Article Text

Abstract
Objective Although COVID-19 is primarily a respiratory illness, there is mounting evidence suggesting that the GI tract is involved in this disease. We investigated whether the gut microbiome is linked to disease severity in patients with COVID-19, and whether perturbations in microbiome composition, if any, resolve with clearance of the SARS-CoV-2 virus.
Methods In this two-hospital cohort study, we obtained blood, stool and patient records from 100 patients with laboratory-confirmed SARS-CoV-2 infection. Serial stool samples were collected from 27 of the 100 patients up to 30 days after clearance of SARS-CoV-2. Gut microbiome compositions were characterised by shotgun sequencing total DNA extracted from stools. Concentrations of inflammatory cytokines and blood markers were measured from plasma.
Results Gut microbiome composition was significantly altered in patients with COVID-19 compared with non-COVID-19 individuals irrespective of whether patients had received medication (p<0.01). Several gut commensals with known immunomodulatory potential such as Faecalibacterium prausnitzii, Eubacterium rectale and bifidobacteria were underrepresented in patients and remained low in samples collected up to 30 days after disease resolution. Moreover, this perturbed composition exhibited stratification with disease severity concordant with elevated concentrations of inflammatory cytokines and blood markers such as C reactive protein, lactate dehydrogenase, aspartate aminotransferase and gamma-glutamyl transferase.
Conclusion Associations between gut microbiota composition, levels of cytokines and inflammatory markers in patients with COVID-19 suggest that the gut microbiome is involved in the magnitude of COVID-19 severity possibly via modulating host immune responses. Furthermore, the gut microbiota dysbiosis after disease resolution could contribute to persistent symptoms, highlighting a need to understand how gut microorganisms are involved in inflammation and COVID-19.
- colonic microflora
- inflammation
- colonic bacteria
Data availability statement
Data are available in a public, open access repository. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA650244. Raw sequence data are available in the Sequence Read Archive (SRA) under BioProject accession PRJNA650244.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
SARS-CoV-2 primarily infects the respiratory tract, however, pathophysiology of COVID-19 can be attributed to aberrant immune responses in clearing the virus.
Several lines of evidence such as replication of SARS-CoV-2 in human enterocytes, detection of viruses in faecal samples and the altered gut microbiota composition in patients with COVID-19 suggest involvement of the GI tract.
COVID-19 gut microbiota surveys are limited and have not examined links between gut microbiome and disease pathophysiology.
What are the new findings?
Composition of the gut microbiota in patients with COVID-19 is concordant with disease severity and magnitude of plasma concentrations of several inflammatory cytokines, chemokines and blood markers of tissue damage.
Patients with COVID-19 were depleted in gut bacteria with known immunomodulatory potential, such as Faecalibacterium prausnitzii, Eubacterium rectale and several bifidobacterial species.
The dysbiotic gut microbiota composition in patients with COVID-19 persists after clearance of the virus.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
These findings suggest that depletion of immunomodulatory gut microorganisms contributes to severe COVID-19 disease.
The dysbiotic gut microbiota that persists after disease resolution could be a factor in developing persistent symptoms and/or multisystem inflammation syndromes that occur in some patients after clearing the virus.
Bolstering of beneficial gut species depleted in COVID-19 could serve as a novel avenue to mitigate severe disease, underscoring importance of managing patients’ gut microbiota during and after COVID-19.
Introduction
Infection by the SARS-CoV-2 induces an immune response to eliminate the virus, but there is mounting evidence suggesting that aberrant responses are responsible for severe outcomes and possibly other inflammatory conditions beyond COVID-19. Patients with severe disease exhibit high blood plasma levels of inflammatory cytokines and inflammatory markers such as IL 6, 8 and 10 as well as C reactive protein (CRP) and lactate dehydrogenase (LDH) reflecting immune response and tissue damage from SARS-CoV-2 infection.1–3 Additionally, some patients develop autoinflammatory symptoms after recovery, most prominent being multisystem inflammatory syndrome and Kawasaki-like disease in children.4–6 Several observations suggest a substantial involvement of the GI tract, such as the ability of SARS-CoV-2 to infect and replicate in human small intestine enterocytes,7 consistent detection of virus RNA in faecal samples8 9 and the altered gut microbiota composition in SARS-CoV-2 infected subjects.10 11 As the GI tract is the largest immunological organ in the body and its resident microbiota are known to modulate host immune responses,12 we hypothesised that the gut microbiota is associated with host inflammatory immune responses in COVID-19. Here, we characterised the gut microbiota and immune response in 100 patients with COVID-19 during hospitalisation and up to 30 days after recovery, showing that gut microbiota composition during hospitalisation is associated with disease severity and plasma concentrations of several cytokines and inflammatory markers. Moreover, gut microbiota composition in recovered patients remained significantly altered compared with non-COVID-19 individuals, which could have important implications in future health problems beyond COVID-19.
