Article Text

Abstract
Objective An understanding of the etiologic heterogeneity of colorectal cancer (CRC) is critical for improving precision prevention, including individualized screening recommendations and the discovery of novel drug targets and repurposable drug candidates for chemoprevention. Known differences in molecular characteristics and environmental risk factors among tumors arising in different locations of the colorectum suggest partly distinct mechanisms of carcinogenesis. The extent to which the contribution of inherited genetic risk factors for CRC differs by anatomical subsite of the primary tumor has not been examined.
Design To identify new anatomical subsite-specific risk loci, we performed genome-wide association study (GWAS) meta-analyses including data of 48 214 CRC cases and 64 159 controls of European ancestry. We characterised effect heterogeneity at CRC risk loci using multinomial modelling.
Results We identified 13 loci that reached genome-wide significance (p<5×10−8) and that were not reported by previous GWASs for overall CRC risk. Multiple lines of evidence support candidate genes at several of these loci. We detected substantial heterogeneity between anatomical subsites. Just over half (61) of 109 known and new risk variants showed no evidence for heterogeneity. In contrast, 22 variants showed association with distal CRC (including rectal cancer), but no evidence for association or an attenuated association with proximal CRC. For two loci, there was strong evidence for effects confined to proximal colon cancer.
Conclusion Genetic architectures of proximal and distal CRC are partly distinct. Studies of risk factors and mechanisms of carcinogenesis, and precision prevention strategies should take into consideration the anatomical subsite of the tumour.
- colorectal cancer
- genetic polymorphisms
- cancer genetics
- cancer susceptibility
- colon carcinogenesis
Data availability statement
Data are available in a public controlled access repository. All genotype data analyzed in this study have been previously published and have been deposited in the database of Genotypes and Phenotypes (dbGaP), which is hosted by the National Center for Biotechnology Information (NCBI) of the US National Institutes of Health (NIH), under accession numbers phs001415.v1.p1, phs001315.v1.p1, phs001078.v1.p1, and phs001903.v1.p1. The UK Biobank resource was accessed through application number 8614. Bioinformatic analyses included public, open access colorectal epigenomic data that were retrieved from the NCBI Gene Expression Omnibus (GEO) database under accession numbers GSE77737 and GSE36401. For all above datasets embargo release dates have passed.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Heterogeneity among colorectal cancer (CRC) tumours originating at different locations of the colorectum has been revealed in somatic genomes, epigenomes and transcriptomes, and in some established environmental risk factors for CRC.
Genome-wide association studies (GWASs) have identified over 100 genetic variants for overall CRC risk; however, a comprehensive analysis of the extent to which genetic risk factors differ by the anatomical sublocation of the primary tumour is lacking.
What are the new findings?
In this large consortium-based study, we analysed clinical and genome-wide genotype data of 112 373 CRC cases and controls of European ancestry to comprehensively examine whether CRC case subgroups defined by anatomical sublocation have distinct germline genetic aetiologies.
We discovered 13 new loci at genome-wide significance (p<5×10−8) that were specific to certain anatomical sublocations and that were not reported by previous GWASs for overall CRC risk; multiple lines of evidence support strong candidate target genes at several of these loci, including PTGER3, LCT, MLH1, CDX1, KLF14, PYGL, BCL11B and BMP7.
Systematic heterogeneity analysis of genetic risk variants for CRC identified thus far, revealed that genetic architectures of proximal and distal CRC are partly distinct, and demonstrated that distal colon and rectal cancer have very similar germline genetic aetiologies.
Taken together, our results further support the idea that tumours arising in different anatomical sublocations of the colorectum may have distinct aetiologies.
How might it impact on clinical practice in the foreseeable future?
Our results provide an informative resource for understanding the differential role that genetic variants, genes and pathways may play in the mechanisms of proximal and distal CRC carcinogenesis.
The new insights into the aetiologies of proximal and distal CRC may inform the development of new precision prevention strategies, including individualised screening recommendations and the discovery of novel drug targets and repurposable drug candidates for chemoprevention.
Our findings suggest that future studies of aetiological risk factors for CRC and molecular mechanisms of carcinogenesis should take into consideration the anatomical sublocation of the colorectal tumour. In particular, our results argue against lumping proximal and distal colon cancer cases.
Introduction
Despite improvements in prevention, screening and therapy, colorectal cancer (CRC) remains one of the leading causes of cancer-related death worldwide, with an estimated 53 200 fatal cases in 2020 in the USA alone.1 CRCs that arise proximal (right) or distal (left) to the splenic flexure differ in age-specific and sex-specific incidence rates, clinical, pathological and tumour molecular features.2–5 These observed differences reflect a complex interplay between differential exposure of colorectal crypt cells to local environmental carcinogenic and protective factors in the luminal content (including the microbiome), and distinct inherent biological characteristics that may influence neoplasia risk, including sex and differences between anatomical segments in embryonic origin, development, physiology, function and mucosal immunology. The precise extrinsic and intrinsic aetiological factors involved, their relative contributions, and how they interact to influence the carcinogenic process remain largely elusive.
