Article Text

Abstract
Objective Microbial exposure is critical to neonatal and infant development, growth and immunity. However, whether a microbiome is present in the fetal gut prior to birth remains debated. In this study, lambs delivered by aseptic hysterectomy at full term were used as an animal model to investigate the presence of a microbiome in the prenatal gut using a multiomics approach.
Design Lambs were euthanised immediately after aseptic caesarean section and their cecal content and umbilical cord blood samples were aseptically acquired. Cecal content samples were assessed using metagenomic and metatranscriptomic sequencing to characterise any existing microbiome. Both sample types were analysed using metabolomics in order to detect microbial metabolites.
Results We detected a low-diversity and low-biomass microbiome in the prenatal fetal gut, which was mainly composed of bacteria belonging to the phyla Proteobacteria, Actinobacteria and Firmicutes. Escherichia coli was the most abundant species in the prenatal fetal gut. We also detected multiple microbial metabolites including short chain fatty acids, deoxynojirimycin, mitomycin and tobramycin, further indicating the presence of metabolically active microbiota. Additionally, bacteriophage phiX174 and Orf virus, as well as antibiotic resistance genes, were detected in the fetal gut, suggesting that bacteriophage, viruses and bacteria carrying antibiotic resistance genes can be transmitted from the mother to the fetus during the gestation period.
Conclusions This study provides strong evidence that the prenatal gut harbours a microbiome and that microbial colonisation of the fetal gut commences in utero.
- intestinal microbiology
Data availability statement
Metagenomic and metatranscriptomic sequencing data for all samples have been deposited in the NCBI Sequence Read Archive database under accession numbers PRJNA601636 (https://www.ncbi.nlm.nih.gov/sra/PRJNA601636) and PRJNA598075 (https://www.ncbi.nlm.nih.gov/sra/PRJNA598075).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
It has long been assumed that the uterus is sterile and that establishment of the fetal microbiota commences at birth. Although microbes have been detected in the placenta, amniotic fluid, fetal membranes and meconium, there is no consensus as to the presence of microbes in the fetal gut prior to delivery.
What are the new findings?
We found that the prenatal gut in lambs harbours a low-diversity and low-biomass microbiome, suggesting that microbial colonisation of the fetal gut commences in utero. We also found multiple microbial metabolites in the fetal gut, suggesting that the prenatal gut microbes are metabolically active. In addition, bacteriophage phiX174, Orf virus and expression of multiple genes involved in antibiotic resistance, were detected in the fetal gut.
How might it impact on clinical practice in the foreseeable future?
Understanding the community composition and function of the fetal gut microbiome may help in the development of strategies to mitigate several diseases and guide the formation of health-promoting microbiota that could be beneficial for the development of healthy offspring.
Introduction
The gut microbial community plays critical roles in host health by providing metabolic products, maintaining metabolic function, developing the immune system and defending against pathogens.1–3 Mounting evidence suggests that dysfunction of the early life gut microbiota can result in immune and metabolic disorders, contributing to the development of non-communicable diseases and obesity.4 However, establishing effective means to reduce disease risk by manipulating early life host–microbe interactions is hindered by considerable gaps in knowledge on the colonisation time and origin of intestinal microbiota.5
Whether there exists colonising microbes in the fetal gut prior to delivery is still controversial. It has long been assumed that the fetus is sterile and that establishment of the fetal microbiota commences during the birthing process.6 Recently, this dogma has been challenged by a growing body of new data. Numerous studies have reported the presence of diverse microbes in samples of human infant meconium,7 8 placenta,9–11 amniotic fluid7 12 and umbilical cord blood13 obtained from healthy normal pregnancies using sequencing-based methods. This suggests that microbial seeding of the fetal gut may initiate in utero before delivery. However, the interpretation of these findings has raised concerns regarding the validity of these conclusions and has primarily focused on methodological challenges, contradictory results and our current immunological understanding.14 As a result, the concept of a ‘sterile womb paradigm’ has persisted, with many scientists contending that microbes detected in utero may be due to underlying contamination issues especially when working with low biomass samples.15–17 For example, several studies assert that microbes detected in the placenta were due to DNA present in laboratory reagents and equipment contaminants.18–20 Thus, it is essential to conclusively determine whether the prenatal gut harbours a microbiome, or whether the purported fetal gut microbiome is merely the result of contamination.
The aim of this study is to provide direct evidences in support of the existence of a microbiome in the prenatal gut. To accomplish this, we used lambs delivered via aseptic hysterectomy as an animal model. Lambs were euthanised immediately after birth via aseptic caesarean section and their cecal content and umbilical cord blood samples were collected. Cecal content samples were assessed using metagenomic and metatranscriptomic sequencing to investigate microbial composition and function, after which both sample types were analysed using metabolomics to detect the presence of microbial metabolites.
Results
Removal of potential contaminating genes and transcripts from the metagenomic and metatranscriptomic datasets
To assess the microbiome of the six in utero lambs used in our study, we performed both metagenomic and metatranscriptomic sequencing of the cecal contents from our study animals. Given concerns from other studies of contaminating DNA/RNA in the reagents used to process samples we also included negative control where nucleic acid-free water without sample DNA was used. From the negative control, we performed a data decontamination step, as described in Methods and online supplemental figure S1.
Supplemental material
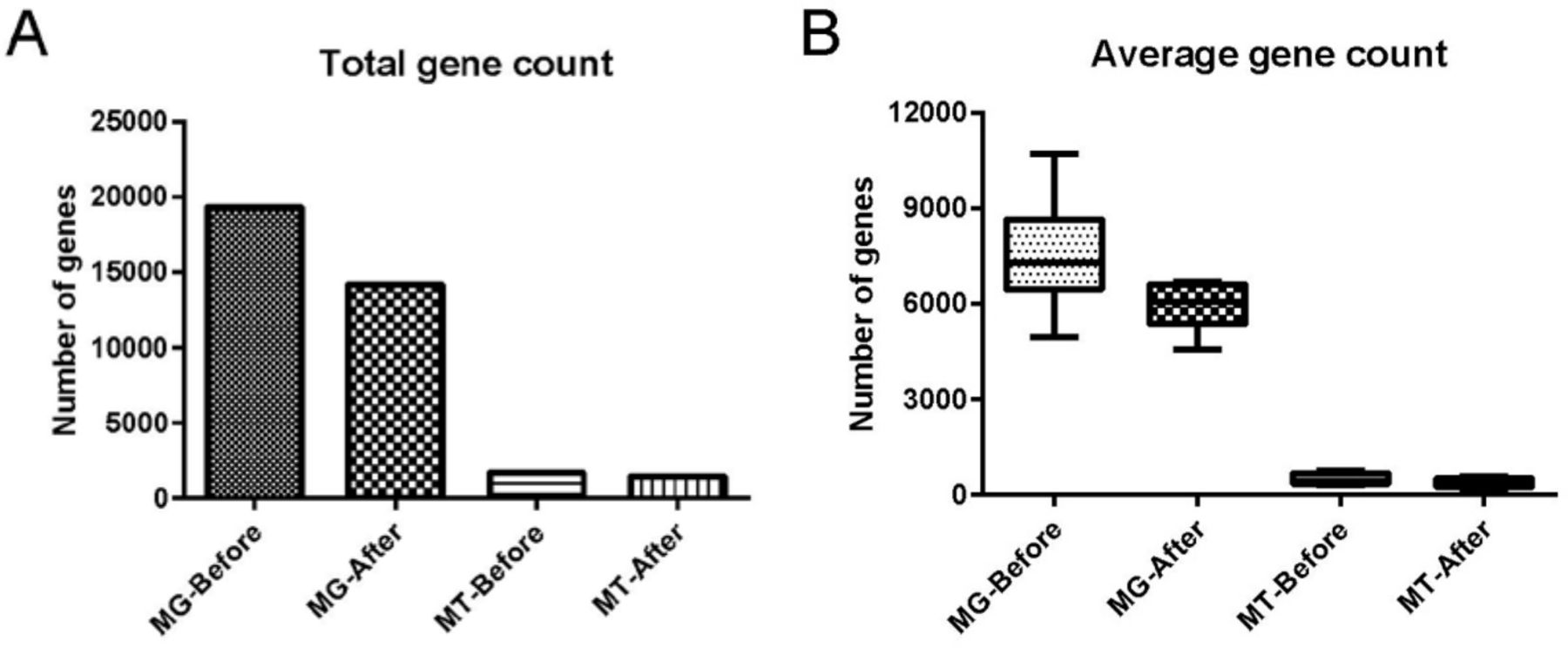
Comparison of the metagenomic dataset (containing 19 320 non-redundant (nr) genes) with the negative control showed that cecal content samples possessed 14 116 unique genes, and that 5204 genes were shared between cecal content samples and negative control samples (online supplemental tables S1 and S2). Of these 5204 shared genes, 83 passed our filtering criteria and remained in our dataset whereas the 5121 remaining genes were discarded as potential contaminants. Comparison of the metatranscriptomic dataset (containing 1691 nr genes) with the negative control indicated that cecal content samples contained 1446 unique genes. Our filtering step identified 245 genes shared between cecal content samples and negative control (online supplemental tables S3 and S4), and of those, 10 genes passed our filtering criteria and thus remained in our dataset. After final quality assessment and data decontamination, the gut microbial metagenomic and metatranscriptomic datasets contained a total of 14 199 genes (5935±711 genes per sample, expressed as mean±SD) and 1456 genes (358±134 genes per sample) for the six cecal content samples, respectively (figure 1, online supplemental tables S5 and S6).