Materials and methods
Subject recruitment and sample collection
The study was conducted in accordance with the declaration of Helsinki. All patients provided written informed consent. As described in our previous study,10 patients with COVID-19 were recruited from the Prince of Wales and United Christian Hospitals in Hong Kong between February and May 2020. These patients were laboratory-confirmed SARS-CoV-2 positive by quantitative reverse transcription PCR (RT-qPCR) performed on nasopharyngeal swabs collected by hospital staff.13 Non-COVID-19 subjects were recruited before COVID-19 as part of a Hong Kong population gut microbiome survey recruited by advertisement14 or non-disease controls from colonoscopy trials,15 whereby subjects had a normal colonoscopy (stools collected several days before bowel preparation). Patients with COVID-19 were classified into four severity groups based on symptoms as reported by Wu et al.16 Briefly, patients were classified as mild if there were no radiographic indications of pneumonia, moderate if pneumonia with fever and respiratory tract symptoms were detected, severe if respiratory rate ≥30 breaths per minute, oxygen saturation ≤93% when breathing ambient air or PaO2/FiO2 ≤300 mm Hg, critical if respiratory failure requiring mechanical ventilation or organ failure requiring intensive care. Blood and stools from in-hospital patients were collected by hospital staff while discharged patients provided stools on day of follow-up or self-sampled at home. Stools were collected in collection tubes containing preservative media (cat. 63700, Norgen Biotek Corp, Ontario Canada) and stored at −80°C until processing. We have previously shown that gut microbiota composition generated from stools collected in this preservative medium was comparable with results obtained from immediate freezing at −80°C.17
Stool DNA extraction and sequencing
Detailed methods are described in Zuo et al.10 Briefly, DNA was extracted from 0.1 g of homogenised faecal samples using the Maxwell RSC PureFood GMO and Authentication Kit and a Maxwell RSC Instrument nucleic acid extraction platform (Promega, Wisconsin, USA) according to manufacturer’s instructions. Sequencing libraries were prepared from extracted DNA using the Nextera DNA Flex Library Prep Kit (Illumina, California, USA) and sequenced on an Illumina NovaSeq 6000 System (2×150 bp) at the Centre for Gut Microbiota Research, Chinese University of Hong Kong. Raw sequence data generated for this study are available in the Sequence Read Archive under BioProject accession PRJNA650244.
Sequence data processing, inferring gut microbiota composition and statistical analysis
Raw sequence data were quality filtered using Trimmomatic V.39 to remove adaptor and low-quality sequences. Following this, microbiota composition profiles were inferred from quality-filtered forward reads using MetaPhlAn218 V.2.7.7 with the V.20 database. The site by species counts and relative abundance tables were input into R19 V.3.5.1 for statistical analysis. Principal component analysis (PCA) ordinations were used to visualise the clustering of samples based on their species level compositional profiles. Associations between gut community composition and patients’ parameters were assessed using permutational multivariate analysis of variance (PERMANOVA) and Procrustes analyses. Associations of specific microbial species with patient parameters were identified using the linear discriminant analysis effect size (LEfSe) and the multivariate analysis by linear models (MaAsLin) statistical frameworks implemented in the Huttenhower Lab Galaxy instance (http://huttenhower.sph.harvard.edu/galaxy/). PCA, PERMANOVA and Procrustes analysis are implemented in the vegan R package20 V.2.4–6.
