Article Text

Abstract
Non-alcoholic fatty liver disease (NAFLD) has become the most common cause of chronic liver disease worldwide. Understanding the pathological and molecular hallmarks from its first description to definitions of disease entities, classifications and molecular phenotypes is crucial for both appropriate clinical management and research in this complex disease. We provide an overview through almost two hundred years of clinical research from the beginnings as a nebulous disease entity of unknown origin in the 19th century to the most frequent and vigorously investigated liver disease today. The clinical discrimination between alcohol-related liver disease and NAFLD was uncommon until the 1950s and likely contributed to the late acceptance of NAFLD as a metabolic disease entity for long time. Although the term ‘fatty liver hepatitis’ first appeared in 1962, it was in 1980 that the term ‘non-alcoholic steatohepatitis’ (NASH) was coined and the histopathological hallmarks that are still valid today were defined. The 2005 NASH Clinical Research Network scoring was the first globally accepted grading and staging system for the full spectrum of NAFLD and is still used to semiquantify main histological features. In 2021, liver biopsy remains the only diagnostic procedure that can reliably assess the presence of NASH and early fibrosis but increasing efforts are made towards non-invasive testing and molecular classification of NAFLD subtypes.
- fatty liver
- fibrosis
- genetics
- obesity
- molecular pathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key message
Obesity has been noticed in antiquity but fatty liver as a disease entity was unknown until the early 19th century.
A causal relationship between hepatic fat accumulation and the development of fibrosis has been observed in 1839 followed by the microscopic finding that ‘fatty degeneration’ develops close to inflammatory deposits and scarring.
The clinical discrimination between alcohol-related and non-alcoholic fatty liver disease (NAFLD) was uncommon until the 1950s. After the introduction of percutaneous biopsies, the association of fatty liver disease with diabetes mellitus became increasingly apparent.
The term ‘fatty liver hepatitis’ first appeared in 1962 in the German literature while the term ‘non-alcoholic steatohepatitis’ (NASH) was coined in 1980, defined by the histopathological hallmarks of steatosis, lobular inflammation, liver cell damage with ballooning and, eventually, appearance of Mallory-Denk bodies.
Grading and staging systems for steatosis and fibrosis emerged in the 1950s but the NASH Clinical Research Network score in 2005 was the first globally accepted histological scoring system that addressed the full spectrum of NAFLD.
Liver biopsy still remains the only diagnostic procedure that can reliably assess the presence of NASH and early fibrosis but increasing efforts are made towards non-invasive testing and molecular classification of disease subtypes.
Introduction
Non-alcoholic fatty liver disease (NAFLD) comprises a wide spectrum of liver damage, ranging from NAFL (simple steatosis; NAFL), to non-alcoholic steatohepatitis (NASH) with inflammation and hepatocyte injury to advanced fibrosis and cirrhosis.1 Although NAFLD histopathology resembles that of alcohol-related liver disease (ALD), the clinical background is different. NAFLD is regarded as one component of the metabolic syndrome, including obesity, insulin resistance or type 2 diabetes mellitus, hypertension and dyslipidaemia. A ‘multiple parallel hits’ hypothesis describes the pathogenesis of this complex disease from simple steatosis to steatohepatitis.2 3 NAFLD affects 10%–24% of the general population worldwide and is the most common cause of chronic liver disease.4
The worldwide number of obese patients tripled from 1975 to 2018.5 The introduction of industrialised food rich in high fructose corn syrup (HFCS) in the late 1960s coincided with the increase in the incidence of obesity, diabetes and metabolic syndrome. Bray et al 6 first hypothesised that HFCS may be directly associated with obesity.6 However, carefully controlled long-term studies are still required to substantiate the aetiological role of HFCS in metabolic syndrome-related diseases, such as NAFLD.7 Given the sheer frequency of patients with obesity, metabolic syndrome and NAFLD, it is remarkable that this disease entity has been overlooked by most clinicians for a long time, except for few pioneers, mostly pathologists, who first described the clinical and pathological characteristics more than 150 years ago. Even as recently as 30 years ago, NASH was familiar only to a small group of experts and the term did not exist in the medical vocabulary until 1980. Today, the lack of awareness of NAFLD by the clinical community has contributed to a lack of NASH-specific drugs and reliable biomarkers. The time has come to take a journey through almost 200 years of clinical research from the beginnings of NASH as a nebulous disease entity of unknown origin in the 19th century to the most frequent and vigorously investigated liver disease today.
