctions.
Supplemental material

Direct contact with neutrophils is required to decrease ITPR3 expression in cholangiocytes. Generation of ‘naked’ neutrophils. (A) Representative immunofluorescence images of untreated neutrophils (intact polymorphonuclear neutrophil (PMN)) or phosphatidylinositol-specific phospholipase C (PI-PLC)-treated neutrophils (naked PMN) stained for the neutrophil surface marker CD16 (green) and the nuclear marker Hoechst (blue). Scale bar: 10 μm. Naked neutrophils demonstrate a marked reduction in surface expression of CD16 relative to intact neutrophils. (B) Flow cytometry analysis of intact neutrophils or naked neutrophils stained with CD16 (left panel). Surface expression of CD16 was markedly reduced in naked neutrophils compared with intact neutrophils. Flow cytometry analysis of intact and naked neutrophils stained with propidium iodide (PI) and annexin V-FITC indicates that nearly all naked neutrophils remain viable (right panel). (C) Transmembrane signalling is impaired in naked neutrophils. Cells were stimulated with fMet-Leu-Phe (fMLP), which activates the G protein-coupled FPR1 receptor in the plasma membrane. Both the percentage of neutrophils that responded to fMLP (left panel), and the amplitude of the fMLP-induced Ca2+ signal in responding cells (right panel) were significantly reduced. Data represent mean±SEM (n=28 intact neutrophils and n=34 naked neutrophils from three independent experiments); *p<0.05, ***p<0.0001, relative to intact neutrophils. (D) Naked neutrophils lose their ability to inhibit ITPR3 expression in the NHC cell line. Representative immunoblot (left panel) and quantitative blot analysis (right panel) of ITPR3 expression in cells co-cultured with control or naked neutrophils for 18 hours. Data represent mean±SEM (n=4); ***p<0.0001 and #p<0.05.

Neutrophil extracellular trap (NET) formation and gap junction communication do not participate in the downregulation of ITPR3 in cholangiocytes. (A) Representative confocal images of neutrophils alone and co-cultured with the normal human cholangiocyte (NHC) cell line. Neutrophils were stained with the NET markers myeloperoxidase (MPO, red) and citrullinated histone H3 (green). Nuclei were stained with DAPI (blue). Lipopolysaccharides (LPS) and phorbol 12-myristate 13-acetate (PMA) were used as positive controls to induce NET formation. Scale bar: 10 μm. (B) The NET constituent neutrophil elastase (NE) does not inhibit ITPR3 expression in the NHC cell line. Cells were co-incubated with varying concentrations of NE for 18 hours and then assessed for ITPR3 protein expression. Representative immunoblot (top panel) and quantitative blot analysis (bottom panel) of ITPR3 expression in NHCs after treatment with NE. Data represent mean±SEM (n=3). (C) High mobility group box-1 (HMGB1), a mediator released during NET formation, does not inhibit ITPR3 expression in the NHC cell line. Representative immunoblot (top panel) and quantitative blot analysis (bottom panel) of ITPR3 expression in NHCs after treatment with HMGB1 for 18 hours. Data represent mean±SEM (n=3). (D) Treatment of the NHC cell line with the gap junction inhibitor, 18α-glycyrrhetinic acid (18α-GA), does not prevent the neutrophil-induced decrease in ITPR3 expression. (Top) Representative immunoblot and (bottom) quantitative blot analysis of ITPR3 in NHCs treated either with vehicle or 18α-GA, either alone or in co-culture for 18 hours with neutrophils. Data represent mean±SEM (n=3); *p<0.05. (E) Knockdown of CX43 in the NHC cell line does not prevent the neutrophil-induced reduction in ITPR3 expression. Representative immunoblot (left) and (right) quantitative blot analysis of ITPR3 expression in cells after CX43 knockdown and co-cultured with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=3); ***p<0.0001.