Measuring SARS-CoV-2 load in stool samples
SARS-CoV-2 virus loads were measured via RT-qPCR as described in Zuo et al.10 RNA was extracted from 0.1 g homogenised stools using the QIAamp Viral RNA Mini Kit (QIAGEN, Hilden Germany) following manufacturer’s instructions. SARS-CoV-2 primer and probe sequences were as provided by the US Centers for Disease Control and Prevention (2019-nCoV_N1-F: 5'-GACCCCAAAATCAGC GAAAT-3', 2019-nCoV_N1-R: 5'-TCTGGTTACTGCCAGTTGAATCTG-3' and 2019-nCoV_N1-P: 5'-FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1-3'). Each one-step RT-qPCR reaction contained 10 µL of extracted RNA, 4 µL TaqMan Fast Virus 1-Step Master Mix (Thermo Fisher Scientific, Massachusetts, USA) in a final reaction volume of 20 µL. Primer and probe concentrations were 0.5 µM and 0.125 µM, respectively. Cycling conditions were 25°C for 2 min, 50°C for 15 min, 95°C for 2 min, followed by 45 cycles of 95°C for 15 s and 55°C for 30 s. Thermocycling was performed on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). Cycle threshold (Ct) values were converted into viral RNA copies based on a standard curve prepared from 10-fold serial dilutions of known copies of plasmids containing the full N gene (2019-nCoV_N_Positive Control, Integrated DNA Technologies, USA). Samples were considered negative if Ct values exceeded 39.9 cycles. The detection limit was 347 copies/mL.
Plasma cytokine measurements
Whole blood samples collected in anticoagulant-treated tubes were centrifuged at 2000 × g for 10 min and the supernatant was collected. Concentrations of cytokines and chemokines were measured using the MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel—Immunology Multiplex Assay (Merck Millipore, Massachusetts, USA) on a Bio-Plex 200 System (Bio-Rad Laboratories, California, USA). Concentration of N-terminal-pro-brain natriuretic peptide (NT-proBNP) was measured using Human NT-proBNP ELISA kits (Abcam, Cambridge, UK).
Patient and public involvement
Patients or the public were not involved in the design, conduct, reporting or dissemination plans of this study.
Results
COVID-19 patient cohort
Between February and May 2020, we collected blood and stool samples from 100 patients with COVID-19 confirmed by positive SARS-CoV-2 RT-qPCR. Demographic and clinical characteristics of these patients and a non-COVID-19 control cohort of 78 adults recruited in Hong Kong before COVID-19 are reported in table 1. There were 47 female and 53 male patients with COVID-19 with mean±SD age of 36.4±18.7 years. Critical, severe, moderate and mild disease16 were observed in 3.0%, 5.0%, 45.0% and 47.0% of patients, respectively. In comparison, the non-COVID-19 cohort was comprised of 45 women and 33 men with mean±SD age of 45.5±13.3 years. Comorbidities in the COVID-19 cohort included hypertension, hyperlipidaemia, diabetes and heart conditions, although there were fewer than five patients each except for hypertension with 11. For the non-COVID-19 cohort, hypertension was the only major comorbidity with 11 individuals. Of the 100 patients with COVID-19, 41 provided multiple stool samples over the duration of their hospital stay and/or follow-up after discharge; 34 and 46 patients received antibiotics and antivirals, respectively, prior to stool collection.
Characteristics of the COVID-19 and non-COVID-19 cohorts
Altered gut microbiota composition in patients with COVID-19