Before autopsy
Obesity has been present in humans, putatively, since the European upper Palaeolithic age. This is illustrated by sculptures discovered across Europe, which are traditionally referred to as ‘Venus figurines’. The most prominent example is the Venus of Willendorf (30 000 BC) found in the Austrian Danube valley (figure 1A).8 In the Neolithic age (7000–3000 BC), clay obese female figurines were still popular in the Mediterranean area (figure 1B).9 Similarly, in ancient Egypt, parts of sub-Saharan Africa, China and south Pacific islands, obesity has been considered a sign of success, prosperity and good health, and in women implied fertility.10

Venus figurines of obese women. (A) Venus of Willendorf. The figurine is estimated to have been created around 30 000 BC in the Austrian Danube Valley near Krems and is displayed at the Museum of Natural History, Vienna, Austria (Natural History Museum Vienna, with permission) (B) Neolithic Venus figurine. Clay naturalistic figurine of a seated obese woman from Farsala, Thessaly (Athanassakeion archaeological museum of Volos, Greece with permission of the Hellenic Ministry of Culture and Sports/Archaeological Resources Fund) middle Neolithic period 5800–5300 BC.9
In antiquity, obesity was first regarded as a pathological state associated with feasts and inactivity by the Indian physician Susruta (circa sixth century BC). Obesity, its comorbidities and their treatment have been described in Ayurvedic classic texts.11 In Europe, Hippocrates of Kos (460–377 BC), recognised the principal importance of healthy nutrition.12 13 Later, Galen of Pergamon (129–216 AD) described obesity as an illness he termed ‘polysarcia’.13 14 Although Galen’s ideas concerning the pathophysiology of obesity are obsolete, some of his treatments such as diet and exercise are relevant today.14
Age of clinical diagnosis and autopsy: term ‘steatosis’
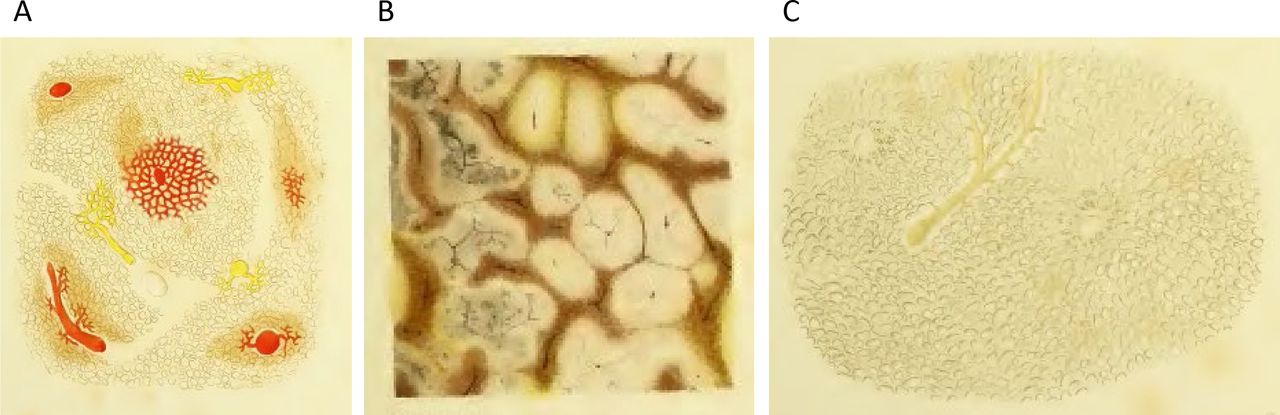
Fatty liver disease was not recognised as a disease entity until the early 19th century. The first autopsy series revealed that hepatic steatosis affected one third of the French and German populations with female preponderance.15–17 The highest frequency of hepatic steatosis in the 1800s was observed in patients affected by tuberculosis.15 17–19 In 1825, the first edition of Louis’ celebrated anatomy and pathology textbook contained the earliest use of the term ‘foie gras’ (fatty liver).15 20 Fatty changes in cirrhosis were noticed as early as 1836 by Addison who introduced the term ‘fatty liver’ in the English medical vocabulary.21 At that time, deposition of free adipose tissue into the liver parenchyma, omentum and mesenteries was regarded as the aetiology of hepatic steatosis.19 The causal relationship between hepatic fat accumulation and fibrosis development was first recognised by the Viennese pathologist Rokitansky in 1839.22
In 1856, Frerichs distinguished between ‘a liver merely abounding in fat’ and ‘one which has undergone fatty degeneration’.17 The general reversibility of hepatic steatosis after diet changes was also recognised.17 In addition, progression to terminal stages with ‘granular’ (cirrhotic) liver, ascites and splenomegaly was observed and liver failure was termed ‘foie inactive’.18 23
Intriguingly, it was already recognised by Frerichs that microscopic liver ‘fatty degeneration’ occurs close to inflammatory infiltrates and fibrosis (figure 2).17 In contrast, ‘simple deposition of fat in cells which were not altered in their other characters’ was defined as ‘fatty infiltration’. This distinction resembles the differentiation between bland steatosis (NAFL) and progressive fatty liver disease (NASH and beyond) used today. By the end of the 19th century, hepatitis had been accepted as ‘the second stage’ thus representing the early recognition of a disease spectrum in NAFLD.24

Historical pathology specimen of a fatty liver from T. Frerichs, Atlas of Pathological Anatomy (Part I, 1861) (A) ‘fatty liver of an advanced grade’. Massive steatosis sparing the vincinity of the central veins; (B) cut surface with pale-yellow parenchyma; (C) after removal of the fat by boiling in ether, only meshes formed by the vessels remain.