Neutrophils inhibit ITPR3 expression in cholangiocytes via integrin β1 signalling
To identify the cholangiocyte surface proteins that interact with neutrophils to mediate loss of ITPR3, we compared RNA-seq data from the NHC cell line with and without neutrophil co-culture. Gene set enrichment analysis of this transcriptome data revealed six pathways in NHCs that were significantly altered by co-culture with neutrophils (figure 5A). Based on this, candidate proteins were restricted to plasma membrane proteins within these six pathways (figure 5B). Integrin signalling was involved in two of these pathways, and ITGB1 was among the most significantly altered proteins (p<1.43×10−24) by RNA-seq as well (figure 5A,B). Expression of ITGB1, ITGB3 and ITGA5 in cholangiocytes was confirmed by real-time PCR and ITGB1 was the most abundant of these integrins (figure 5C). Confocal immunofluorescence staining confirmed that ITGB1 is expressed in the NHC cell line (figure 5D). ITGB1 protein expression was markedly induced in the NHC cell line by co-culture with neutrophils (figure 6A), and immunohistochemical staining of human liver biopsy specimens demonstrated a marked increase in ITGB1 labelling of bile ducts in patients with AH, relative to what was observed in normal controls (figure 6B). To establish a functional role for ITGB1, the NHC cell line was co-cultured with neutrophils in the presence of anti-ITGB1 neutralising antibody (ITGB1Ab), and this markedly attenuated the decrease in ITPR3 expression (figure 6C). A similar rescue effect was seen in cells transfected with ITGB1‐specific siRNA prior to co-culture with neutrophils (figure 6D). Endotoxin can decrease ITPR3 in cholangiocytes via toll-like receptor 4 (TLR4)/NFκB,14 but the effect of neutrophils on the NHC cell line was not reduced by blocking TLR4 (online supplemental figure S6A) or by knocking down NFκB (online supplemental figure S6B,C). The effect of neutrophils on ITPR3 also was not associated with apoptosis or necrosis in the NHC cell line (online supplemental figure S7).
Supplemental material
Supplemental material

RNA-seq analysis implicates integrin β1 in neutrophil-cholangiocyte interactions and integrin β1 is abundantly expressed in the normal human cholangiocyte (NHC) cell line. (A) Analysis of RNA-seq transcriptome data with ingenuity pathways analysis (IPA) software identifies the six signalling pathways that are significantly altered in the NHC cell line when co-cultured with neutrophils. Bars represent −log(p value) of significance level for each pathway; orange indicates upregulated and blue indicates downregulated pathways. (B) Volcano plot of RNA-seq analysis of NHC proteins that are affected by exposure of the cells to neutrophils. RNA was extracted from the NHC cell line alone (n=3) and co-cultured with neutrophils (n=4), and both groups were analysed by Affymetrix High-Throughput Transcriptomics Array. A total of 5818 mRNA genes were differentially expressed (grey circles), including plasma membrane proteins (orange circles) and integrin β1 (ITGB1) (blue circle). (C) RT-PCR demonstrates that ITGB1 is more heavily expressed than ITGB3 or ITGA5 in the NHC cell line (n=4). mRNA expression levels were normalised to ACTB. (D) Representative immunofluorescence images of NHCs stained for ITGB1 (green). Hoechst 33342 is used to stain the nuclei (blue). Scale bar: 10 μm.

Neutrophils inhibit ITPR3 expression in cholangiocytes via integrin β1 (ITGB1) signalling. (A) ITGB1 expression is increased in the normal human cholangiocyte (NHC) cell line when it is co-cultured with neutrophils. Representative immunoblot (left panel) and quantitative blot analysis (right panel) of ITGB1 expression in cells in the absence or presence of neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=5); **p<0.01 relative to NHCs alone. (B) Representative immunohistochemical staining of bile ducts for ITGB1 in liver biopsy specimens from patients with alcoholic hepatitis and normal controls (left panel). ITGB1-positive cells are brown. Scale bars: 20 μm. Quantitative analysis of ITGB1 staining (right panel) demonstrates that ITGB1 labelling is significantly increased in bile ducts of patients with alcoholic hepatitis (n=8) relative to what is seen in normal controls (n=9). Values are mean±SEM, ***p<0.0001. (C) An anti-ITGB1 neutralising antibody (ITGB1Ab) mitigates the inhibitory effect of neutrophils on ITPR3 expression in the NHC cell line. Representative immunoblot (top panel) and quantitative blot analysis (bottom panel) of ITPR3 expression in NHCs after blocking of ITGB1 and co-cultured for 18 hours with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=3); **p<0.01 relative to NHCs alone, #p<0.05 relative to NHC+polymorphonuclear neutrophil (PMN). (D) Knockdown of ITGB1 in the NHC cell line by small interfering RNA (siRNA) protects against neutrophil-induced down-regulation of ITPR3 expression. Cells were transfected with human ITGB1-specific siRNA for 24 hours and then co-cultured with neutrophils for an additional 18 hours. Representative immunoblot (top panel) and quantitative blot analysis (bottom panel) of ITPR3 expression in NHCs after treatment with scrambled or ITGB1 siRNA and co-culture with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=4); *p<0.05 relative to NHCs treated with scrambled siRNA and #p<0.05 relative to cells treated with scrambled siRNA+PMN.