An individual’s genetic background plays an important role in the initiation and development of CRC. Based on twin registries, heritability is estimated to be around 35%.6 Since genome-wide association studies (GWASs) became possible just over a decade ago, over 100 independent common genetic variant associations for overall CRC risk have been identified, over half of which were identified in the past few years.7–10 Three decades ago, based on observed similarities between Lynch syndrome and proximal CRC, and between familial adenomatous polyposis and distal CRC, Bufill proposed the existence of two distinct genetic categories of CRC according to the location of the primary tumour.2 However, given that genetic variants that influence CRC risk typically have small effect sizes, until very recently, sample sizes did not provide adequate statistical power to conduct meaningful subsite analyses. As a consequence, GWASs to detect genetic associations specific to CRC case subgroups defined by primary tumour anatomic subsite have not been reported yet. Similarly, a comprehensive analysis of the extent to which allelic risk of known GWAS-identified variants differs by primary tumour anatomic subsite is lacking.
To address the major gap in our knowledge of the differential role that genetic variants, genes and pathways play in mechanisms of proximal and distal CRC carcinogenesis, we analysed clinical and genome-wide genotype data for 112 373 CRC cases and controls. First, to discover new loci and genetic risk variants with site-specific allelic effects, we conducted GWASs of case subgroups defined by the location of their primary tumour within the colorectum. Next, we systematically characterised heterogeneity of allelic effects between primary tumour subsites for new and previously identified CRC risk variants to identify loci with shared and site-specific allelic effects.
Methods
Detailed methods are provided in online supplemental materials.
Supplemental material
Samples and genotypes
This study included clinical and genotype data for 48 214 CRC cases and 64 159 controls from three consortia: Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), Colorectal Cancer Transdisciplinary Study (CORECT) and Colorectal Cancer Family Registry (CCFR). Online supplemental table 1 provides details on sample numbers and demographic characteristics by study. All study participants were of genetically inferred European-ancestry. Across studies, participant recruitment occurred between the early 1990s and the 2010s. Details of genotype data sets, genotype QC, sample selection and studies included in this analysis have been published previously.7 8 11 12 All participants provided written informed consent, and each study was approved by the relevant research ethics committee or institutional review board.
Supplemental material
Colorectal tumour anatomic sublocation definitions
We defined proximal colon cancer as any primary tumour arising in the cecum, ascending colon, hepatic flexure or transverse colon; distal colon cancer as any primary tumour arising in the splenic flexure, descending colon or sigmoid colon; and rectal cancer as any primary tumour arising in the rectum or rectosigmoid junction. For the GWAS discovery analyses, we analysed five case subgroups based on primary tumour sublocation. In addition to the three afore-mentioned mutually exclusive case sets (proximal colon, distal colon and rectal cancer), we defined colon cancer and distal/left-sided colorectal cancer case sets. Colon cancer cases comprised combined proximal colon and distal colon cancer cases, and additional colon cases with unspecified site. In the distal/left-sided colorectal cancer cases analysis, we combined distal colon and rectal cancer cases based on the different embryonic origins of the proximal colon versus the distal colon and rectum. Online supplemental figure 1 and table 1 summarise distributions of age of diagnosis by sex and primary tumour site.
Supplemental material
Statistical analysis
GWAS meta-analyses
We imputed all genotype datasets to the Haplotype Reference Consortium panel.13 In brief, we phased all genotyping array data sets using SHAPEIT214 and used the Michigan Imputation Server15 for imputation. Within each dataset, variants with an imputation accuracy r2≥0.3 and minor allele count ≥50 were tested for association with CRC case subgroup. Variants that only passed filters in a single dataset were excluded. We assumed an additive model using imputed genotype dosage in a logistic regression adjusted for age, sex and study or genotyping project-specific covariates, including principal components to adjust for population structure. Details of covariate corrections have been published previously.8 Because Wald tests can be anticonservative for rare variants, we performed likelihood ratio tests and combined association summary statistics across sample sets via fixed-effects meta-analysis employing Stouffer’s method, implemented in the METAL software.16 Reported p values are based on this analysis. Reported combined OR estimates and 95% CIs are based on an inverse variance-weighted fixed-effects meta-analysis.
Heterogeneity in allelic effect sizes between tumour anatomic sublocations
To characterise tumour subsite-specificity and effect size heterogeneity across tumour subsites for new loci, and for established loci for overall CRC, we examined association evidence in three different ways. First, for each index variant we created forest plots of OR estimates from GWAS meta-analyses for proximal colon, distal colon and rectal cancer. Second, we tested for heterogeneity using multinomial logistic regression. In brief, after pooling of datasets, we performed a likelihood ratio test comparing a model in which ORs for the risk variant were allowed to vary across tumour subsites, to a model in which ORs were constrained to be the same across tumour subsites. Third, inspired by reference,17 we used a multinomial logistic regression-based model selection approach to assess which configuration of tumour subsites is most likely to be associated with a given variant. For each variant, we defined and fitted 11 possible causal risk models specifying variant effect configurations that vary or are constrained to be equal among subsets of tumour subsites (online supplemental table 2). We then identified and report the best fitting model using the Bayesian information criterion (BIC). For each model i we calculated ∆BIC i =BIC i −BICmin, where BICmin is the BIC value for the best model. Models with ∆BIC i ≤2 were considered to have substantial support and indistinguishable from the best model.18 For these variants, we do not report a single best model. Analyses were carried out using the VGAM R package.19 The list of index variants for previously published CRC risk signals is based on Huyghe et al.8
Supplemental material
Genomic annotation of new GWAS loci and gene prioritisation
We annotated all new loci with five types of functional and regulatory genomic annotations: (i) cell-type-specific regulatory annotations for histone modifications and open chromatin, (ii) nonsynonymous coding variation, (iii) evidence of transcription factor binding, (iv) predicted functional impact across different databases, (v) colocalisation with expression quantitative trait loci (eQTL) signals. Genes were further prioritised based on biological relevance, colorectal tissue expression, presence of associated non-synonymous variants predicted to be deleterious, evidence from functional studies, somatic alterations or familial syndromes. Details are in online supplemental materials.