In total, 274 stool samples were sequenced generating an average of 6.8 Gbp (47 386 950 reads) per sample. First, we compared gut microbiota compositions of the first stool samples of each patient with COVID-19 collected during hospitalisation (n=87; 13 of the 100 patients with COVID-19 only provided stools after recovery) (40 women to 47 men, 35.6±18.8 years old (mean±SD)) with non-COVID-19 subjects (45 women to 33 men, 45.5±17.4 years old (mean±SD)) to assess whether gut microbiota composition was altered in this COVID-19 cohort. At the phylum level, members of the Bacteroidetes were more relatively abundant in patients with COVID-19 compared with non-COVID-19 individuals (mean 23.9% vs 12.8%, p<0.001, Mann-Whitney test), whereas Actinobacteria were more relatively abundant in non-COVID-19 individuals (26.1% vs 19.0%, p<0.05, Mann-Whitney test) (figure 1A). At the species level, we identified significant associations with disease (COVID-19 vs non-COVID-19) and antibiotics (figure 1B) (p<0.05, PERMANOVA) but not stool SARS-CoV-2 load, antiviral (lopinavir/ritonavir, ribavirin or oseltamivir in 39 of 87 patients), corticosteroids and proton pump inhibitor use (online supplemental table S1). Without controlling for use of antibiotics, compositional differences in the gut microbiota of COVID-19 were primarily driven by enrichment of species including Ruminococcus gnavus, Ruminococcus torques and Bacteroides dorei and depletion of Bifidobacterium adolescentis, Faecalibacterium prausnitzii and Eubacterium rectale (p<0.05, LEfSe) (table 2, online supplemental table S2). When antibiotic effects were examined, differences between cohorts were primarily linked to enrichment of taxa such as Parabacteroides, Sutterella wadsworthensis and Bacteroides caccae and depletion of Adlercreutzia equolifaciens, Dorea formicigenerans and Clostridium leptum in COVID-19 relative to non-COVID-19 (p<0.05, MaAsLin) (online supplemental table S3), although most of the implicated taxa comprised less than 0.1% average relative abundance in these samples. While the overall gut microbiota composition was distinct between the 87 COVID-19 and 78 non-COVID-19 subjects, there were no significant differences in species richness and Shannon diversity (p>0.05, Mann Whitney test) (online supplemental figure S1). Within the COVID-19 cohort, gut microbiota composition sampled during hospitalisation (n=87) was most significantly associated with disease severity (mild, moderate, severe, critical) followed by antibiotics with descending size of effect (p<0.05, PERMANOVA) (online supplemental table S1). In identifying microbial species associated with disease severity, we found that F. prausnitzii and Bifidobacterium bifidum were negatively correlated with severity after adjusting for antibiotic use and patients’ age (p<0.05, ordinal regression). Relative abundances of several other microbial species typically abundant in the human gut including B. adolescentis and E. rectale also showed reductions with increasing disease severity although these were not statistically significant (online supplemental table S4).
Supplemental material
Supplemental material

Compositional differences in gut microbiota between patients with COVID-19 and non-COVID-19 subjects. (A) Average relative abundances of microbial phyla detected in stools from in-hospital patients with COVID-19, patients discharged after negative RT-qPCR for viral RNA in nasopharyngeal swabs, and non-COVID-19 individuals. (B) Principal component analysis of gut microbiota composition of patients with COVID-19 with and without antibiotics compared with non-COVID-19 subjects. Filled circles represent the first stool samples (if serial samples are available) of in-hospital patients whereas crosses represent non-COVID-19 subjects. Group centroids are indicated by the group labels.
Bacteria species associated with COVID-19 patients during hospitalisation
Plasma concentrations of inflammatory cytokines, chemokines and tissue damage markers are correlated with gut microbiota composition
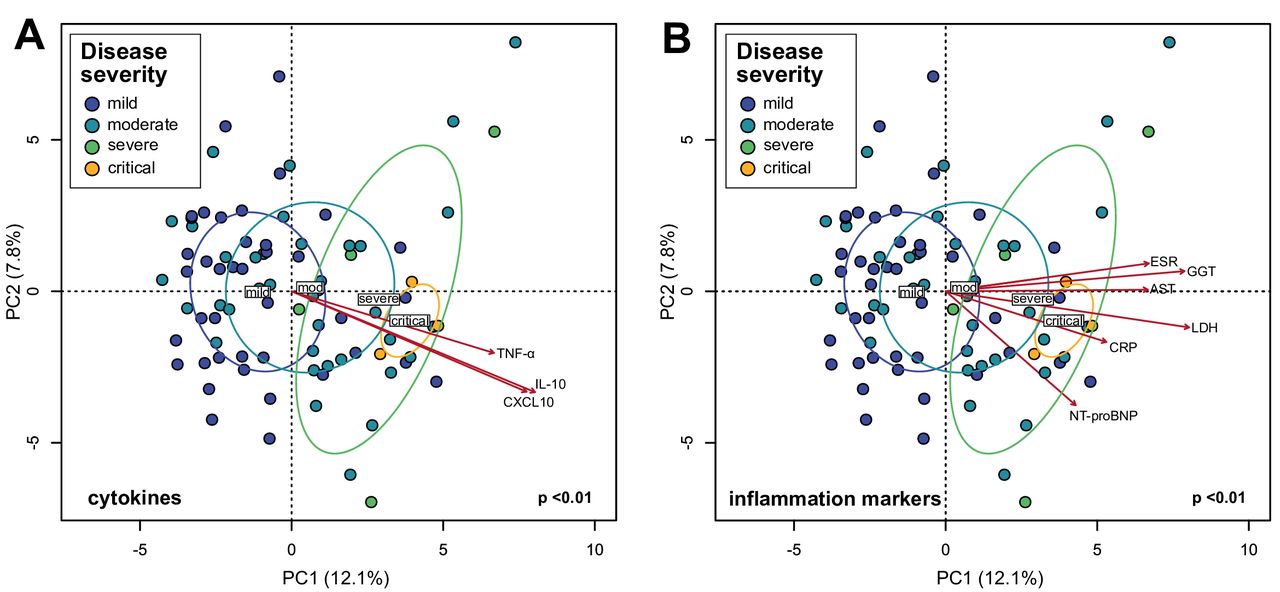