Frerichs described ‘distended cells’17 while Lereboullet25 coined the term ‘cloudy swelling’.26 The morphological description is similar to what is now regarded as hepatocellular ballooning. Grading of steatosis, assessed as percentage of affected hepatocytes, was established by Frerichs.17
These histological observations were made on autopsy material. A clinical diagnosis could only be made in vivo by palpation of a rounded margin of the enlarged liver.16 The clinical association of fatty liver with diabetes and obesity were first made by Pepper27 and Bartolow,28 respectively.
Pathological classification and clinical descriptions of the disease: first biopsy series
Most early reports on fatty liver disease came from pathologists.29 Hanssen, a clinician at the Steno Memorial Hospital in Copenhagen, Denmark described hepatomegaly due to fat accumulation in diabetic patients30 but it was Connor, a pathologist at the University of California San Francisco, USA who in 1938 described the histopathological features of fatty liver disease in diabetics and its association with development of cirrhosis indicating for the first time an aetiological link31 and put the stage for many other studies. In 1950s, possible link(s) of fatty liver with morbid obesity were discussed.32 33
Liver biopsies for the assessment of fatty liver disease were rarely performed before 1950 and often interpreted by clinicians themselves. Serial biopsies for the assessment of fatty liver started in the early 1950s and documented the potentially progressive nature of the disease.34–38 The ‘one second biopsy of the liver’ introduced by Menghini in the late 1950s contributed to the rapid dissemination of percutaneous biopsy as a routine procedure.39
During this time, the association of fatty liver disease with diabetes mellitus became increasingly apparent and most biopsy series included diabetic patients.40 However, discrimination between ALD and NAFLD was uncommon and many investigators analysed mixed-patient cohorts.38
Notably, the grading and staging systems for steatosis and fibrosis in the 1950s differed from current standards. In the 1950s, livers were considered steatotic when more than 10% (today 5%) of the parenchymal cells were fat-ladden. An accepted grading scale extended from grade 1 (10% steatosis) to grade 4 (approaching 100%).41 Prevalence rates of steatosis were around 60% in diabetic subjects42–44 compared with between 18% and 36% among patients with non-diabetes.42 43
There was a major discrepancy in the literature regarding the incidence of advanced fibrosis and cirrhosis and the terms ‘cirrhosis’ or ‘portal cirrhosis’ were used as synonyms for advanced fibrosis. The fibrosis staging scale ranged from 1, denoting increased periportal connective tissue extending into the lobules, up to stage 4 with ‘severe distortion of lobular architecture’. Popper suggested stages A–D similar to current fibrosis staging systems.38 He concluded that subdivision by septa (stage B beginning, stage C connecting) is the basic mechanism of cirrhosis formation.