Results
The final analyses included data for 48 214 CRC cases and 64 159 controls of European ancestry. To discover new loci and genetic risk variants with site-specific allelic effects, we conducted five genome-wide association scans of case subgroups defined by the location of their primary tumour within the colorectum: proximal colon cancer (n=15 706), distal colon cancer (n=14 376), rectal cancer (n=16 212), colon cancer, in which we omitted rectal cancer cases (n=32 002), and distal/left-sided CRC, in which we combined distal colon and rectal cancer cases (n=30 588). Next, we systematically characterised heterogeneity of allelic effects between tumour subsites for new and previously identified CRC risk variants to identify loci with shared and site-specific allelic effects.
New colorectal cancer risk loci
Across the five CRC case subgroup GWAS meta-analyses, a total of 11 947 015 single nucleotide variants (SNVs) were analysed. Inspection of genomic control inflation factors and quantile–quantile plots of test statistics indicated no residual population stratification issues (online supplemental materials and figure 2). Across tumour subsites, we identified 13 loci that mapped outside regions previously implicated by GWASs for overall CRC risk (closest known locus 3.1 megabases away) and that reached genome-wide significance (p<5×10−8) in at least one of the meta-analyses (table 1, figure 1, online supplemental figures 3 and 4). Seven of the new loci passed a Bonferroni-adjusted genome-wide significance threshold correcting for five case subgroups analysed (table 1). All lead variants were well imputed (minimum average imputation r2=0.788), had minor allele frequency (MAF) >1%, and displayed no significant heterogeneity between sample sets (Cochran’s Q heterogeneity test p>0.05; table 1).
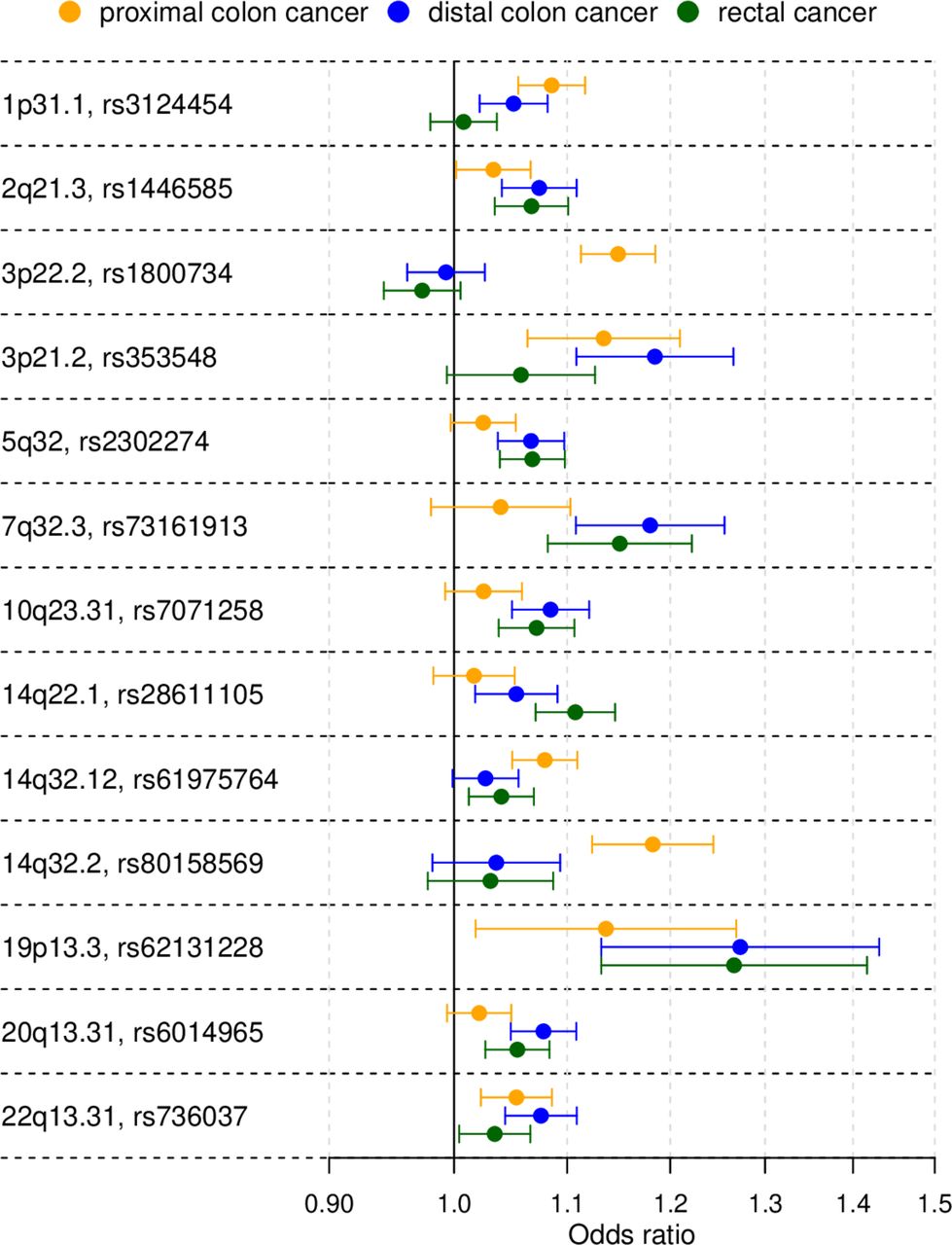
Primary tumour site-specific associations for the lead single nucleotide polymorphisms (SNPs) of the 13 colorectal cancer risk loci not reported in previous genome-wide association studies. The forest plot shows the (log-additive) OR estimates together with 95% CIs. For clarity, this figure only shows results for the proximal colon, distal colon and rectal cancer case subgroup analyses.