In COVID-19 infection, the immune system produces inflammatory cytokines in response to virus infection. In some cases, the inflammatory response can be overaggressive (ie, ‘cytokine storm’) and results in widespread tissue damage, septic shock and multi organ failure.1 Based on the observation that the gut microbiota is altered in patients with COVID-19 (figure 1) and the associations of several species with disease severity (online supplemental table S4), we hypothesised that these compositional changes play a role in exacerbating disease by contributing to dysregulation of the immune response. PCA visualisation of gut microbiota composition in the cohort of 87 patients with COVID-19 during hospitalisation revealed a continuum along the mild, moderate, severe and critical disease severity groups (figure 2), indicating a stratification of gut microbiota composition associated with disease severity. We then fitted plasma concentrations of cytokines (measured at admission; median 2 days before stool samples) and inflammation markers onto the PCA and observed that C-X-C motif ligand 10 (CXCL10), IL-10, tumour necrosis factor-α (TNF-α), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), CRP, LDH, NT-proBNP and erythrocyte sedimentation rate were significantly associated with microbiota composition (figure 2) (p<0.05, Procrustes analysis). Notably, these measurements increased concomitant with microbiota composition representing more severe disease states. Since CXCL10, IL-10, TNF-α, AST, GGT, CRP, LDH and NT-proBNP are typically elevated in more severe COVID-19,2 21–23 these results suggest that gut microbiota composition is associated with the magnitude of immune response to COVID-19 and subsequent tissue damage and thus could play a role in regulating disease severity. We then assessed which specific species enriched or depleted in patients with COVID-19 correlated with cytokine concentrations. From the list of most relatively abundant species in table 2, six species depleted in the COVID-19 cohort negatively correlated with CXCL10, five with IL-10 and two each with TNF-α and C-C motif ligand 2 (CCL2) (figure 3A–D) (p<0.05, Spearman correlation). These included B. adolescentis, E. rectale and F. prausnitzii known to play immunomodulatory roles in the human GI system.24–26 Conversely, two species enriched in the COVID-19 cohort B. dorei and Akkermansia muciniphila were positively correlated with IL-1β, IL-6 and C-X-C motif ligand 8 (CXCL8) (figure 3E–G). Correlations with other relatively less abundant gut bacteria are shown in online supplemental table S5).

Associations between gut microbiota composition of in-hospital patients with COVID-19 and plasma concentrations of inflammatory cytokines and blood inflammation markers. (A) Principal component analysis (PCA) of gut microbiota composition and association with plasma concentrations of cytokines/chemokines. (B) PCA of gut microbiota composition and association with blood inflammation markers. Statistical correlations were determined using Procrustes tests. Only cytokines and inflammation markers significantly correlated with gut microbiota composition are shown. Red arrows represent gradients of the corresponding cytokines/inflammation marker concentrations and point to the direction of greatest increase in these measures. Colour of the circles represents disease severity groups, and ellipses represent SD of the group centroid. Group centroids are indicated by the group labels. AST, aspartate aminotransferase; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; GGT, gamma-glutamyl transferase; LDH, lactate dehydrogenase; NT-proBNP, N-terminal-pro-brain natriuretic peptide; TNF, tumour necrosis factor.

Correlations between COVID-19 enriched/depleted gut microbial taxa and plasma concentrations of (A) CXCL10, (B) IL-10, (C) TNF-α, (D) CCL2, (E) CXCL8, (F) IL-1β and (G) IL-6. Only statistically significant correlations are shown. Linear regression lines are shown in each scatter plot in blue, and shaded regions represent 95% CIs. CCL, C-C motif ligand; CXCL, C-X-C motif ligand; TNF, tumour necrosis factor.