Adhesion molecules on neutrophils interact with integrin β1 on cholangiocytes to decrease ITPR3 expression by a RAC1/JNK/c-Jun pathway
Integrins can bind to integral membrane proteins of the immunoglobulin superfamily including ICAM-1 and VCAM-1,42 so we investigated whether these particular adhesion molecules contributed to binding of neutrophils to cholangiocytes. Blockade of either VCAM-1 or ICAM-1 on the surface of neutrophils by neutralising antibodies for anti-VCAM-1 or anti-ICAM-1 partially rescued the reduced ITPR3 expression in the NHC cell line, and this effect was potentiated by simultaneous blockade of both VCAM-1 and ICAM-1 (figure 7A). Integrins link to a variety of intracellular signalling cascades,43 and our bioinformatic analysis implicated the RAC1-JNK signalling pathway in particular in cholangiocytes (figure 5A), so we examined whether neutrophil-cholangiocyte interactions trigger this signalling cascade. Co-culture with neutrophils significantly increased both RAC1 protein expression and phospho-JNK/JNK in the NHC cell line (figure 7B), consistent with their roles as intermediaries in elevating the inflammatory mediator c-Jun. Furthermore, immunohistochemistry of human liver specimens demonstrated that c-Jun expression was low in normal control bile ducts but was markedly increased in bile ducts of patients with AH (figure 7C).

Interaction of vascular cell adhesion molecule-1 (VCAM-1)/intercellular adhesion molecule-1 (ICAM-1) on neutrophils with integrin β1 (ITGB1) on cholangiocytes activates a RAC1/JNK/c-Jun cascade to suppress ITPR3 expression. (A) An anti-VCAM-1 antibody (VCAM-1Ab) and anti-ICAM-1 antibody (ICAM-1Ab) each attenuate the inhibitory effect of neutrophils on ITPR3 expression in the normal human cholangiocyte (NHC) cell line. Representative immunoblot (top panel) and quantitative blot analysis (bottom panel) of ITPR3 expression in NHCs after blocking of VCAM-1 and/or ICAM-1 on surface of neutrophils and co-cultured with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=3); ***p<0.0001 relative to the NHC cell line alone, #p<0.05, ##p<0.001 relative to NHC+polymorphonuclear neutrophil (PMN). (B) Co-culture of neutrophils and NHCs activates RAC1-induced JNK activation and decreases ITPR3 expression. Representative immunoblot (left panel) and quantitative blot analysis of RAC1 (right top panel) and JNK activation (pJNK/JNK) (right bottom panel) in the NHC cell line alone and co-cultured with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=3); *p<0.05 relative to the NHC cell line alone. (C) c-Jun expression is increased in bile ducts of patients with alcoholic hepatitis. Representative images of immunohistochemistry staining of c-Jun in human liver samples. In each panel, a bile duct is indicated by the asterisk and shown under higher magnification in the inset in the lower left corner. Images are representative of what was observed in four to six patients in each category. Original magnification, ×20. (D) c-Jun inhibits ITPR3 promoter activity. A human ITPR3 promoter construct (p-1812/+326 Luc) was co-transfected with a c-Jun expression plasmid in the NHC cell line. Data represent mean±SEM (n=5); ***p<0.0001 relative to cells transfected with empty vector, and ###p<0.0001 relative to cells co-transfected with c-Jun. (E) c-Jun mediates the neutrophil-induced downregulation of ITPR3 expression in the NHC cell line. Quantitative blot analysis of ITPR3 expression in NHCs after treatment with scrambled or c-Jun small interfering RNA (siRNA) and co-culture with neutrophils. GAPDH is used as an internal loading control. Data represent mean±SEM (n=6); **p<0.01 relative to cells treated with scrambled siRNA, #p<0.05 relative to cells treated with scrambled siRNA+PMN.