{kind=link}
{kind=link}
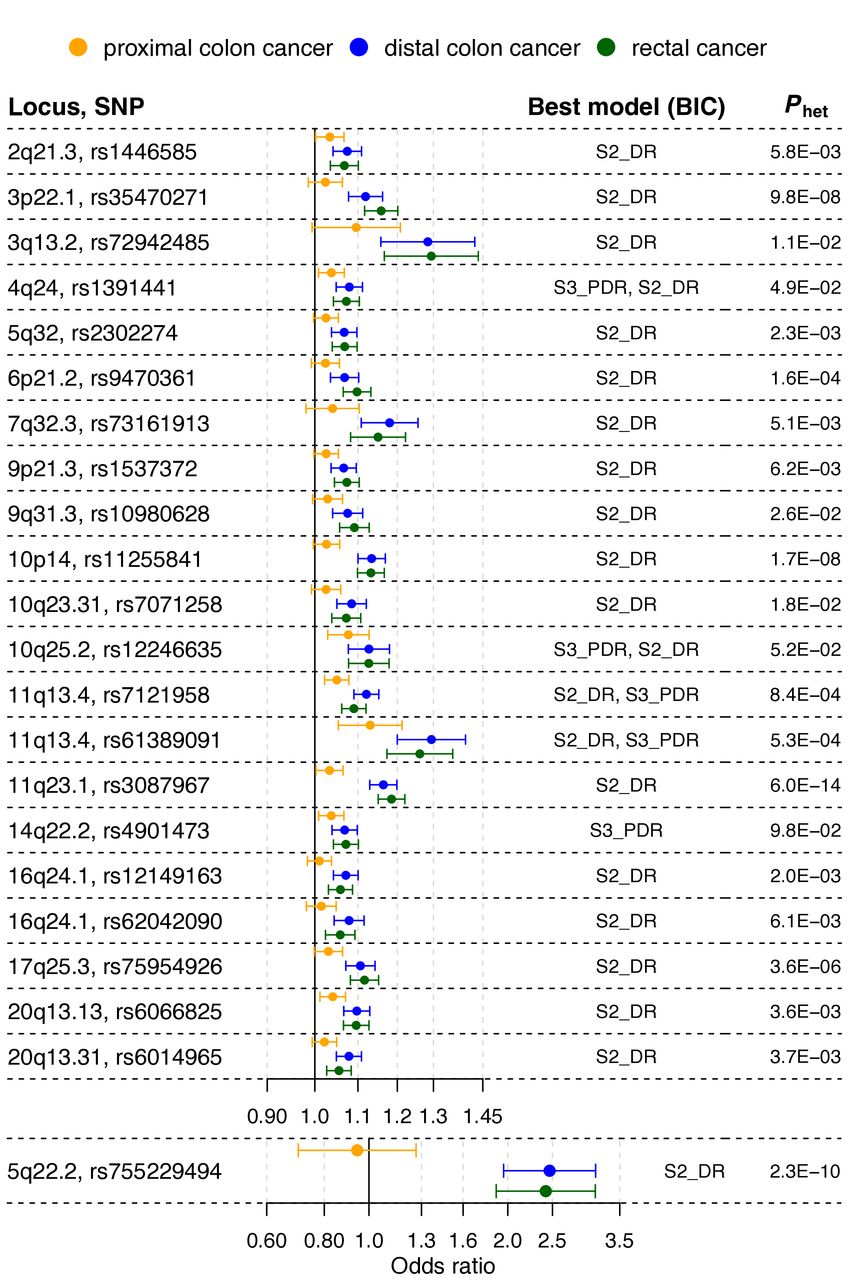
Loci showing association with risk of distal colorectal cancer (ie, distal colon+rectal), but attenuated or no evidence for association with proximal colon cancer risk. The forest plot shows the (log-additive) OR estimates for the lead single nucleotide polymorphisms (SNPs) at the loci, together with 95% CIs, from the genome-wide association study meta-analyses of case subgroups defined by primary tumour anatomical subsite for proximal colon, distal colon and rectal. Best model is the best-fitting multinomial logistic regression model according to the Bayesian information criterion (BIC). Models are defined in online supplemental table 2. Phet is the p value from a test for heterogeneity of allelic effects across tumour subsites.