From the turn of the century until 1955, general autopsy series in the United States revealed a continuous increase in the prevalence of cirrhosis from 13% to approximately 20%, with the majority representing the ‘fatty nutritional type’ which included both alcoholic and NAFL.44 The prevalence of cirrhosis in diabetic autopsy series in the 1930s to 1950s ranged from 12.7% to as high as 44% and indicated that advanced fibrosis was frequent in poorly controlled patients with diabetes.40 42 45 In parallel to these observations, a continuous increase of primary liver carcinoma was noticed in ‘fatty nutritional cirrhosis’ thus completing the full spectrum of NAFLD as it is appreciated today.44
Regarding pathophysiology of fatty liver disease, mitochondriopathy was first described in 1952 in a French series of liver biopsies.37 As a pathological correlate to mitochondrial dysfunction, the authors observed mitochondrial degeneration and rarefication in progressive disease. Megamitochondria within hepatocytes in NASH are currently regarded as morphological equivalents of mitochondrial dysfunction; indeed, NAFLD and NASH can be considered as mitochondrial diseases in view of important function impairment of mitochondria and their role as reactive oxygen species (ROS) sources.46 47
Attempts to correlate clinico-chemical test results with liver histology failed, with particularly poor results for early disease stages.35 40 43 48 Frequently, before routine liver transaminase testing was introduced, even the combination of four different liver function tests was normal.42
Definition of NASH: the emergence of ‘steatohepatitis’
With the emergence of overnutrition after World War II and increasing clinical relevance of obesity and related comorbidity, attention was finally directed to NAFLD as a part of the metabolic syndrome.49 50 Over time it became increasingly clear that the underlying pathophysiology of NAFLD was fundamentally different from alcoholic steatosis. Thaler in Vienna, Austria showed that steatosis affected half of obese subjects, whereas the frequency in a random series of 10 900 liver biopsies was 26.5%.51 The degree of steatosis in diabetic patients correlated with the extent of obesity.52 Although earlier studies had shown an increased prevalence of cirrhosis in diabetics,41 53 it was Thaler who clearly noted ‘a cirrhotogenic role of diabetes mellitus’.50
The term ‘fatty liver hepatitis’ as a surrogate of ‘steatohepatitis’ first appeared in 1962 in the German literature to describe fatty liver with necroinflammation.49 In this seminal paper, Thaler described the presence of inflammatory infiltrates after exclusion of concurrent diseases, which could be responsible for the pathological picture. His early reports on steatohepatitis included separate series of both alcohol-related and non-alcoholic aetiology, highlighting the similarity in histopathological appearance but differences in their clinical course. Even today, we struggle with a substantial overlap in real life, as reflected by the emergence of new acronyms, such as BASH and BAFLD for ‘both’ ALD and NAFLD.
Histopathological studies in obese or patients with diabetes with symptomatic liver disease28 54–70 documented lesions already known in ALD that are now diagnosed as NAFL/NASH.71
Despite considerable similarities, NAFLD/NASH and ALD differ in certain morphological aspects. For example, central veno-occlusive lesions are usually absent in NAFLD/NASH in contrast to ALD, and the presence of abundant, large, well-formed, Mallory-Denk bodies (MDBs) with surrounding neutrophil infiltration (satellitosis) points to an alcohol-related aetiology.72 73 Previously, others generated the hypothesis that the observed inflammatory changes could be caused by other pathogenetic influences.35 Thaler recognised that inflammation evolved independently of the degree of fatty change and he first used the term ‘non-alcoholic’.74 75 Over time, the term ‘fatty liver hepatitis’ gained broader acceptance.54
The term ‘NASH’ in the English language was coined by Jurgen Ludwig from the Mayo Clinic, Rochester, Minnesota, USA in 1980, who defined the ‘hitherto unnamed liver disease that histologically mimics alcoholic hepatitis and that also may progress to cirrhosis’.76 However, the term ‘non-alcoholic’ was already in use by European and Japanese authors to describe patients with this disease.63 77 The Mayo team described the histopathological hallmarks characterised by the presence of lobular hepatitis, focal necrosis with mixed inflammatory infiltrates, and in most instances MDBs and fibrosis. For the first time, the disease had been clearly linked to a pathophysiological scenario of obesity and comorbidities such as diabetes mellitus. In 1988, Thaler added the term ‘ballooning’ for what had previously been described as ‘cloudy swelling’ of damaged hepatocytes to complete the NASH criteria of today’s scoring systems.75 Still, in 1990, both terms ‘fatty liver hepatitis’ and ‘steatohepatitis’ were used.78 These descriptions triggered an exponential growth of research on NASH since 1980 (figure 3). Recently, the term metabolic dysfunction-associated fatty liver disease (MAFLD) has been proposed to emphasise this association but unfortunately it may also cover other forms of metabolic liver disease with steatosis.1

Numbers of annual PubMed/MEDLINE references for the term ‘NASH’ from 1980 to 2019. References were identified for the search term ‘NASH’ (excluding author names), accessed on 8 April 2020. NASH, non-alcoholic steatohepatitis.