To examine the functional significance of this increase in biliary c-Jun expression, the role of c-Jun in the regulation of ITPR3 expression was investigated. A 2.1‐kb sequence of the human ITPR3 promoter region was analysed using MatInspector, and putative AP-1 binding sites where c-Jun could bind were identified (online supplemental figure S8). To determine if these AP-1 sites repress human ITPR3 transcription, reporter gene assays were performed in the NHC cell line co-transfected with a 2.1 kb human ITPR3 luciferase construct and a c-Jun expression plasmid. c-Jun co-transfection with the ITPR3 promoter led to significant downregulation of reporter gene activity (figure 7D). Furthermore, c-Jun protein expression was markedly induced when ITPR3 expression was decreased in cells co-cultured with the neutrophils (online supplemental figure S9A), and knockdown of c-Jun in NHCs prevented the neutrophil-induced reduction in ITPR3 expression (figure 7E and online supplemental figure S9B). Moreover, expression of c-Jun was significantly increased, whereas ITPR3 was significantly decreased, in livers of ethanol-fructose-LPS-treated mice (online supplemental figure S9C,D).
Supplemental material
Supplemental material
Cholangiocytes secrete CXCL8 to attract neutrophils
Interleukin-6 (IL-6), CXCL1 and CXCL8 are elevated in AH44 45 and this is associated with disease severity and mortality.46 Hepatocytes secrete CXCL1 and CXCL8,47 each of which attracts neutrophils,48 so we investigated whether these cytokines and chemokines also are involved in recruiting neutrophils to bile ducts. Neutrophils stimulated expression of IL-6, CXCL1 and CXCL8 in the NHC cell line, and neutrophils from patients with AH were more potent than neutrophils from controls, especially for CXCL8 (figure 8A–C). Treatment of the NHC cell line with LPS was similarly able to induce IL-6, CXCL1 and particularly CXCL8 expression (figure 8D–F). Pretreatment of NHCs with LPS also induced neutrophils to migrate to them, and this effect was markedly attenuated by a CXCL8 antagonist (figure 8G). CXCL8 without NHCs also was sufficient to induce neutrophil migration (figure 8G).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neutrophils are recruited to cholangiocytes by CXCL8. Human neutrophils increase the expression of (A) the pro-inflammatory cytokine interleukin-6 (IL-6), (B) the pro-inflammatory chemokines CXCL1 and (C) the pro-inflammatory chemokine CXCL8 in the normal human cholangiocyte (NHC) cell line. IL-6, CXCL1 and CXCL8 expression each is significantly increased in the NHC cell line co-cultured with neutrophils from healthy control subjects, and the increase is even greater when co-cultured with neutrophils from patients with AH. Data represent mean±SEM (n=4), *p<0.05. Lipopolysaccharide (LPS) increases the expression of (D) IL-6, (E) CXCL1 and (F) CXCL8 in the NHC cell line in a dose-dependent manner. Data represent mean±SEM (n=4) (**p<0.01; ***p<0.0001). Note that the increase in CXCL8 expression is much more than that of IL-6 or CXCL1 in response to either neutrophils or LPS. (G) CXCL8 is responsible for recruiting neutrophils to NHCs. Left: representative differential interference contrast (DIC) and CMTMR fluorescence images of neutrophil migration assay (left). Neutrophils (polymorphonuclear neutrophil (PMN)) were stained with CMTMR CellTracker (red) and co-cultured with NHCs in the presence of either LPS or CXCL8 antagonist. Recombinant CXCL8 was used as a positive control for neutrophil migration. LPS and recombinant CXCL8 each stimulate migration of neutrophils through Transwell membranes, and the CXCL8 antagonist inhibits LPS-induced neutrophil migration. Original magnification ×20. Right: quantitative assessment of neutrophil migration across a permeable Transwell chamber. Data represent mean±SEM (n=5), ***p<0.0001. (H) Proposed mechanism by which neutrophils interact with cholangiocytes to cause cholestasis in alcoholic hepatitis. Endotoxin (LPS) stimulates cholangiocytes to produce IL-6, CXCL1 and CXCL8, thus recruiting neutrophils to bile ducts. Vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) on the neutrophils interact with integrin β1 (ITGB1) on cholangiocytes, which triggers RAC1 signalling to phosphorylate JNK. Phospho-JNK (pJNK) then enters the nucleus to phosphorylate the transcription factor c-Jun/AP-1, which in turn binds to AP-1 sites on the promoter of the ITPR3 gene to suppress expression of ITPR3. Loss of ITPR3 expression results in loss of Ca2+-mediated biliary bicarbonate secretion, which contributes to cholestasis.