New genome-wide significant colorectal cancer risk loci identified by genome-wide association analysis of case subgroups defined by primary tumour anatomic subsite
The novel associations showing the strongest statistical evidence were obtained for proximal colon cancer and mapped near MLH1 on 3p22.2 (rs1800734, p=3.8×10−18) and near BCL11B on 14q32.2 (rs80158569, p=8.6×10−11). These loci showed strongly proximal cancer-specific associations. The proximal colon analysis also yielded a locus on 14q32.12 (rs61975764, p=2.8×10−8) that showed attenuated effects for other tumour subsites (figure 1 and online supplemental table 3). Most new loci (six) were discovered in the left-sided CRC analysis: 2q21.3 (rs1446585, p=3.3×10−8), near CDX1 on 5q32 (rs2302274, p=4.9×10−9), near KLF14 on 7q32.3 (rs73161913, p=1.3×10−9), 10q23.31 (rs7071258, p=8.4×10−9), 19p13.3 (rs62131228, p=2.4×10−8) and near BMP7 on 20q13.31 (rs6014965, p=4.5×10−9). The rectal cancer analysis identified an additional locus near PYGL on 14q22.1 (rs28611105, p=4.7×10−9) that showed an attenuated effect for distal colon cancer (figure 1 and online supplemental table 3). No additional new loci were detected in the distal colon analysis. The colon cancer analysis identified three new loci: near PTGER3 on 1p31.1 (rs3124454, p=1.4×10−8), 3p21.2 (rs353548, p=1.3×10−8) and 22q13.31 (rs736037, p=2.8×10−8).
Genomic annotations and most likely target gene(s) at new loci
To gain insight into molecular mechanisms underlying new association signals, and to identify candidate causal variants and target gene(s), we annotated signals with functional and regulatory genomic annotations, assessed colocalisation with eQTLs, and performed literature-based gene prioritisation. Results for all new signals are given in online supplemental tables 4 and 5, and candidate target genes are also given in table 1. Notable and strong candidate target genes include PTGER3, LCT, MLH1, CDX1, KLF14, PYGL, RIN3, BCL11B and BMP7. Strong candidate causal variants were identified at loci 2q21.3 (rs4988235; LCT), 3p22.2 (rs1800734; MLH1), 14q32.12 (rs61975764; RIN3) and 14q32.3 (rs80158569; BCL11B). A detailed interpretation of candidate causal variants and target genes is deferred to the Discussion section.
Risk heterogeneity between tumour anatomical sublocations
Multinomial logistic regression modelling of 96 known and 13 newly identified risk variants showed the presence of substantial risk heterogeneity between cancer in the proximal colon, distal colon and rectum. For 61 variants, the heterogeneity p value (phet) was not significant (phet>0.05). For 51 of those variants, a multinomial model in which ORs were identical for the three cancer sites provided the best fit, and for 8 of the remaining 10 variants, this model did not significantly differ from the best fitting model (online supplemental tables 2, 3 and 7; figure 5).
Among the 109 known or new variants, 48 showed at least some evidence of heterogeneity with phet<0.05, and after Holm-Bonferroni correction for multiple testing, 14 variants showing strong evidence of heterogeneity remained significant (phet<4.6×10−4). These included 10 variants previously reported in GWASs for overall CRC risk.
For 17 out of the 48 variants with phet<0.05, the best-fitting model supported an effect limited to left-sided CRC (figure 2 and online supplemental tables 3 and 7). Of these 17 variants, 6 were in the list of variants with the strongest evidence of heterogeneity (phet<4.6×10−4), including the following previously reported loci: C11orf53-COLCA1-COLCA2 on 11q23.1 (phet=6.0×10−14), APC on 5q22.2 (phet=2.3×10−10), GATA3 on 10p14 (phet=1.7×10−8), CTNNB1 on 3p22.1 (phet=9.8×10−8), RAB40B-METRLN on 17q25.3 (phet=3.6×10−6) and CDKN1A on 6p21.2 (phet=1.6×10−4). Inspection of forest plots and association evidence also suggest stronger risk effects for left-sided tumours for the following additional five known loci: TET2 on 4q24, VTI1A on 10q25.2, two independent signals near POLD3 on 11q13.4, and BMP4 on 14q22.2.
For 5 out of the 49 variants with phet<0.05, a model with association with colon cancer risk, but no association with rectal cancer risk, provided the best fit (online supplemental tables 3 and 7). These involve the following loci: PTGER3 on 1p31.1, STAB1-TLR9 on 3p21.2, HLA-B-MICA/B-NFKBIL1-TNF on 6p21.33, NOS1 on 12q24.22 and LINC00673 on 17q24.3. Association evidence also suggests stronger risk effects for colon tumours for one of two independent signals near PTPN1 on 20q13.13.