Supplemental material

Effect of data decontamination on gut microbial metagenomic and metatranscriptomic gene counts. (A) Total gene count for microbial metagenome (MG) and metatranscriptome (MT) before and after data decontamination in samples of fetal cecal content. ‘Before’ indicates gene counts before data decontamination; ‘after’ indicates gene counts after data decontamination. (B) Average gene count in microbial MG and MT per cecal content sample before and after data decontamination. In (B), the boxes represent IQRs between the first and third quartiles, and the horizontal line inside the box indicates the median; whiskers represent the minima or maxima within 1.5× IQR from the first or third quartiles.
Taxonomic composition identified by metagenomic analysis
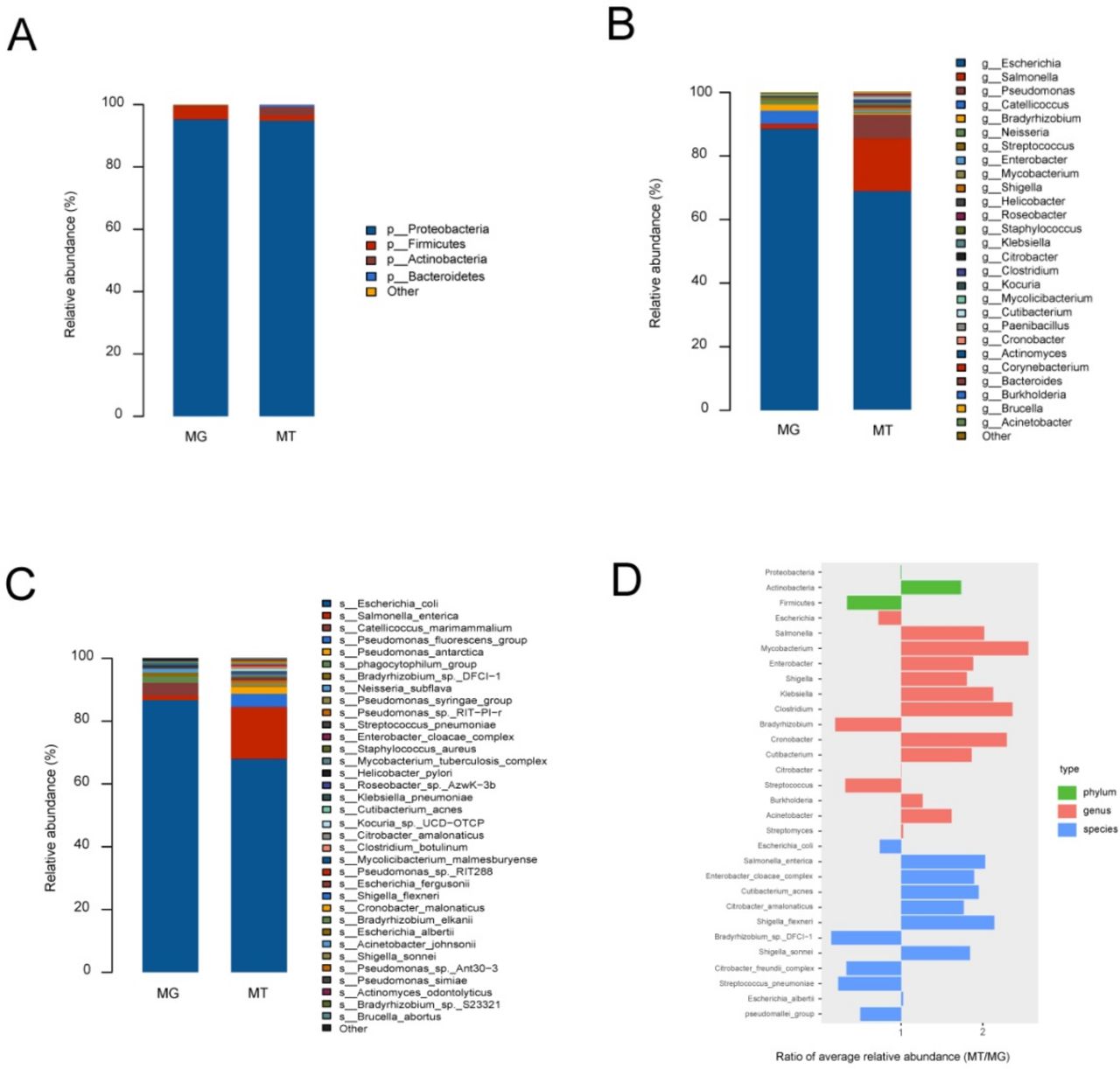
To identify microbial community structures of the fetal gut at a metagenomic level, we performed a metagenomic sequencing of cecal content samples obtained from six lambs delivered via aseptic hysterectomy. We aligned the metagenomic genes against the nr nucleotide database of National Center of Biotechnology Information (NCBI) for taxonomic assignment. Microbial taxa were defined as being present in at least three out of six samples. We found that the fetal gut of lambs showed low alpha diversity (online supplemental figure S2), indicating that bacterial richness in the fetal gut was low. Four major phyla were identified in cecal content samples: Proteobacteria (95.30%±2.19%), Firmicutes (4.85%±1.71%), Actinobacteria (0.53%±0.22%) and Thaumarchaeota (0.02%±0.01%) (figure 2A, online supplemental table S7). At the genus and species levels, 33 genera and 50 species were detected in cecal content samples. Escherichia (88.76%±2.04%) and Catellicoccus (4.19%±1.65%) were the main bacterial genera (figure 2B, online supplemental table S7), while Escherichia coli (86.89%±2.21%) and Catellicoccus marimammalium (4.11%±1.61%) were the most abundant species (figure 2C, online supplemental table S7). We also detected high levels of bacteriophage phiX174 (52.29%±5.55% of all mapped reads) and Orf virus (0.03%±0.01% of all mapped reads) in all cecal contents samples (online supplemental figure S3A) and 22 antibiotic resistance genes in the fetal gut (online supplemental table S8).

Microbiome composition of metagenome (MG) and metatranscriptome (MT) in fetal gut. (A–C) Represent phylum-level, genus-level and species-level bacterial composition of the fetal gut microbiome (relative abundance ≥0.1%), respectively. Microbiome composition shows average relative abundance of bacterial presence in all samples. n=6. (D) Ratios of average relative microorganism abundance in MT relative to the MG (MT/MG).