Bile acids do not alter the effects of neutrophils on cholangiocytes
Serum bile acids are increased in cholestasis and this may contribute to inflammation.49 To determine whether bile acids play a role in the effects of neutrophils on cholangiocytes, the NHC cell line was co-cultured with neutrophils pretreated with chenodeoxycholic acid or glycochenodeoxycholic acid, alone or in combination. Neutrophils had the same effect on ITPR3 expression in the NHC cell line regardless of pretreatment with bile acids (online supplemental figure S10).
Supplemental material
Discussion
There is an evolving appreciation that cholestasis is an important component of AH and that worsening cholestasis is associated with a worse prognosis.11–14 The current work integrates new clinical, translational and basic observations to provide mechanistic insight into this manifestation of AH. The clinical-pathological finding that neutrophils associate with bile ducts in AH and that this association relates to the presence and degree of cholestasis suggests a causal role for this interaction. The fact that a similar relationship was observed in PSC raises the unexpected idea that there may be similarities in the pathophysiology of these two seemingly different diseases. Furthermore, neutrophils reduced expression of ITPR3 in co-cultured cholangiocytes, which is important because fluid and electrolyte secretion in cholangiocytes depends on expression of this subapical intracellular calcium release channel,17 and loss of ITPR3 is part of a final common pathway in a variety of cholestatic disorders of the bile ducts.14 18 26 27 Neutrophils from patients with severe AH more potently reduced ITPR3 than neutrophils from either healthy controls or abstinent patients with alcoholic cirrhosis, suggesting that this loss of ITPR3 specifically contributes to cholestasis in AH. Studies in naked neutrophils suggested that neutrophils bind directly to cholangiocytes to reduce ITPR3 expression, and bioinformatic analysis helped identify that the binding is specifically between VCAM-1/ICAM-1 on neutrophils and ITGB1 on cholangiocytes. ITGB1 in turn inhibited ITPR3 expression through a RAC1/JNK/c-Jun pathway, which has not previously been recognised as a signalling pathway to inhibit ITPR3 expression. Although LPS can directly decrease ITPR3 expression in cholangiocytes, the current work suggests that neutrophils are attracted to cholangiocytes because LPS also induces the cholangiocytes to secrete inflammatory mediators that attract the neutrophils. The components of this cholestatic signalling pathway are summarised in figure 8H.
AH generally has been attributed to hepatocellular damage, and the newly appreciated cholestatic component of this disease might be attributable to impaired secretory function of either hepatocytes or cholangiocytes. Both hepatocellular and cholestatic diseases can elevate serum bilirubin, and all of the static or dynamic scores that are used to determine whether to treat patients with AH with steroids include serum bilirubin.50 The current work suggests the novel idea that the clinically severe form of AH is due at least in part to loss of ITPR3 from the cholangiocyte, which is a cell type that previously was not thought to play a major role in this disease. This interpretation is reinforced by our observation that a mouse model of AH was able to recapitulate key aspects of this cholestatic mechanism, including elevated alkaline phosphatase, neutrophils in proximity to bile ducts and increased c-Jun along with loss of ITPR3 in the cholangiocytes.