Evidence from the three approaches (figure 1; online supplemental tables 3 and 7) indicates that only two loci are strongly proximal colon cancer-specific: MLH1 on 3p22.2 (phet=5.4×10−19), and BCL11B (phet=1.5×10−5) on 14q32.2. Finally, for only one variant, at one of two independent loci near SATB2 on 2q33.1, a model with a rectal cancer-specific association provided the best fit, but association evidence shows attenuated effects for proximal and distal colon cancer. OR estimates also suggest stronger risk effects for rectal cancer at the known loci LAMC1 on 1q25.3, and CTNNB1 on 3p22.1, and at new locus PYGL on 14q22.1.
Pathway enrichment analyses
To explore whether biological pathways play different roles in tumourigenesis of proximal and distal CRC, we conducted pathway enrichment analyses of GWAS summary statistics. There was no clear and strong evidence for differential involvement of pathways; pathways that were Bonferroni-significant for one anatomical subsite, reached at least suggestive significance levels for other subsites (online supplemental table 8). Several of the Bonferroni-significant pathways related to transforming growth factor β (TGFβ) signalling.
Discussion
It has long been recognised that CRCs arising in different anatomical segments of the colorectum differ in age-specific and sex-specific incidence rates, clinical, pathological and tumour molecular features. However, our understanding of the aetiological factors underlying these medically important differences has remained scarce. This study aimed to examine whether the contribution of common germline genetic variants to CRC carcinogenesis differs by anatomical sublocation. The large sample size comprising 112 373 cases and controls provided adequate statistical power to discover new loci and variants with risk effects limited to tumours for certain anatomical subsites, and to compare allelic effect sizes across anatomical subsites.
Our CRC case subgroup meta-analyses identified 13 additional genome-wide significant CRC risk loci that, due to substantial allelic effect heterogeneity between anatomical subsites, were not detected in larger, previously published GWASs for overall CRC risk.8 9 In fact, the only way to discover certain loci and risk variants with case subgroup-specific allelic effects is via analysis of homogeneous case subgroups.24 For example, p values for rs1800734 and rs80158569 were ~18 and~5 powers of 10, respectively, more significant in the proximal colon analysis compared with in our overall CRC analysis. While follow-up studies are needed to uncover the causal variant(s), biological mechanism and target gene, multiple lines of evidence support strong candidate target genes at many of the new loci, including genes MLH1, BCL11B, RIN3, CDX1, LCT, KLF14, BMP7, PYGL and PTGER3.
At the MLH1 gene promoter region on 3p22.2, associated to proximal colon cancer, previous studies have reported strong and robust associations between the common single nucleotide polymorphism (SNP) rs1800734, and CRC with high microsatellite instability (MSI-H).25 26 Rare deleterious nonsynonymous germline mutations in the DNA mismatch repair (MMR) gene MLH1 are a frequent cause of Lynch syndrome (OMIM #609310). The risk allele of the likely causal SNP rs1800734 is strongly associated with MLH1 promoter hypermethylation and loss of MLH1 protein in CRC tumours.26 The mechanisms of MLH1 promoter hypermethylation and subsequent gene silencing may account for most CRC tumours with defective DNA MMR and MSI-H.27
At the highly localised, proximal colon-specific association signal on 14q32.2, lead SNP rs80158569 is located in a colonic crypt enhancer and overlaps with multiple transcription factor binding sites, making it a strong candidate causal variant. Nearby gene BCL11B encodes a transcription factor that is required for normal T cell development,28 29 and that is a SWI/SNF complex subunit.30 BCL11B acts as a haploinsufficient tumour suppressor in T-cell acute lymphoblastic leukaemia.31 32 Experimental work suggests that impairment of Bcl11b promotes intestinal tumourigenesis in mice and humans through deregulation of the Wnt/β-catenin pathway.33
At locus 14q32.12, lead SNP rs61975764 showed the strongest association evidence in the proximal colon analysis and attenuated effects for other tumour locations. Genotype-Tissue Expression (GTEx) data show that rs61975764 is an eQTL for gene Ras and Rab interactor 3 (RIN3) in transverse colon tissue. RIN3 functions as a RAB5 and RAB31 guanine nucleotide exchange factor involved in endocytosis.34 35
At locus 5q32, associated with left-sided CRC, the intestine-specific transcription factor caudal-type homeobox 1 (CDX1) encodes a key regulator of differentiation of enterocytes in the normal intestine and of CRC cells. CDX1 is central to the capacity of colon cells to differentiate and promotes differentiation by repressing the polycomb complex protein BMI1 which promotes stemness and self-renewal. The repression of BMI1 is mediated by microRNA-215 which acts as a target of CDX1 to promote differentiation and inhibit stemness.36 CDX1 has been shown to inhibit human colon cancer cell proliferation by blocking β-catenin/T-cell factor transcriptional activity.37
In a region of extensive LD on locus 2q21.1, lead SNP rs1446585, associated with left-sided CRC, is in strong LD with functional SNP rs4988235 (LD r2=0.854) in the cis-regulatory element of the lactase (LCT) gene. In Europeans, the rs4988235 genotype determines the lactase persistence phenotype, or the ability to digest lactose in adulthood. The p value for functional SNP rs4988235 under an additive model was 7.0×10−7. The allele determining lactase persistence (T) is associated with decreased CRC risk. This is consistent with a previously reported association between low lactase activity defined by the CC genotype and CRC risk in the Finnish population.38 The protective effect conferred by the lactase persistence genotype is likely mediated by dairy products and calcium which are known protective factors for CRC.39 When we tested for association with left-sided CRC assuming a dominant model, associations for rs1446585 and rs4988235 became more significant with p values of 4.4×10−11 and 1.4×10−9, respectively. For functional SNP rs4988235, the OR estimate for having genotype CC versus CT or TT, and left-sided CRC was 1.14 (95% CI 1.09 to 1.19). Because this region has been under strong selection, it is particularly prone to population stratification.40 However, we adjusted for genotype principal components, and the association showed a consistent direction of effect across sample sets (online supplemental table 6), suggesting this association is not spurious.