NASH is a progressive disorder characterised by steatosis, hepatocyte damage, lobular inflammation and fibrosis with centrilobular (zone 3) pattern of injury in adult patients (figure 4). In paediatric NASH, portal predominance of the characteristic lesions or mixed forms are more frequent than the zone 3 pattern which is commoner in older children.79 Together with lobular infiltration by neutrophils and/or mononuclear cells, enlarged ‘ballooned’ hepatocytes with lightly stained, rarified cytoplasm and often a hyperchromatic nucleus with enlarged nucleolus are a diagnostically decisive feature of NASH; they often, but not always, contain irregular eosinophilic cytoplasmic inclusions resembling MDBs.80 81 MDBs are, however, not specific for NASH and can occur in other chronic liver diseases, such as chronic cholestatic and metabolic disorders, and in hepatocellular carcinoma (HCC).82 83 MDBs, originally described in 1911 by Mallory, a pathologist at Boston City Hospital, MA, USA in alcoholic cirrhosis,84 display an irregular filamentous ultrastructure and consist of partially degraded, misfolded and cross-linked keratins (particularly keratin 8), ubiquitin, stress and adaptor protein sequestosome1/p62, and heat-shock proteins, as revealed by chemical and immunohistochemical analyses.81–83 Diminution or even disappearance of the keratin intermediate filament cytoskeleton, as demonstrated by immunohistochemistry, is a characteristic feature of ballooned hepatocytes and may compromise cell stability and intracellular organisation; impaired secretory capacity and retention of proteins and fluid may also be involved.82 85 Disturbance of the keratin system adversely affects hepatocyte viability and function, since keratins are regarded as ‘guardians of the cell’. Morphologically, ballooned hepatocytes resemble stress-induced (oxidative) senescent cells with abnormal synthesis of stress proteins (keratins, heat shock protein, p62 and others) as adaptation and survival strategies. They may contribute to inflammation and fibrosis.83 86
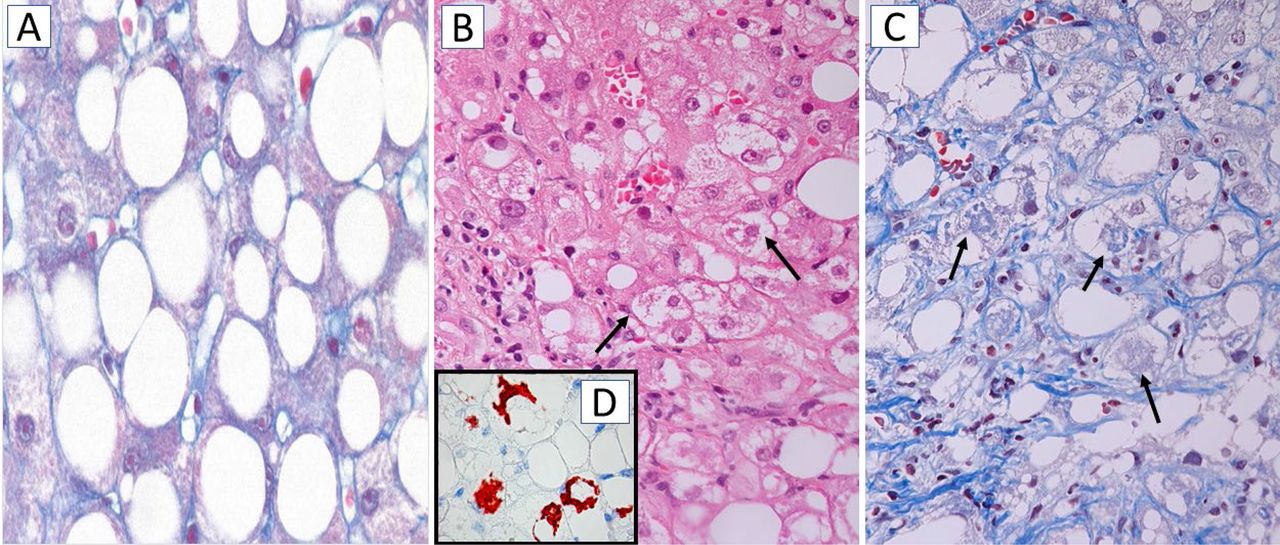
Histology of steatosis and non-alcoholic steatohepatitis (NASH). (A) Macrovesicular steatosis. The hepatocytes are distended by large fat droplets; chromotrope-aniline blue, x200. (B) NASH characterised by ballooned hepatocytes with lightly stained cytoplasm, some of them containing indistinct cytoplasmic inclusions resembling Mallory-Denk bodies (MDBs; arrows); H&E stain, x200. (C) NASH with ballooned hepatocytes, some of them containing MDBs (arrows) most hepatocytes are surrounded by pericellular fibrosis (blue RIMs around hepatocytes); chromotrope-aniline blue stain x200. (D, inset) Immunohistochemistry using antibodies to sequestosome1/p62 reveals p62-containing MDBs (red), x200.