The molecular mechanism for cholestasis that is proposed to occur in this clinical condition is new as well. Oxidative and LPS-induced damage both are known to occur in AH, but the principal target is thought to be hepatocytes.51 However, cholangiocytes also are involved in the cholestasis that occurs in AH,13 as in other cholangiopathies,18 and in each case this is due to loss of ITPR3.14 Several mechanisms regulate ITPR3 expression in cholangiocytes,14 26 27 but none of these mechanisms appear to be responsible for the neutrophil-induced loss of ITPR3 that occurs in AH.14 Similarly, a role for neutrophils in the pathogenesis of AH has always been presumed, and in fact their presence in the hepatic lobule is part of the criteria for histological diagnosis.12 However, to our knowledge, it has not previously been considered that neutrophils interact with cholangiocytes in this disease (except for with reactive bile ducts),52 nor that their interaction must include direct contact. By providing evidence that neutrophils bind to cholangiocytes via ITGB1 in AH and that the degree of interaction reflects the degree of jaundice (figures 1 and 6), the current work provides unexpected new cell and molecular targets for therapy. A role for blocking integrins has established therapeutic benefit in several other diseases mediated by leucocyte-tissue interactions, most notably in multiple sclerosis and IBD.53 It remains to be established whether a similar strategy can be exploited to treat AH.
There are several limitations to the current study. First, although the clinical-histological correlations we observed (figure 1) are highly statistically significant, the sample sizes are small and so these observations would benefit from validation from additional patient cohorts. Second, although we observed that neutrophils from patients with clinically severe AH more potently affect cholangiocytes than neutrophils from control groups (figure 2), we did not distinguish between patients with AH with and without cholestasis. Third, although the current work implicates cholangiocytes in the cholestasis that occurs in AH, the relative contributions to cholestasis of hepatocytes and cholangiocytes cannot be determined from our study. Finally, although the current work implicates a novel neutrophil-cholangiocyte signalling pathway in the development of cholestasis in AH, it remains to be proven that disruption of this pathway would ameliorate the cholestasis, and if so, that this would improve survival. However, the mouse model introduced here may provide a next step in addressing these important clinical questions.
Supplemental material
Acknowledgments
The authors would like to thank Albert Mennone for assistance with microscopy, and Scott Roberts for advice and assistance with neutrophil isolation and FACS analysis. The authors would also like to thank the Thailand Research Fund and Mahidol University for the support (RSA6080072).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MHN and JW are joint senior authors.
Twitter @MichaelNathanso
Contributors MT performed the majority of experiments, analysed the data and wrote the first draft of the manuscript. PTV participated in histological analysis of biopsy specimens. MTG helped with imaging of NETs and super-resolution imaging. MH contributed to clinical data collection for each patient group. MER provided access to relevant human liver biopsy specimens and contributed to clinical data of all patients in which liver biopsies were obtained. MO-M contributed to the clinical study of alcoholic hepatitis and cirrhosis. SA assisted with RNA-seq analysis. TK assisted with cell culture studies. RK assisted with neutrophil calcium signalling. EK assisted with NFκB silencing and TLR4 inhibitors. RRR obtained clinical specimens, including all blood samples. JMB provided assistance related to the NHC cell line. MHN conceived of the idea, designed the research project, supervised the study and edited the manuscript. JW designed the research, planned and conducted experiments, analysed the data, supervised the study, generated figures and edited the manuscript.
Funding This work was supported by the Gladys Phillips Crofoot Professorship and grants from the National Institutes of Health (P01-DK57751, P30-DK34989, R01-DK114041, R01-DK112797 and S10-OD023598).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was conducted under the auspices of protocols approved by the Institutional Review Board on the Protection of the Rights of Human Subjects (Yale University). The Human Investigation Committee protocol numbers are HIC-2000025846 and HIC-1304011763. Written informed consent was obtained from all participants. The study protocol conformed to the ethical guidelines of the Helsinki Declaration. Animal studies were approved by Yale University’s Institutional Animal Care and Use Committee (IACUC).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The RNA-seq data have been deposited in the Gene Expression Omnibus under the accession number GSE146899.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.