Taxonomic composition identified by metatranscriptomic analysis
To examine the potential activity of the detected microbes, we performed a metatranscriptomic analysis of RNA extracted from cecal content samples. A taxonomic classification of the metatranscriptomic data revealed a total of bacterial phyla, 26 bacterial genera and 32 bacterial species. The four bacteria phyla included the Proteobacteria (94.73%±3.66%), Actinobacteria (2.75%±2.52%), Firmicutes (1.69%±1.13%) and Bacteroidetes (0.67%±0.62%) (figure 2A, online supplemental table S9). At the genus and species levels, Escherichia (64.23%±19.30%) and Salmonella (15.51%±6.84%) were the main bacterial genera (figure 2B, online supplemental table S9), while E. coli (64.14%±19.17%) and Salmonella enterica (15.60%±6.87%) were the most abundant species (figure 2C, online supplemental table S9). Metatranscriptomic analysis of the cecal content samples also indicated high levels of bacteriophage phiX174 (58.88%±14.73% of all mapped reads) (online supplemental figure S3B).
To infer relative microbial activity at the time of sample collection, we analysed microbial gene expression in the samples based on the ratio of microbial functional gene transcripts to genes.21 MT/MG ratios were defined as ratios of average relative microbe abundance in the metatranscriptomes, relative to those in the corresponding metagenomes.21 Members of the Actinobacteria, such as the genera Mycobacterium and Cutibacterium, showed increased MT/MG ratios (figure 2D). Members of the Firmicutes, such as the genus Streptococcus, exhibited decreased MT/MG ratios, but the MT/MG ratios of Clostridium were increased. The MT/MG ratios of the Proteobacteria were near 1, but members belonging to this phylum varied greatly. For example, members of the genera Escherichia and Citrobacter complex showed decreased MT/MG ratios, whereas other microbes belonging to the Proteobacteria showed increased MT/MG ratios.
Quantification of microbes in samples of cecal content and in the controls
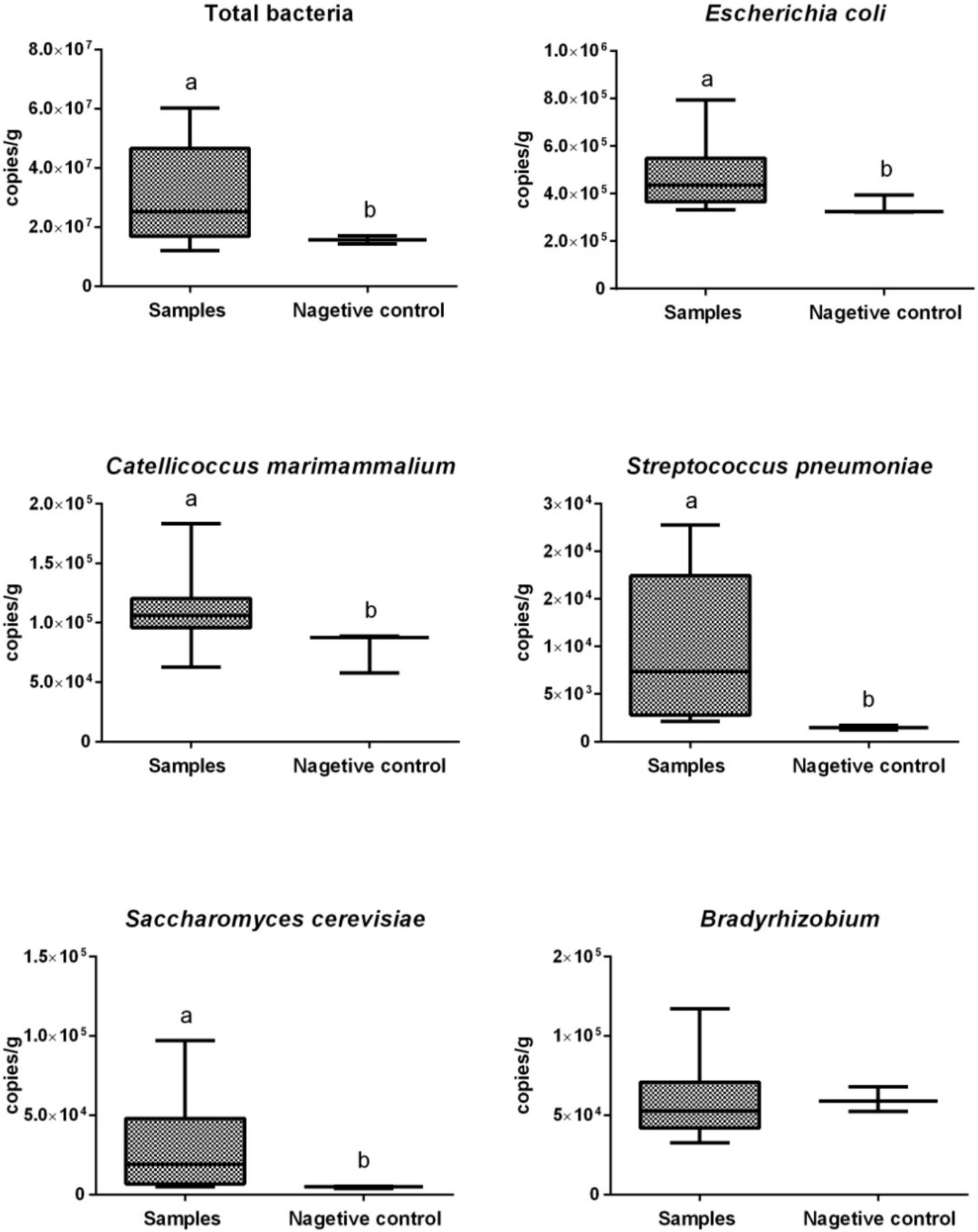
We performed absolute quantitative real-time PCR (qPCR) to quantify the copy numbers of total bacteria and five selected microbes (figure 3) showing a high relative abundance in our metagenomic data. Copy numbers per gram of total bacteria in cecal content samples (expressed as mean±SD) and negative controls were 4.6×107 ± 3.4×107 and 1.6×107 ± 1.1×106, respectively. Cecal content samples showed significantly higher (p<0.05) copy numbers per gram of total bacteria than did negative controls. Copy numbers per gram of E. coli, C. marimammalium, Streptococcus pneumoniae and Saccharomyces cerevisiae were significantly higher (p<0.05) in cecal content samples than those in the negative controls, whereas copy numbers per gram of Bradyrhizobium did not differ significantly between these samples.

Copy numbers of total bacteria and selected microbes per gram cecal content. The boxes represent IQRs between the first and third quartiles, and the horizontal line inside the box indicates the median; whiskers represent the minima or maxima within 1.5× IQR from the first or third quartiles. Boxes with different letters above their whiskers are significantly different at p<0.05.
Functional characterisation of the cecal content microbiome
To functionally characterise the cecal microbiome, we aligned nr gene catalogues of the metagenomes and metatranscriptomes against the Kyoto Encyclopaedia of Genes and Genomes (KEGG) and evolutionary genealogy of genes: non-supervised orthologous groups (eggNOG) databases. Of the metagenomic genes, 5458 and 8749 genes were annotated using KEGG and eggNOG, respectively. KEGG pathways belonging to ‘Signal transduction’, ‘Carbohydrate metabolism’, ‘Amino acid metabolism’ and ‘Energy metabolism’ were highly enriched in the microbiome of fetal gut (figure 4A). Similarly, microbial genes involved in ‘Amino acid transport and metabolism’, ‘Carbohydrate transport and metabolism’, ‘Energy production and conversion’ and ‘Signal transduction mechanisms’ pathways were highly enriched in eggNOG functional categories. Furthermore, pathways involved in ‘Transcription’, ‘Posttranslational modification, protein turnover, chaperones’ and ‘Intracellular trafficking, secretion and vesicular transport’ were also highly enriched in eggNOG functional categories (figure 4C).

KEGG and eggNOG classifications of the fetal gut metagenome and metatranscriptome. (A, B) KEGG classifications for the fetal gut metagenome and metatranscriptome, respectively. (C, D) eggNOG functional classifications for the fetal gut metagenome and metatranscriptome, respectively. eggNOG, non-supervised orthologous groups; KEGG, Kyoto Encyclopaedia of Genes and Genomes.