By the 1990s, the field was prepared to further study the clinical course of this newly named, old disease. It soon became clear that NASH is not just a disease in obese women with diabetes.87 Bacon et al at Saint Louis University Hospital, Missouri, USA described a substantial proportion of males and non-obese patients with normal glucose and lipid levels in their cohort. This report emphasised ‘the need to consider NASH as a definite diagnostic possibility in an expanded group of patients’.87 While earlier reports described this disease entity as a rather benign and only slowly progressive disorder,88 89 the progressive nature of NASH had become clear from combined analyses of cohorts, where 43% of patients developed fibrosis.87–89 These observations were then challenged by Day’s group at Newcastle University, UK, who observed normal liver enzymes in the majority NAFLD patients and no progression to cirrhosis or liver-related death at a median follow-up of 11 years.90 This challenge was further supported by observations in a cohort of 1515 morbidly obese patients where cirrhosis prevalence was only 3% despite an 80% steatosis rate.91 By the end of the 1990s, pathophysiological studies in rodents and humans supported the importance of specific events like oxidative stress and endotoxin-mediated cytokine release triggering the development of necroinflammation in NASH patients.92 Still, a likely contributor to the confusion was due to the term ‘NASH’ being frequently used not in the specific context of histopathological steatohepatitis.93
As liver enzymes were introduced into routine automated testing platforms, investigating apparently healthy patients with abnormal liver blood tests became a relevant issue.90 Two distinct groups of adult NAFLD patients emerged: those with simple fatty liver who had an excellent prognosis and those with progressive NASH and often fibrosis.93 Poorer outcomes were observed more frequently in patients with ballooning, MDBs or fibrosis.94 Matteoni et al from the Mayo Clinic showed that more than 20% of patients with these lesions together with inflammation developed cirrhosis over a 10-year follow-up in contrast to only 4% of those with simple steatosis.94
In early 2000, the first studies on paediatric NAFLD documented progression to cirrhosis in 3% of the cases.95 96 Most cases are aetiologically related to the metabolic syndrome but some are due to inherited syndromes characterised by obesity and insulin resistance.71 97 The first histological autopsy study, conducted at the University of California San Diego, USA in 2006, showed a 9.6% prevalence of fatty liver in children.98
Despite the emerging awareness among specialised liver centres, surprisingly little attention was being paid to NASH in regular clinical practice.93 One ongoing challenge is the lack of a predictable correlation between abnormal standard liver tests (such as aminotransferases) and the severity of histological lesions.99 100 This has contributed to the under-recognition of NASH, particularly in patients with so-called cryptogenic cirrhosis.101 102 Diagnosis of NAFLD in lean subjects is of particular importance since 20% of these patients have NASH, >F2 fibrosis and carotid atherosclerosis.102 Furthermore, NAFLD is recognised as the most common cause of cryptogenic cirrhosis but the diagnosis frequently appears to be delayed.103
Semiquantification of NASH: histological scoring systems
From the study of Matteoni et al, 94 it became clear that specific histopathological features discriminated between two prognostically different patient groups.94 Different scoring systems were established to quantify the lesions and assign qualitative and semiquantitative numerical values to the histopathological diagnoses of steatosis alone, steatohepatitis and fibrosis. In 1999, Brunt et al published a histological system for assessment of ‘activity’ grade and staging fibrosis in NASH.104 NASH grades (1–3) were based on the combination of steatosis, hepatocellular ballooning, lobular and portal inflammation. Fibrosis staging recognised early zone 3 sinusoidal and/or pericellular ‘chickenwire’ fibrosis either alone (stage 1) or in combination with periportal (stage 2) or bridging fibrosis (stage 3), while stage 4 was cirrhosis.104