Candidate genes at left-sided CRC loci 7q32.2 and 20q13.31 are involved in TGFβ signalling. At 7q32.3, gene Krüppel-like factor 14 (KLF14) is a strong candidate. We previously reported loci at known CRC oncogene KLF5 and at KLF2.8 The imprinted gene KLF14 shows monoallelic maternal expression, and is induced by TGFβ to transcriptionally corepress the TGFβ receptor 2 (TGFBR2) gene.41 A cis-eQTL for KLF14, uncorrelated with our lead SNP rs73161913, acts as a master regulator related to multiple metabolic phenotypes,42 43 and a nearby independent variant is associated to basal cell carcinoma.44 For both reported associations, effects depended on parent-of-origin of risk alleles. The association with metabolic phenotypes also depended on sex. We did not find evidence for strong sex-dependent effects (men: OR=1.13, 95% CI 1.07 to 1.20; women: OR=1.17, 95% CI 1.09 to 1.25). Further investigation is warranted to analyse parent-of-origin effects. At 20q13.31, gene bone morphogenetic protein 7 (BMP7) is a strong candidate. BMP7 signalling in TGFBR2-deficient stromal cells promotes epithelial carcinogenesis through SMAD4-mediated signalling.45 In CRC tumours, BMP7 expression correlates with parameters of pathological aggressiveness such as liver metastasis and poor prognosis.46
On 14q22.1, the single locus identified only in the rectal cancer analysis, GTEx data show that, in gastrointestinal tissues, lead SNP rs28611105 colocalises with a cis-eQTL coregulating expression of genes PYGL, ABHD12B and NIN. We reported an association between genetically predicted glycogen phosphorylase L (PYGL) expression and CRC risk in a transcriptome-wide association study.47 This glycogen metabolism gene plays an important role in sustaining proliferation and preventing premature senescence in hypoxic cancer cells.48
At 1p31.1, identified in the colon cancer analysis, PTGER3 encodes prostaglandin E receptor 3, a receptor for prostaglandin E2 (PGE2), a potent pro-inflammatory metabolite biosynthesised by cyclooxygenase-2 (COX-2). COX-2 plays a critical role in mediating inflammatory responses that lead to epithelial malignancies. The anti-inflammatory activity of non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen operates mainly through COX-2 inhibition, and long-term NSAID use decreases CRC incidence and mortality.49 PGE2 is required for the activation of β-catenin by Wnt in stem cells,50 and promotes colon cancer cell growth.51 PTGER3 plays an important role in suppression of cell growth and its downregulation was shown to enhance colon carcinogenesis.52
Previous CRC GWASs had already reported allelic effect heterogeneity between tumour sites, including for 10p14, 11q23 and 18q21 but only contrasted colon and rectal tumours, without distinguishing between proximal and distal colon.53 54 Sample size and timing of the present study enabled systematic characterisation of allelic effect heterogeneity between more refined tumour anatomical sublocations, and for a much expanded catalogue of risk variants. Our analysis revealed substantial, previously unappreciated allelic effect heterogeneity between proximal and distal CRC. Results further show that distal colon and rectal cancer have very similar germline genetic aetiologies. Our findings at several loci are consistent with CRC tumour molecular studies. Consensus molecular subtypes (CMSs), which are based on tumour gene expression, are differentially distributed between proximal and distal CRCs. The canonical CMS (CMS2) is enriched in distal CRC (56% vs 26% for proximal CRC) and is characterised by upregulation of Wnt downstream targets.55 We found that variant associations near Wnt/β-catenin pathway genes APC and CTNNB1 were confined to distal CRC. We also found that associations for variants near genes BOC and FOXL1, members of the Hedgehog signalling pathway, were confined to distal CRC, suggesting that Wnt and Hedgehog signalling may contribute more to the development of distal CRC tumours. However, pathway enrichment analyses did not provide clear evidence for differential involvement of pathways, suggesting perhaps that associations for proximal and distal CRC mostly converge on the same pathways. Pathway analysis results should, however, be interpreted taking into consideration the limitations of available approaches. Genetic variants were mapped to the nearest gene which is often not the target gene.
The precise intrinsic or extrinsic effect modifiers explaining observed allelic effect heterogeneity between anatomical subsites remain unknown and further research is needed. Short-chain fatty acids, in particular butyrate, produced by microbiota through fermentation of dietary fibre in the colon may be involved. Concentrations of butyrate, which plays a multifaceted antitumorigenic role in maintaining gut homoeostasis, are much higher in proximal colon.56 Moreover, the known chemopreventive role of butyrate may involve modulation of signalling pathways including TGFβ and Wnt.57 This may contribute to possible differences between anatomical segments in colorectal crypt cellular dynamics.