For the metatranscriptomic data, 912 and 1168 genes were annotated using KEGG and eggNOG, respectively. KEGG pathways involving the ‘Carbohydrate metabolism’, ‘Energy metabolism’, ‘Amino acid metabolism’, ‘Membrane transport’ and ‘Signal transduction’ were highly enriched in the microbiome of fetal gut (figure 4B). The most active eggNOG functional categories in the fetal gut were ‘energy production and conversion,’ ‘amino acid transport and metabolism’ and ‘carbohydrate transport and metabolism,’ followed by ‘translation, ribosomal structure and biogenesis’ and ‘cell wall/membrane/envelope biogenesis’ (figure 4D).
Metabolomic profiling of cecal content and umbilical cord blood
To further analyse microbial activity in the fetal gut, we conducted a metabolomic analysis to detect microbial metabolites in our samples. The concentrations of short chain fatty acids (SCFAs) were measured and used as a proxy for microbial metabolic activity in the gut. For this study, we used a detection limit of 5×10−5 µg/mg for acetic, propionic and isobutyric acids and a detection limit of 5×10−6 µg/mg for butyric, isovaleric, valeric and hexanoic acids. This range allowed for the detection of all SCFAs in our cecal content samples, except for propionic, isobutyric and isovaleric acids. The concentrations of acetic, butyric, valeric and hexanoic acids were 1.88×10−3 ± 4.05×10–4, 1.01×10−4 ± 2.43×10−5, 2.71×10−5 ± 4.56×10−6 and 1.02×10−4 ± 3.01×10−5 µg/mg, respectively. To trace the possible sources of these SCFAs in the gut, we examined SCFAs in umbilical cord blood. The concentrations of acetic, propionic, butyric, isobutyric, valeric, isovaleric and hexanoic acids in umbilical cord blood were 6.23±2.94, 1.69±0.55, 0.72±0.24, 0.56±0.19, 0.24±0.09, 0.04±0.03 and 0.41±0.24 µg/mL, respectively (online supplemental table S10).
We also performed non-targeted metabolomic profiling using high-performance liquid chromatography-quadruple time-of-flight-mass spectrometry (UHPLC-QTOF-MS) to detect other metabolites in our samples. In total, 2808 peaks were obtained from cecal content samples, including 1271 and 1537 peaks in positive ion mode (ESI +) and negative ion mode (ESI −), respectively. These peaks were aligned using Human Metabolome Database (HMDB), PubChem and the KEGG databases and we were able to annotate 1990 total peaks (937 peaks in ESI+ and 1053 peaks in ESI −) (online supplemental tables S11 and S12). Of these 1990 metabolites, 151 were annotated to 180 KEGG pathways, whereas the other 1839 metabolites were not assigned to a specific pathway. These KEGG pathways were then divided into host-associated (ie, Ovis aries) pathways and non-host associated pathways (ie, all KEGG pathways not shared with Ovis aries KEGG pathways). Of these 151 total metabolites, 143 were involved in 127 host-related KEGG pathways (online supplemental table S13), and 77 were involved in 53 non-host microbially related KEGG pathways (online supplemental table S14). The 53 non-host microbially related metabolic pathways contained eight unique metabolites including deoxynojirimycin, mitomycin, tobramycin, p-benzoquinone, cyclohexylsulfamate, daidzein, chlorogenate and hesperetin.
We note that all 8 of these metabolites are associated with known microbial metabolic pathways. Specifically, deoxynojirimycin, mitomycin and tobramycin are involved in ‘biosynthesis of antibiotics’, tobramycin is involved in ‘neomycin, kanamycin and gentamicin biosynthesis’, p-benzoquinone and cyclohexylsulfamate are involved in ‘microbial metabolism in diverse environments’, daidzein and chlorogenate are involved in ‘Biosynthesis of secondary metabolites’ and hesperetin is involved in ‘flavonoid biosynthesis’ (online supplemental table S14). To trace the possible sources of these microbial metabolites in the gut, we performed non-targeted metabolomic profiling to detect metabolites in umbilical cord blood, but did not find the presence of deoxynojirimycin, mitomycin or tobramycin (online supplemental tables S15 and S16).
Associations between cecal microbiome and cecal content metabolites
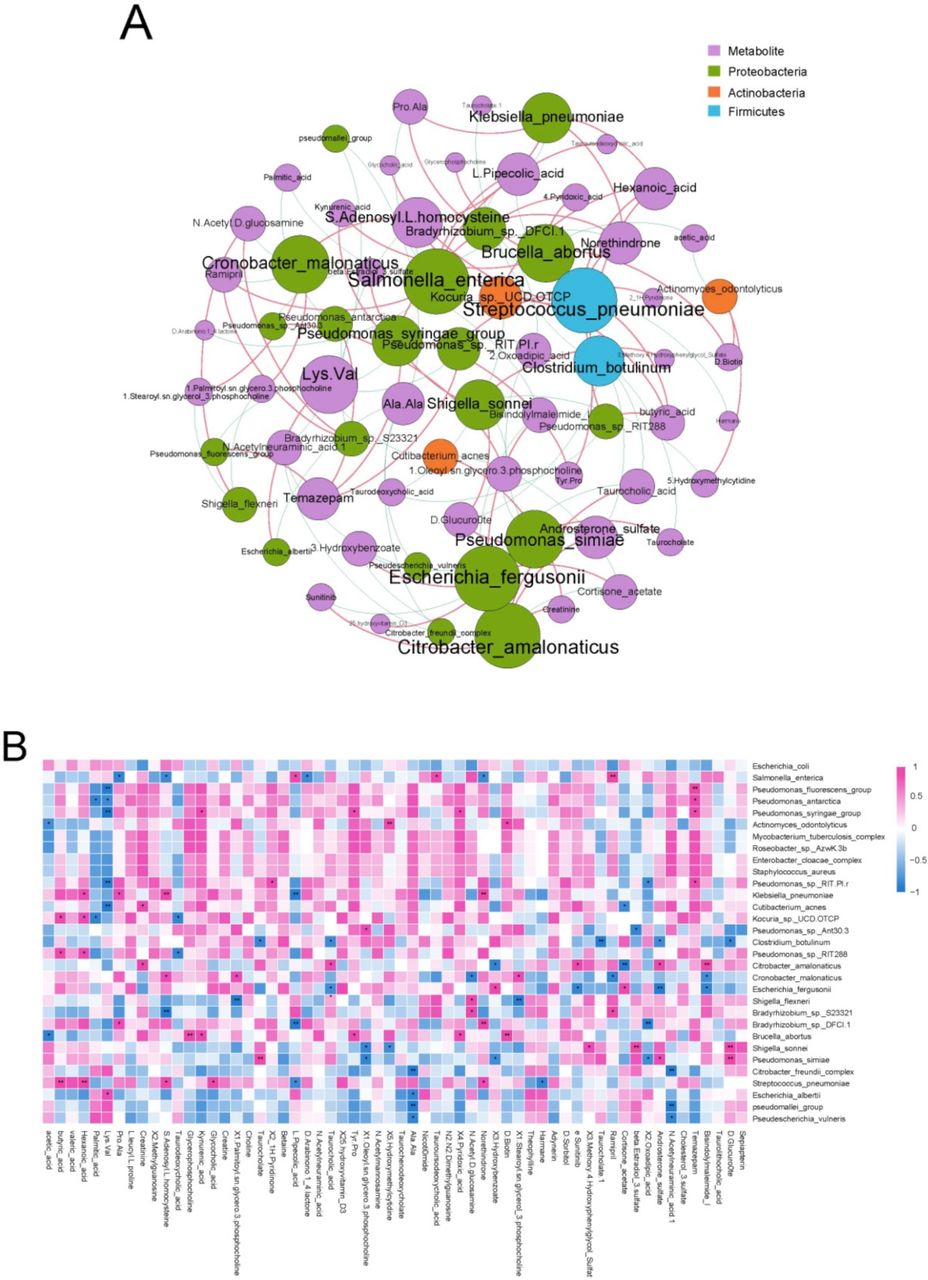
To identify the correlations between cecal microbiome and cecal content metabolites, we performed co-occurrence network and heat map analyses using Spearman’s rank correlation coefficient. In the co-occurrence network and heat map, butyric and hexanoic acids concentrations showed positive correlations with the relative abundances of S. pneumoniae and Kocuria spp. UCD.OTCP (Spearman’s correlation value >0.6, p<0.05; figure 5A,B). Furthermore, hexanoic acid concentration was positively associated with the relative abundance of Klebsiella pneumoniae. Norethindrone concentration was also found to be positively correlated with the relative abundances of Klebsiella pneumoniae and S. pneumoniae, but negatively correlated with that of Salmonella enterica. In addition to the correlations identified between cecal microbiome and cecal content metabolites, we also found that the cecal content metagenome, macrotranscriptome and metabolome shared numerous KEGG pathways. Of the 376 and 198 KEGG pathways in our metagenomic and metatranscriptomic data, respectively, 152 and 95 KEGG pathways, respectively, were shared with the metabolome (online supplemental tables S17 and S18).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Associations of cecal microbiome with cecal content metabolites. (A) Analysis of co-occurrence networks between cecal content metabolites and microbial species in the fetal gut metatranscriptome. Nodes represent metabolites and microbial species. An increase in the number of lines through a node increases the size of a node. Red edges indicate positive correlations between metabolites and microbial species; Spearman’s rank correlation coefficient >0.6, p<0.05. Blue edges indicate negative correlations between metabolites and microbial species; Spearman’s rank correlation coefficient < −0.6, p<0.05. (B) Heat map of Spearman’s rank correlation coefficients between cecal content metabolites and microbial species in the fetal gut metatranscriptome. Spearman’s rank correlation coefficient >0.6 or < −0.6, *p<0.05, **p<0.01. Red and blue colours denote positive and negative correlations, respectively. Colour intensity is proportional to Spearman’s rank correlation values.