The necessity to define a widely accepted scoring system for NASH was highlighted by a National Institutes of Health symposium which fostered further efforts.105 The NASH Clinical Research Network (NASH CRN) established and validated the NASH CRN score as the first globally accepted, scoring system that addressed the full spectrum of NAFLD lesions and proposed the summative NAFLD activity score (NAS) to semiquantify disease activity in clinical trials.106 The NAS (range 0–8) is calculated by summing-up semi-quantitative scores for three of the most important histological features of NAFLD: steatosis (0–3), lobular inflammation (0–2) and hepatocellular ballooning (0–2) Kleiner et al of the NASH CRN Pathology Committee initially observed that NAS>5 correlated with NASH diagnosis whereas biopsies with NAS scores of <3 correlated with ‘not NASH’. Subsequent work from the same group made clear, however, that these NAS threshold values were not always associated with the underlying histological diagnosis of simple steatosis or steatohepatitis and, therefore, could not replace histopathological assessment.107 The NASH CRN pathologists have also coined the pattern-based ‘borderline NASH’ category for those cases that could not be binary classified. They also devised a staging system for assessing fibrosis in NAFLD based on Brunt’s 5-tier staging (0–4)104 with only modification being the subdivision of stage 1 into 3 substages.106
In 2011, a research workshop of the American Association for the Study of Liver Diseases reached consensus on key endpoints and the design of clinical trials for adult NASH.108 It was agreed that “definite steatohepatitis” is characterised by zone 3 accentuation of macrovesicular steatosis of any grade, hepatocellular ballooning of any degree, and lobular inflammation of any degree and recommended that the NAS should be used to semiquantify disease activity.109
Grading and staging of morphological features enhance information particularly in clinical trials. NAFLD heterogeneity reflects individual variability in response to metabolic stress and susceptibility to hepatocyte lipotoxicity, depending on genetic and environmental factor.110 As shown by pioneers in the field, NAFLD displays a continuous spectrum of hepatocytic, inflammatory and fibrous lesions. Therefore, the binary categorisation of NAFLD into NASH and ‘not NASH’ is artificial in a continuous disease process.110 Bedossa et al developed a simple algorithm to standardise the histological diagnosis of NASH and reduce inter-observer variability. The diagnostic algorithm was informed by scores for steatosis (S0–S3), activity grade (A0–A4 by adding scores for ballooning (0–2) and lobular inflammation (0–2)) and fibrosis stage (F0–F4).111 A group of expert hepatopathologists and general pathologists from the Fatty Liver Inhibition of Progression (FLIP) consortium further validated the FLIP algorithm.112 The SAF scoring system (Steatosis, Activity, Fibrosis) includes the same categories as NAS for the semiquantitation of liver injury but the diagnostic FLIP algorithm requires the simultaneous presence of steatosis, ballooning and lobular inflammation for NASH diagnosis.1 110 111
Despite the use of widely accepted minimal diagnostic criteria for diagnosing NASH, the issue of interobserver variability for assessing the characteristic histological features still remains. In order to increase the reliability of NASH diagnosis, both NASH CRN and FLIP SAF scoring systems are now simultaneously used in clinical trials and registries worldwide and can be performed by expert and general pathologists equally well if they are properly trained.112 113
The role of fibrosis as prognostic indicator
The prognostic relevance of these histopathological scores has been validated in several large registries with up to four decades follow-up. Fibrosis starts in zone 3 (perivenular, sinusoidal, pericellular). In later stages, portal, periportal and bridging fibrosis leads, finally, to cirrhosis.71–73 In 2015, it became clear that evaluation of fibrosis stage may be even more fundamental than scoring necroinflammation or diagnosing NASH since it emerged as the main prognostic factor.114 115 In two independent cohorts, the American PRELHIN study and the cohort from Karolinska and Linköping University Hospital, Sweden, NAS score alone was unable to predict overall mortality, whereas fibrosis stage predicted both overall and disease-specific mortality.115 Similar results were obtained for SAF score, which was not associated with increased mortality in NAFLD after adjustment for fibrosis.116 In the largest retrospective cohort study from Karolinska and Linköping University Hospital, Sweden from 1971 to 2009, the presence of NASH alone did not significantly increase the risk of liver-specific morbidity or overall mortality during a mean 20-year follow-up.117 In recent systematic meta-analyses, the risk of liver-related mortality increased exponentially with increasing fibrosis stage,118 while biopsy-confirmed fibrosis was associated with overall mortality risk in NAFL/NASH patients after adjusting for confounding factors.119 Further dissecting the natural course of advanced disease, a multinational study of 458 patients documented that patients with NAFLD cirrhosis have predominantly liver-related events, whereas those with bridging fibrosis experience mainly extrahepatic malignancy and cardiovascular events.120 In 475 NASH patients enrolled in two negative phase 2b trials, the primary determinant of clinical disease progression was fibrosis extent, fibrosis at baseline and its change over time.121