One limitation of our study is that we have not performed GWAS analyses of case subgroups based on more detailed anatomical sublocations. However, given current sample size, such analyses would result in reduced statistical power owing to reduced sample sizes and the aggravated multiple testing burden. As another limitation, our study was based on European-ancestry subjects and it remains to be determined whether findings are generalisable to other ancestries.
In conclusion, germline genetic data support the idea that proximal and distal colorectal cancer have partly distinct aetiologies. Our results further demonstrate that distal colon and rectal cancer have very similar germline genetic aetiologies and argue against lumping proximal and distal colon cancer in studies of aetiological factors. Future genetic studies should take into consideration differences between primary tumour anatomical subsites. A better understanding of differing carcinogenic mechanisms and neoplastic transformation risk in proximal and distal colorectum can inform the development of novel precision treatment and prevention strategies through the discovery of novel drug targets and repurposable drug candidates for treatment and chemoprevention, and improved individualised screening recommendations based on risk prediction models incorporating tumour anatomical subsite.
Data availability statement
Data are available in a public controlled access repository. All genotype data analyzed in this study have been previously published and have been deposited in the database of Genotypes and Phenotypes (dbGaP), which is hosted by the National Center for Biotechnology Information (NCBI) of the US National Institutes of Health (NIH), under accession numbers phs001415.v1.p1, phs001315.v1.p1, phs001078.v1.p1, and phs001903.v1.p1. The UK Biobank resource was accessed through application number 8614. Bioinformatic analyses included public, open access colorectal epigenomic data that were retrieved from the NCBI Gene Expression Omnibus (GEO) database under accession numbers GSE77737 and GSE36401. For all above datasets embargo release dates have passed.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @dan_buchanan, @scastellvibel, @mazda_j
Deceased Albert de la Chapelle is deceased.
Contributors JRH, TAH, SAB, HH, JCF, SLS, DVC, JAB, AJC, BD, DD, SH, LI, VP, AP-C, LCS, FRS, MLS, AET, FJBvD, BVG, AA, DA, MHA, KA, CA-C, VA, SIB, SB, DTB, JB, HBoeing, M-CB-R, HBrenner, SBrezina, SBuch, DDB, AB-H, BJC, PTC, PC, AC, SC-B, ATC, JC-C, SJC, AdlC, DFE, DRE, EJMF, MG, SJG, WJG, GGG, PJG, WMG, JSG, AG, MJG, RWH, JH, MH, JLH, W-YH, TJH, MJ, MAJ, ADJ, TOK, CK, TK, SK, LLM, FL, CIL, LL, WL, AL, NML, SM, SDM, RLM, LM, NM, RN, KO, SO, SP, PSP, RP, PDPP, AIP, EAP, JDP, RLP, LQ, LR, GR, HSR, ER, CS, RES, DS, MS, CMT, SNT, DCT, AT, CMU, KV, PV, LV, VV, KW, SJW, EW, AW, MOW, AHW, GRA, DAN, PCS, AK, GC, SBG, LH, VM, RBH, PAN and UP conceived and designed the study. JRH, TAH, SAB, SLS, DVC, SC, CQ, YL, RB, HMK, DML, FRS, BB, KRC, W-LH, Y-RS, AK, LH and UP analysed the data. JRH, TAH, HH, JCF, JAB, AJC, BD, SH, LI, HMK, VP, AP-C, LCS, MLS, AET, FJBvD, BVG, AA, DA, MHA, KA, CA-C, VA, MCB, SIB, SB, DTB, JB, HBoeing, M-CB-R, HBrenner, SBrezina, SBuch, DDB, AB-H, BJC, PTC, PC, AC, SC-B, ATC, JC-C, SJC, AdlC, DFE, DRE, EJMF, MG, SJG, WJG, GGG, PJG, WMG, JSG, AG, MJG, RWH, JH, MH, JLH, W-LH, W-YH, TJH, MJ, MAJ, ADJ, TOK, CK, TK, SK, LLM, FL, CIL, LL, WL, AL, NML, SM, SDM, RLM, LM, NM, RN, KO, SO, SP, PSP, RP, PDPP, AIP, EAP, JDP, RLP, LQ, LR, GR, HSR, ER, CS, RES, MS, Y-RS, CMT, SNT, DCT, AT, CMU, KV, PV, LV, VM, KW, SJW, EW, AW, MOW, AHW, GRA, DAN, PCS, AK, GC, SBG, VM, RBH, PAN and UP contributed reagents/materials/analysis tools. JRH, TH and UP wrote the first draft. All authors reviewed the manuscript for intellectual content and approved the final version of the manuscript. UP supervised the study.
Funding This work was supported by grants from the National Cancer Institute (NCI), National Institutes of Health (NIH), US Department of Health and Human Services (U01 CA164930, U01 CA137088, R01 CA059045, R21 CA191312, R01 CA201407, P30 CA015704). Genotyping services were provided by the Center for Inherited Disease Research (CIDR; X01-HG008596 and X01-HG007585). CIDR is fully funded through a federal contract from the NIH to the Johns Hopkins University, contract HHSN268201200008I. The full list of funding and acknowledgements can be found in the supplemental file.
Disclaimer Where authors are identified as personnel of the International Agency for Research on Cancer/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/WHO.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.