Discussion
In this study, we used lambs delivered by aseptic hysterectomy and multi-omics based metagenomic, metatranscriptomic and metabolomic analyses to investigate the presence of a microbiome in the prenatal fetal gut. In order to ensure the accuracy of our results, we undertook many measures to eliminate the risk of contamination from environmental sources and handing. First, the lambs used in our animal model were delivered via aseptic hysterectomy, thereby preventing microbial contamination from the maternal vaginas and faeces. Second, the euthanasia, open surgery and collection of cecal content samples from these lambs, as well as total DNA and RNA extractions, were performed in strictly controlled, decontaminated and sterilised biosafety cabinets to avoid potential environmental contamination. Importantly, the negative control was performed and treated identically during sample collection, extraction of genetic material, sequencing and data analysis to eliminate (via data filtration) potential contamination from DNA and RNA found in extraction kits and other laboratory reagents and equipment.15 22
After potentially-contaminating microbial genes were removed from our metagenomic and metatranscriptomic sequencing datasets, a diverse and metabolically active microbiota was detected in the prenatal fetal gut. The fetal gut microbiome was predominantly composed of Proteobacteria, Actinobacteria and Firmicutes, with Proteobacteria showing the highest abundance and accounting for more than 90% of all sequences in the different samples. E. coli showed the highest relative abundance in all the samples, accounting for more than 86% and 64% of the total sequences in our metagenomic and metatranscriptomic sequencing datasets, respectively. Additionally, other species, such as C. marimammalium, Salmonella enterica, S. pneumoniae, Enterobacter cloacae, Escherichia albertii and Shigella sonnei, were detected in the fetal gut. These bacterial taxa are similar to previous reports from humans, particularly in term and preterm placentas without intraamniotic infection.9 11 These observations suggest that bacterial colonisation is consistent across species.
Numerous studies have also identified microbes using non-sequencing methods in utero in the absence of immunopathology. For example, intact placental microbes were visualised and localised by 16S rRNA in situ hybridisation and traditional histological techniques in term and preterm placentas without intraamniotic infection11; gram-positive and—negative intracellular bacteria were identified in the basal plate of human placenta without clinical or histologic evidence of chorioamnionitis based on morphological evidence23; probiotic bacterial strains administered to mothers before delivery were detected using qPCR in all placenta of full-term infants delivered by caesarean section.24 Our observations are consistent with these studies, suggesting that seeding of the microbiome likely commences in utero prior to delivery.
Importantly, our qPCR results showed that the copy numbers per gram of total bacteria, E. coli, C. marimammalium, . pneumoniae and S. cerevisiae were significantly higher in the cecal content samples than in our negative control, further providing evidence for the presence of a microbiome in the prenatal fetal gut. However, the copy numbers per gram of these microbes in the cecal content were low, with an order of magnitude of 103–105. Compared with our previous reports on the gut microbiota of a 3-day-old lambs, the fetal gut of in utero lambs contained significantly lower bacterial diversity and biomass,25 indicating that bacterial richness and biomass were low in the prenatal fetal gut. Our findings further support several previously published reports,5 9 11 which used metagenomic sequencing to demonstrate the existence of low-abundance, low-biomass and sparse microbial communities in utero.
In addition to our transcriptomics analysis, which provides support that the microbiome present in the prenatal fetal gut are active, we also conducted a metabolomics analysis. We detected several microbial metabolites including SCFAs, deoxynojirimycin, mitomycin and tobramycin in fetal cecal content samples. SCFAs are the major end products of microbial fermentation in the gut and act as a proxy for inferring microbial metabolic activity.26 Previous studies have found SCFAs, such as acetic, propionic and butyric acids, in the infant first-pass meconium,7 27 which is consistent with our findings. Nojirimycin, a precursor to deoxynojirimycin, is known to inhibit various microbial glucosidases28 and is produced by several strains of Streptomyces. This secondary compound has robust biological activity against several drug-resistant bacterial strains including Sarcina lutea, Shigella flexneri and Xanthomonas oryzae.29 30 Mitomycins, which are important antibiotics also produced by members of the Streptomyces, show reduced toxicity and broad spectrum activity against tumours and thus are widely used in the clinic.31 Tobramycin is an aminoglycoside antibiotic derived from nebramycin, a broad-spectrum antibiotic also produced by members of the Streptomyces.32 It shows excellent antibacterial activity against Pseudomonas, Staphylococcus aureus and most of the Enterobacteriaceae.33 Our metabolite analysis using KEGG revealed that deoxynojirimycin, mitomycin and tobramycin are involved in the pathway ‘biosynthesis of antibiotics’. Tobramycin is also involved in ‘neomycin, kanamycin and gentamicin biosynthesis’. In this study, we detected sequences belonging to the Streptomyces in both metagenomic and metatranscriptomic data from fetal cecal contents and it is possible that the presence of the Streptomyces and the metabolites deoxynojirimycin, mitomycin and tobramycin is not coincidental. One hypothesis is that members of the Streptomyces present in the fetal microbiome are producing these secondary compounds as a way to outcompete other microbes in the community.
In order to trace the possible sources of these microbial metabolites in the fetal gut, we also investigated metabolites present in umbilical cord blood. The developing fetus is supported by the umbilical cord in mammals, through which almost all nutrients, excretion products, respiratory gases and xenobiotics are passed.34 In this study, we detected the presence of SCFAs in umbilical cord blood, but it is unknown if SCFAs in the fetal gut are from the fetal gut microbiome or from the ewe. It is possible that these SCFAs are the product of the gut microbiota of the ewe that are transferred from the gut, across the placenta and into the fetus.34 It is important to note that we did not detect the presence of deoxynojirimycin, mitomycin and tobramycin in the umbilical cord blood, which were present in the fetal gut. Although with detected antibiotics, they could be from the bacteria locally or transmitted previously from mothers. These data further demonstrated that the prenatal gut not only harbours the presence of a microbiome, but that it may also be metabolically active.