Gut microbiota dysbiosis persists after clearance of SARS-CoV-2
As some patients with COVID-19 report persistent symptoms after recovery and/or subsequently develop multisystem inflammation,27 we hypothesised that the dysbiotic gut microbiota seen in patients with COVID-19 persists after recovery and could contribute to these conditions. To assess gut microbiota composition following recovery from COVID-19, 42 stool samples were collected from 27 patients (13 women to 14 men, 45.6±17.6 years old (mean±SD)) up to 30 days (median 6 days, IQR 14 days) after their nasopharyngeal aspirates or swabs tested negative for SARS-CoV-2 via RT-qPCR. Compared with non-COVID-19 subjects, gut microbiota composition of the 27 recovered patients remained significantly distinct irrespective of whether they had received antibiotics (p<0.05, PERMANOVA) (14 received antibiotics, 13 did not), although composition in patients who had received antibiotics were more dissimilar relative to non-COVID-19 subjects than patients who did not (figure 4). Gut microbiota of recovered patients were enriched in species including Bifidobacterium dentium and Lactobacillus ruminis irrespective of whether they had received antibiotics and depleted in E. rectale, R. bromii, F. prausnitzii and Bifidobacterium longum (p<0.05, LEfSe) (table 3; online supplemental table S6). To determine whether antibiotics were associated with improved disease outcomes in patients with COVID-19, we examined its use in the moderate disease cohort in which the number of patients who had/had not received antibiotics was comparable (21 of 45 moderate disease patients received antibiotics). Representation in other disease states was less balanced (table 1). We found no difference in the number of days from onset of COVID-19 symptoms until discharge from hospital with or without antibiotics (p>0.05, Mann-Whitney test). Additionally, since there were no records of bacteraemia or blood cultures in the 45 patients and all but one had procalcitonin <0.2 ng/mL during admission, these findings indicated that antibiotics are unlikely to be associated with improved patient outcomes assuming no bacterial coinfections but in contrast could exacerbate and prolong gut microbiota dysbiosis in patients with COVID-19.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Principal component analysis of gut microbiota composition in recovered patients with COVID-19 who had or had not received antibiotics compared with non-COVID-19 subjects. Patients were considered recovered following negative quantitative reverse transcription PCR (RT-qPCR) tests for SARS-CoV-2 RNA in nasopharyngeal swabs. Filled circles represent all stools collected after discharge from hospital whereas crosses represent non-COVID-19 subjects.
Bacterial species associated with patients with COVID-19 after recovery
Discussion
The pathophysiology of SARS-CoV-2 infection is characterised by aggressive inflammatory responses that are strongly implicated in the causation of multiorgan dysfunction seen in some patients, and thus disease severity is likely due to not only viral infection but also host immune responses.1 21 28–30 Here we show that gut microbiota composition of patients with COVID-19 during hospitalisation is correlated with plasma concentrations of several cytokines, chemokines and inflammation markers, suggesting that the gut microbiota could play a role in modulating host immune response and potentially influence disease severity and outcomes. Specifically, the depletion of several bacterial species in the COVID-19 cohort was linked to increased concentrations of TNF-α, CXCL10, CCL2 and IL-10 consistent with immunological studies of patients with COVID-19,2 indicating that these depleted taxa may have a role in preventing overaggressive inflammation. In support of this inference, depleted gut commensals such as B. adolescentis, F. prausnitzii, E. rectale, R. (Blautia) obeum and D. formicigenerans have been separately linked to reduced host inflammatory response in other inflammatory-related diseases.24–26 For example, F. prausnitzii has been shown to induce priming of human colonic regulatory T cells that secrete the anti-inflammatory cytokine IL-10,31 high relative abundances of E. rectale in the gut are linked to reduced inflammation in Alzheimer’s disease,32 and B. adolescentis is able to suppress activation of nuclear factor κB that promotes expression of proinflammatory cytokines.33 In addition, the enrichment of Ruminococcus gnavus, Ruminococcus torques, Bacteroides dorei and Bacteroides vulgatus in COVID-19 is also consistent with the inference of a microbial-mediated immune dysregulation. R. gnavus and R. torques in the gut have been reported to co-occur with inflammatory bowel disease,34 35 and B. dorei and B. vulgatus are implicated in several inflammatory gut diseases such as irritable bowel disease and ulcerative colitis.36 However, it remains unknown as to whether inflammatory-associated gut microorganisms enriched in COVID-19 indeed play an active part in disease or simply flourish opportunistically due to a depletion of other gut microorganisms.