The available evidence suggests that microbial colonisation of the fetal gut is likely established by ascension from the vagina or through the hematogenous spread of the maternal gut or oral microbiome. It is well established that certain vaginal microbes can ascend into the uterus and invade the amniotic cavity during the pregnancy.35–37 The amniotic fluid contains microbes that are ingested by the fetus during pregnancy, which ultimately colonise the fetal gut.38 39 Microbial DNA orally ingested by pregnant mice was discovered in various organs of fetuses, and a transplacental pathway was proposed as the route of transmission.40 Genetically labelled bacteria introduced into the gut of pregnant mice were detected using culture-based and PCR-based methods in the placenta and meconium of healthy neonates born by sterile caesarean section.13 41 The possible mechanism of microbial transport from the maternal gut to the developing fetus is bacterial translocation from the maternal gut epithelium into the bloodstream followed by colonisation in the placenta.42 Several species of microbiota identified in the placenta with DNA-based technology have been found in the maternal oral cavity, but not in the maternal gut and vagina.43–45 Moreover, significant similarities between human oral and placenta microbial communities were previously reported,9 in which the placental microbiota was more similar to the oral microbiota than that in any other body site (including the vagina and gut). Some of these oral microbes such as Fusobacterium nucleatum can bind to the vascular endothelium and increase permeability, allowing other microbial species such as E. coli to cross endothelial barriers, facilitating hematogenous transmission during the placentation.46
Prenatal seeding of a metabolically active microbiome may be clinically important for the developing fetus. First, several members of the microbiota or microbial metabolites may drive immune system development and intestinal epithelium maturation in the fetus. For example, the probiotics Bifidobacterium lactis and Lactobacillus rhamnosus GG, when supplemented to pregnant mothers, were detected in the placenta and amniotic fluid, and the expression of Toll-like receptor-related genes in the fetal gut were significantly modulated, suggesting that microbial contact in utero was associated with changes in fetal intestinal innate immune development.24 The inflammatory cytokines levels of the amniotic fluid were associated with the microbiome presence within the amniotic fluid, which may influence the developing fetal immune system.7 In an experimental model of transient gestational colonisation, germ-free pregnant female mice were treated with a transiently colonising live E. coli strain that could not replicate in vivo.3 Compared with the pups born to the control germ-free mothers, germ-free pups born to transiently colonised mothers harboured increased numbers of intestinal group 3 innate lymphoid cells and F4/80+CD11c+ mononuclear cells. Maternal colonisation of E. coli also reshaped the intestinal transcriptional profiles in the pups, including the increased expression of antibacterial peptides and increased cell division and differentiation of epithelial cells.3 In this study, we detected high levels of E. coli in the fetal gut, suggesting that fetuses were exposed to bacterial-derived immune stimulation prior to birth. Thus, the E. coli identified in our study may be involved in supporting fetal immune system development in utero. Microbial metabolites SCFAs serve as important energy sources and signalling molecules and are involved in the augmentation of energy metabolism, maintenance of the immune system, induction of reactive oxygen species, modulation of chemotaxis and phagocytosis, inhibition of histone deacetylases, mediation of cell proliferation and differentiation, and enhancement of intestinal barrier integrity.26 47 48 SCFAs contribute to host immune maturation by activating G protein-coupled receptors on the immune cell surface or by inhibiting lysine deacetylases.14 In the current study, we identified various SCFAs in the fetal gut, although the sources of the SCFAs were uncertain; however, the presence of these SCFAs might serve to modify fetal immune function and development.26 Second, the microbial biomass must be maintained at extremely low levels in the intrauterine environment and excessive colonisation by microbes or potential pathogens in utero may result in intrauterine infection, causing adverse pregnancy outcomes such as miscarriage, preterm delivery or stillbirth.35 For example, transmission of Streptococcus agalactiae from mothers to fetuses during pregnancy can lead to neonatal sepsis20; oral Fusobacterium nucleatum colonises the mouse placenta, proliferates quickly to reach 107 colony forming unit per gram tissue in 72 hours, and spreads to the fetus and amniotic fluid, causing term stillbirth45 49; some bacterial species identified in the placenta, including Campylobacter, E. coli, Leptotrichia, Neisseria, Peptostreptococcus and Streptococcus, are associated with preterm birth, fetal death and neonatal sepsis.45 Furthermore, perinatal transmission of bacteria carrying antibiotic resistance genes to fetuses may make newborns resistant to certain antibiotics after birth. Screening for certain bacteria carrying antibiotic resistance genes or potential pathogens during pregnancy and targeted use of antibiotics can prevent adverse pregnancy outcomes and reduce neonatal morbidity and mortality.35 50
To our knowledge, this study is the first to provide direct evidence in support of the existence of a microbiome in the prenatal gut. However, several issues remain to be resolved. First, using the metatranscriptomic sequencing and metabonomics analysis techniques, we demonstrated the presence of metabolically active microbes in the prenatal gut, but this is not equivalent to capturing live microbes. Culture-based methods are required in the future to confirm the presence of live microbes. Second, we identified numerous microbial species and metabolites in the fetal gut. Since the knowledge regarding the functional role of these microbial species and metabolites remains limited, the effects of these on the fetal development needs to be explored. Third, our association analysis can not directly infer that these metabolites are produced by the microbes we detected, but merely suggests the relatedness between cecal content metabolites and the microbiome. For instance, we found that the butyric and hexanoic acids concentrations showed positive correlation with the relative abundance of S. pneumoniae. However, S. pneumoniae is not known to produce butyric and hexanoic acids.51 Furthermore, our study did not investigate the timing and origins of fetal gut microbiome colonisation. The fetal microbiome may begin triggering and educating the fetal immune system far earlier than previously thought. Finally, owing to the intrinsic differences between humans and animal models, the composition and function of the gut microbiome in fetal lambs likely differ from that in humans. Clearly, such experiments would be infeasible in humans, but this study does provide some insight into the process of microbial colonisation in the fetal gut.
In conclusion, using lambs delivered by aseptic hysterectomy as an animal model and multiomics based analyses, our study provides strong evidence that the prenatal gut harbours a low-diversity and low-biomass metabolically active microbiome, and that the fetal gut microbiome is seeded antenatally. These findings advance our understanding of the fetal gut microbiome, which is helpful in designing clinical therapies aimed at improving human health and treating non-communicable diseases associated with deregulation of the gut microbiota. However, further studies are required for gaining an in-depth understanding of the origin, composition, function, dynamics and colonisation time of the fetal gut microbiome, and for elucidating the effects of the fetal gut microbiome and their metabolites on fetal development and health throughout the early stages of life.
Methods
Establishment of animal model and study design
In this study, we used healthy newborn lambs delivered by aseptic hysterectomy as an animal model to investigate the presence of a microbiome in the prenatal fetal gut. Healthy nulliparous pregnant ewes approaching delivery were used to obtain healthy lambs via aseptic hysterectomy. Newborn lambs were euthanised using a penetrative captive bolt and their cecal content and umbilical cord blood samples were collected aseptically. Cecal content samples were analysed using metagenomic and metatranscriptomic sequencing to determine the composition and function of microbes. Cecal content and umbilical cord blood samples were assessed using metabolomics analysis to detect microbial metabolites.
Procedures involving animals and sample collection
All surgeries were performed under sodium pentobarbital anaesthesia and all efforts were made to minimise the suffering of animals.
Healthy nulliparous pregnant ewes with the same expected date of parturition were selected from the same herd. These ewes were fed the same diet without administration of antibiotics or probiotics during pregnancy. Offspring was obtained from the ewes via aseptic hysterectomy performed when the ewes exhibited obvious signs of parturition, such as swollen vulva and udders. Ewes showing the onset of labour, rupture of membranes or signs of maternal infection were excluded from the study. This resulted in a total of six ewes that were finally selected for use in this study.
Preoperative preparation of the ewes and surgical facility, as well as aseptic hysterectomy were performed as described previously52 using a standardised procedure by a team of trained technicians. Briefly, the ewes were narcotised when parturition was imminent and the disinfected surgical area of donor females was adhered to the sterile chamber of the surgical isolator. Hysterectomy was performed in a sterile surgical isolator using sterile disposable surgical instruments. After opening the abdominal cavity, the intact pregnant uterus and its entire contents were harvested, clamped, introduced into a germicidal bath, and transferred into a sterile disposable transfer isolator. As soon as the intact pregnant uterus was introduced into the transfer isolator, the connection between the transfer isolator and the surgery isolator was sealed to avoid contamination.
Samples were collected as previously described,11 by pathology-trained personnel wearing surgical masks and sterile gloves and using sterile disposable surgical instruments. The uterus was opened without affecting the large vessels. Umbilical cord blood was sampled using a sterile needle and syringe and centrifuged at 4°C to obtain serum samples. The lamb was removed to a chemical and ultraviolet sterilised, class II B2 biosafety cabinet and placed into an autoclaved and blaze-disinfected salver. The lamb was then euthanised, and the abdominal cavity was opened with a sterile scalpel and forceps. We chose the cecum as the proxy of the intestinal tract. The cecum is the main fermentation organ in the intestine and is rich in microorganisms and microbial metabolites. Moreover, the saclike structure of the cecum has a large amount of enteral content, which is conducive to sample detection. Cecal content samples were collected with new sterile disposable scalpel and forceps. To avoid potential cross-contamination among lambs, cecal content samples, and instruments, none of the salvers or instruments were reused for any collection or dissection. All samples were collected within 1 hour of delivery under clean and sterile conditions. The serum and cecal content samples were collected into sterile closed vials, snap-frozen in liquid nitrogen, and stored at −80°C until further analysis. A negative control (nucleic acid-free water rather than samples) was subjected to the sample collection procedure.
DNA extraction and metagenomic sequencing
To reduce the risk of environmental contamination of samples, all experimental procedures (total DNA and RNA isolation, and qPCR) were performed in a stringent clean and sterile class II B2 biosafety cabinet. Total genomic DNA was extracted from cecal content samples using a DNeasy PowerSoil Kit (Cat. No.12888, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The negative control during sample collection and an extraction control composed of E. coli (positive control) were subjected to the same protocols for DNA extraction, sequencing, and data analysis as the lamb-derived samples. DNA integrity was evaluated using agarose gel electrophoresis. DNA quality was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and DNA concentration was measured using a Qubit 2.0 Fluorimeter (Invitrogen, Carlsbad, California, USA). Construction of the metagenomic library was performed using a TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, California, USA). The quantity of each metagenomic library was evaluated with a Qubit V.2.0 Fluorimeter. Metagenomic sequencing was conducted using an Illumina HiSeq 4000 platform (paired-end; insert size, 300 bp; read length, 150 bp).
RNA extraction and metatranscriptomic sequencing
Total RNA was extracted from cecal content samples using a RNeasy PowerSoil Total RNA Kit (Cat. No.12866, Qiagen) according to the manufacturer’s protocols. Negative and positive controls were the same as those described for DNA extraction and metagenomic sequencing and were processed alongside cecal content samples throughout RNA extraction and metatranscriptomic sequencing. RNA concentration was measured using a Qubit V.2.0 Fluorimeter and RNA quality was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, USA). rRNA in each sample was removed using Ribo-Zero rRNA Removal Kits (Bacteria) and Ribo-Zero Magnetic Gold Kit (Yeast) (Epicentre, San Diego, California, USA) as per the manufacturer’s instructions. Enriched mRNAs were used to construct an mRNA-enriched metatranscriptomic library using a NEBNext Ultra RNA Library Prep Kit (New England Biolabs, Ipswich, Massachusetts, USA). The metatranscriptomic library was sequenced using an Illumina HiSeq 4000 (paired-end; insert size, 300 bp; read length, 150 bp).
Quality control of metagenomic and metatranscriptomic datasets
Metagenomic and metatranscriptomic sequencing generated 113.60 Gbp (18.93±2.24 Gbp per sample; data are expressed as mean±SD) and 64.86 Gbp (10.81±8.52 Gbp per sample) raw reads for the six cecal content samples, respectively. Quality control of the metagenomic and metatranscriptomic raw datasets was performed using Trimmomatic (V.0.35)53 to trim adapters, filter out reads with >1% of uncertain bases, and remove low-quality bases (quality scores <20) and short reads (<150 bp). After quality control of the sequencing datasets, the remaining reads were mapped to the sheep (Ovis aries) genome using bowtie254 to eliminate potential host DNA and RNA contamination. A total of 10 544 549 and 56 746 269 high-quality pair-end clean reads for the six cecal content samples were acquired from the metagenome (1757, 425±5 62 944 clean reads per sample; 0.53±0.17 Gbp per sample) and metatranscriptome (9457, 711±5 982 821 clean reads per sample; 2.84±1.79 Gbp per sample), respectively.
Construction of a reference gene catalog for microbial metagenome and metatranscriptome analysis
High-quality metagenomic and metatranscriptomic reads were de novo assembled into contigs using MEGAHIT (V.1.0.6)55 and Trinity,56 respectively. This generated a total of 64 220 contigs with an average length of 864 bp (maximum 415 711 bp) and N50 length of 794 bp for the metagenomics data, and a total of 24 858 contigs with an average length of 398 bp (maximum 21 634 bp) and N50 length of 356 bp for the metatranscriptomics data. MetaGeneMark (V.2.7) was used to predict open reading frames (ORFs). All predicted ORFs were clustered via CD-HIT57 using the following criteria: identity >95% and coverage >90%. Gene sequences with the greatest number of hits in each cluster were selected to represent the cluster, while other genes in the cluster were regarded as redundant and were not considered in downstream analyses. Metagenomic and metatranscriptomic gene catalogues containing 19 320 and 1691 nr genes, respectively, were constructed. Of the 19 320 genes identified in the metagenome, 5204 genes were shared between the cecal content samples and negative control, and 14 116 genes were unique to the samples (online supplemental tables S1 and S2). Of the 1691 genes identified in the metatranscriptome, 245 genes were shared between the cecal content samples and negative control, and 1446 genes were unique to the samples (online supplemental tables S3 and S4).
Filtering of potential contaminating genes from metagenomic and metatranscriptomic reference gene catalogs
Before phylogenetic and functional annotation of the metagenome and metatranscriptome, potential contaminant genes (originating from reagents or instruments) were removed from all sample sequencing datasets. Filtering was done by comparing the abundance of shared nr genes between each sample and the negative control. First, all shared nr genes in each sample and the negative control were selected; second, high-quality reads from each sample and the negative control were remapped to the shared genes using the RSEM software, as described previously.58 The abundance of shared genes in cecal content samples and in the negative control were calculated using the fragments per kilobase of transcript per million fragments method59 based on the number of uniquely mapped reads. DESeq260 was used to identify differentially expressed genes between each sample and the negative control. A log2FoldChange>2 was used as the threshold for identifying contaminant genes in cecal content samples.61 After data decontamination, the gut microbial metagenome and metatranscriptome gene catalogues contained 14 199 and 1456 nr genes, respectively (online supplemental tables S5 and S6).
Taxonomic assignment and functional annotation of the metagenome and metatranscriptome
To estimate the taxonomic profiles of the gut microbial metagenome and metatranscriptome, the predicted nr genes were aligned against NCBI’s nr database using DIAMOND blastx62 (V.0.8.23, default parameter except that evalue=1e-5). Bacterial compositional profiles were summarised at the phylum, genus and species levels. To identify metagenomic and metatranscriptomic functional categories, the predicted nr genes were aligned against protein sequences in the KEGG and eggNOG databases using DIAMOND blastx62 (V.0.8.23, default parameter except that evalue=1e-5, top hit=1, max-target-seqs/k=1). Antibiotic-resistance genes were annotated by alignment against the CARD database via BLASTN (V.2.9.0, default parameter except that evalue=1e-10).
α-diversity analysis
Only microbial taxa detected in at least three out of six samples were considered as being present, and were used for α-diversity analysis. Within-sample diversity (α-diversity) was used to estimate the diversity and evenness of microbial taxa contained in a sample according to the Shannon index and based on the microbial taxa profile of the sample.