The potential role played by gut microorganisms in COVID-19 could allow the use of a microbiome-based risk profile to identify individuals at risk of severe disease or downstream inflammatory symptoms such as multisystem inflammation and Kawasaki-like disease in children.4 6 37 Based on several patients surveyed in this study for up to 30 days after clearing SARS-CoV-2, the gut microbiota is likely to remain significantly altered after recovery from COVID-19. In light of reports that a subset of recovered patients with COVID-19 experience persistent symptoms such as fatigue, dyspnoea and joint pains, some over 80 days after initial onset of symptoms,27 38 39 we posit that the dysbiotic gut microbiome could contribute to immune-related health problems post-COVID-19. Our short follow-up period does not permit extrapolation of gut microbiota composition to long-term persistent symptoms. As such, longer follow-up of patients with COVID-19 (eg, 3 months to 1 year after clearing the virus) is needed to address questions related to the duration of gut microbiota dysbiosis postrecovery, link between microbiota dysbiosis and long-term persistent symptoms and whether the dysbiosis or enrichment/depletion of specific gut microorganisms predisposes recovered individuals to future health problems.
Our study has several shortcomings, including heterogeneous patient clinical management which could confound microbial signatures associated with COVID-19. A homogenous case–control cohort with adequate representation in each disease severity group and minimal variability across treatments is needed for a properly controlled study, although these ideals may not be possible in the real world. As such, it is unclear to what extent the gut microbiota composition as an outcome of COVID-19 is influenced by clinical management. Moreover, the observed gut microbiota composition could simply be a response to patients’ health and immune states rather than a direct involvement in disease severity, as such it may not be directly applicable to predicting disease susceptibility in non-COVID-19 subjects. Caution is also needed in interpreting patient outcomes associated with antibiotic use. It was estimated that half to three quarters of patients with COVID-19 received antibiotics empirically despite less than 7% presenting bacterial infections.40 41 We found no difference in outcomes with or without antibiotics that supports calls to curb unnecessary antibiotics in managing patients with COVID-19, but our comparison was limited to patients with moderate disease as patient numbers in this subcohort were more comparable. While our finding suggests that antibiotics does not improve patient outcomes, it is still possible that a higher prevalence of antibiotic administration in severe and critical patients could worsen inflammation.42 Finally, gut microbiota composition is highly heterogeneous across human populations and changes in compositions reported here may not necessarily be reflected in patients with COVID-19 from other biogeographies. Nevertheless, this survey of gut microbiota alterations in association with immune dysregulation revealed that gut microorganisms are likely involved in the modulation of host inflammatory responses in COVID-19. With mounting evidence that gut microorganisms are linked with inflammatory diseases within and beyond the gut,12 43 these findings underscore an urgent need to understand the specific roles of gut microorganisms in human immune function and systemic inflammation.
Data availability statement
Data are available in a public, open access repository. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA650244. Raw sequence data are available in the Sequence Read Archive (SRA) under BioProject accession PRJNA650244.
Ethics statements
Ethics approval
This study has been approved by the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (reference number 2020.076). Written informed consent was obtained from all participants prior to collecting stool samples.
Acknowledgments
We would like to thank all healthcare workers working in isolation wards of the Prince of Wales and United Christian Hospitals, Hong Kong SAR, China. We thank Miu Ling Chin, Apple CM Yeung, Wendy CS Ho, Rity Wong, Vickie Li, Ida MT Chu and other staff/students for their technical contribution in this study including sample collection, inventory and processing, and Hui Zhan, Yating Wan, Nan Chen for assistance with DNA extraction.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
PKC and SCN are joint senior authors.
Twitter @Tao_Zuo_, @Siew_C_Ng
YKY, TZ and CKW contributed equally.
Contributors SCN, GC-YL, ET, KSCF, VC, LL recruited study subjects. AYLL, RWYN, TCFY, GL-HW procured and collated patients’ clinical data. PC organised sample inventory and processing. TZ, QL, FZ, AC, CPC performed laboratory work including extracting DNA and generating sequence data. CKW performed cytokine and chemokine measurements. YKY and SCN analyzed and interpreted the data, and wrote the manuscript. GJ, DS-CH and other authors reviewed the manuscript. SCN, PC and FKLC designed and supervised the study.
Funding This study was supported by the Health and Medical Research Fund, the Food and Health Bureau, The Government of the Hong Kong Special Administrative Region (COVID190111), and donations from Hui Hoy & Chow Sin Lan Charity Fund Limited, Pine and Crane Company Limited, Mr. Hui Ming, and The D.H. Chen Foundation.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.