QPCR analysis
QPCR analysis was performed to detect the copy numbers of total bacteria, and those of five selected microbes showing a high relative abundance in our metagenomic analysis; the primers used in qPCR are shown in online supplemental table S19. A standard curve was constructed for total bacteria and for each individual strain. qPCR was performed in triplicate using 20 μL reaction mixtures containing 10 μL 2×qPCR Master Mix (SinoGene, Beijing, China), 0.5 μL each primer (10 μM), 8.0 μL ddH2O and 1 μL 10 ng DNA templates. PCR cycling parameters were as follows: initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 20 s, annealing at 60°C for 30 s, extension at 72°C for 30 s and a final extension at 72°C for 10 min. Copy numbers of total bacteria and those of five selected microbes, per gram of cecal content were then calculated.
SCFAs profiling of cecal content and umbilical cord blood samples
Approximately 100 mg cecal content was vortexed with 1 mL HPLC-grade water. The mixture was then homogenised in a ball mill for 3 min at 45 Hz and centrifuged at 6000×g and 4°C for 10 min. The supernatant fluid (0.3 mL) or serum sample of umbilical cord blood (0.3 mL) was combined with 0.4 mL internal standard (25 µg/mL 2-methylvaleric acid) and 0.1 mL 50% H2SO4, vortexed for 30 s, and ultrasonicated in ice water for 5 min. After centrifuging at 13,000×g and 4°C for 10 min, the supernatant (60 μL) was transferred into a fresh 2 mL glass vial for gas chromatography-mass spectrometry (GC-MS) analysis. Standard solutions containing acetic, propionic, isobutyric, butyric, isovaleric, valeric and hexanoic acids (Sigma, St.Louis, Missouri, USA) were used for calibration. The concentration of SCFAs was measured using an Agilent 7890 GC system (Agilent Technologies, Santa Clara, California, USA) equipped with an HP-FFAP capillary column (30 m × 250 μm × 0.25 μm, Agilent Technologies) coupled with an Agilent 7000D mass spectrometer. Sample aliquots (1 µL) were injected using a split mode (5:1). Helium gas was used as a carrier at a flow rate of 1 mL/min. The oven temperature was initially maintained at 100°C for 1 min, increased to 150°C at a rate of 5°C/min and maintained for 0 min, further increased to 240°C at a rate of 20°C/min, and finally maintained at 240°C for 5 min. Temperatures used for injection, transfer line, quad and ion source were 260°C, 280°C, 150°C and 230°C, respectively. MS data were acquired in multiple reaction monitoring mode with a solvent delay of 3 min. SCFAs quantities are expressed as micrograms per milligram cecal content.
Metabolomics profiling of cecal content and umbilical cord blood samples
Sample preparation
Approximately 50 mg cecal content or 100 μL serum was placed in an Eppendorf tube, to which 1 mL ice-cold extraction solution (V methanol: V acetonitrile: V water=2:2:1) containing an internal standard was added; the resulting mixture was vortexed for 30 s. The mixture was then homogenised in a ball mill for 4 min at 45 Hz and ultrasonicated in an ice water bath three times for 5 min each time (This step was not performed for serum samples). The mixture was incubated at −20°C for 1 hour. After centrifuging at 13,000×g and 4°C for 15 min, 750 µL of the supernatant was transferred into an Eppendorf tube and dried in a vacuum concentrator without heating. The residue was reconstituted in 200 µL extraction solution (V acetonitrile: V water=1:1), vortexed for 30 s, and ultrasonicated in an ice water bath for 10 min. After centrifuging at 13,000×g and 4°C for 15 min, 60 μL of the supernatant was transferred into a fresh 2 mL liquid chromatography-MS glass vial for UHPLC-QTOF-MS analysis. The quality control sample was prepared by mixing an equal aliquot of the supernatants from all samples.
UHPLC-QTOF-MS analysis
UHPLC-QTOF-MS metabolomics analysis was performed using an Agilent 1290 UHPLC system coupled with a QTOF mass spectrometer (Triple TOF 5600, AB Sciex, Framingham, Massachusetts, USA). A Waters ACQUITY UPLC BEH Amide column (1.7 μm × 2.1 mm × 100 mm, Milford, Massachusetts, USA) was used for chromatographic separation and the column temperature was maintained at 25°C. The mobile phase consisted of 25 mM NH4OAc and 25 mM NH4OH in deionised water (pH 9.75; A) and acetonitrile (B); the elution gradient was as follows: 0–0.5 min, 95% B; 0.5–7 min, 95% B; 7–8 min, 65% B; 8–9 min, 40% B; 9–9.1 min, 40% B; 9.1–12 min, 95% B, delivered at 0.5 mL/min. The sample injection volume was 4 µL. Sample analysis was performed using electrosprayelectrospray ionisation (ESI) source in positive and negative ion modes. The TOF mass scanning range was m/z 50–1200 Da. MS spectra were acquired using Analyst TF 1.7 (AB Sciex) in information-dependent basis. In each cycle, precursor ions with an intensity greater than 100 were selected for fragmentation at a collision energy of 30 eV (15 MS/MS events with product ion accumulation time of 50 ms each). ESI source parameters were as follows: ion source gas 1, 60 psi; ion source gas 2, 60 psi; curtain gas, 35 psi; source temperature, 650°C; ion spray voltage floating, 5000 V in positive mode, and –4000 V in negative mode.
Data analysis and metabolite identification
MS raw data files were converted into mzXML format using ProteoWizard, and then processed using R package XCMS (V.3.2). Pretreatment of the acquired MS data included: correction of retention time (RT); peak identification, selection and grouping; integration of the peak area; and peak alignment and peak normalisation. Preprocessing results were used to generate a data matrix containing RT, mass-to-charge ratio (m/z) values and peak intensity. R package CAMERA was used for peak annotation after XCMS data processing. An in-house MS2 database was used for metabolite identification. Metabolites were identified and validated by aligning the molecular mass data (m/z) using the online HMDB (http://www.hmdb.ca), PubChem (https://pubchem.ncbi.nlm.nih.gov/) and KEGG (http://www.genome.jp/kegg/) databases.
Correlations between the cecal microbiome and cecal content metabolites
Correlations between cecal bacterial species and cecal content metabolites were calculated using Spearman’s rank correlation coefficient and visualised by co-occurrence networks and heat map. Cecal content metabolites were considered to associate with cecal bacterial species if |Spearman’s rank correlation coefficient|>0.6 and p<0.05.
Statistical analyses
The results of qPCR analysis of total bacteria and five selected microorganisms in the cecal content samples and negative control were analysed by one-way analysis of variance using SAS V.9.4 (SAS Institute). Statistical significance was set to p<0.05.
Data availability statement
Metagenomic and metatranscriptomic sequencing data for all samples have been deposited in the NCBI Sequence Read Archive database under accession numbers PRJNA601636 (https://www.ncbi.nlm.nih.gov/sra/PRJNA601636) and PRJNA598075 (https://www.ncbi.nlm.nih.gov/sra/PRJNA598075).
Ethics statements
Ethics approval
Animal care and use for all experiments were approved by and performed following the guidelines of the Animal Ethics Committee of Feed Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China.
Acknowledgments
The authors thank Beijing Allwegene Co., Ltd. for performing metagenomic and metatranscriptomic sequencing. The authors thank Professor Leluo Guan in Department of Agricultural, Food and Nutritional Science of University of Alberta for technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YT, SL and QD contributed equally.
Contributors The study was conceived by QD and designed by SL and YT. The experiments were performed by YB, SW, FZ and NZ. Data analysis was performed by YB, DS and GS. Paper writing was conducted by YB. All authors read and approved the final manuscript.
Funding This study was supported by the National Key Research and Development Programme of China (2017YFD0500500), National Natural Science Foundation of China (31702136), earmarked fund for the China Agriculture Research System (CARS-38) and Fundamental Research Funds for Central Non-profit Scientific Institution (1610382021008, Y2019CG08).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.