Although cirrhosis is the major risk factor for HCC development in NAFLD, there is increasing evidence that NAFLD-associated HCC frequently occurs in the absence of cirrhosis.122 123
The role of NASH as a precursor lesion for fibrosis emerged as patients with NASH developed severe liver disease slightly earlier than patients without NASH in the Swedish cohort.117 Recently, two positive NASH CRN clinical trials demonstrated a strong association between improvements in fibrosis and resolution of steatohepatitis.124 In addition, an NASH CRN prospective study has shown that changes in NAFLD activity are positively associated with changes in fibrosis.125 The general reversibility of ballooning, lobular inflammation and fibrosis after weight loss has been documented in patients undergoing bariatric surgery.126 Whether histopathological improvement from liver biopsy data in fact translates into a reduction in overall mortality and liver-related events is currently being investigated in phase 3 studies.
In 2021, liver biopsy still remains the only diagnostic procedure that can reliably assess the various NAFLD patterns, and particularly to diagnose NASH and early fibrosis.71 113
Genetic basis of NAFLD phenotypes
At the turn of 21st century, large-scale sequencing techniques became more widely available at reasonable cost. Genome-wide association (GWAS) and candidate gene studies have contributed to our understanding of interindividual variation in the progression and outcome of ALD and NAFLD.127 Genetic variants influence the risk and fate of NAFLD, particularly intensity and effects of oxidative stress, severity of steatosis and fibrosis, response to endotoxin, release of cytokines/chemokines and immune response. In 2008, Romeo and colleagues published the first GWAS in NAFLD.128 Of relatively modest size by current standards, this study examined 9229 nonsynonymous single-nucleotide polymorphisms (SNPs) in a North American population of diverse ethnicity from the Dallas Heart Study and identified a single highly significant association between increased hepatic triacylglycerol accumulation measured using non-invasive proton MR spectroscopy and the patatin-like phospholipase domain-containing 3 (PNPLA3) gene locus. Ethnic differences were described for the p.I148M SNP that was most common in Hispanics, lower in subjects of European ancestry (0.23) and lowest in African-Americans.128 Together with further SNPs associated with increased hepatic triacylglycerol content, PNPLA3 has been confirmed in a subsequent GWAS meta-analysis using 2.4 million SNPs from 7176 individuals of European ancestry by Speliotes et al.129 This variant may explain, at least in part, special phenotypes like paediatric and lean NAFLD.130 131 The search for further fatty liver genes revealed transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulatory protein gene (GCKR), and HSD17B13, encoding for a retinol dehydrogenase, among an increasing list of associated loci.127 132–134 Combined effects of these risk alleles were observed in replication cohorts.133 In recent years, additional data supported the particular contribution of PNPLA3 and other gene loci to the development of fibroinflammation and HCC.127 In the largest histology-based NAFLD GWAS in a cohort of 1483 European patients with biopsy-proven NAFLD and 17 781 genetically matched controls, PNPLA3 was confirmed as risk factor for the full histological spectrum of NAFLD, while TM6SF2, GCKR and HSD17B13 were also confirmed as NAFLD risk modifiers.135 Combining the number of inherited NAFLD risk genes with the multitude of exogenous NAFLD risk factors (exposome), three-dimensional risk-space models allow visualisation of disease trajectory in NAFLD risk gene carriers over time.136 It is tempting to speculate that genotyping will guide our clinical practice in the future but it has not as yet entered clinical practice because the effect magnitude of PNPLA3 risk allele for NASH development or liver cancer is rather modest.137 138
Towards non-invasive diagnosis of NASH and fibrosis in NAFLD: future trends
Research on NAFLD has made remarkable progress over the past two centuries (figure 5) but major issues remain. Despite progress in non-invasive tests (NITs) for the evaluation of liver fibrosis in NAFLD, such as elastography devices and blood tests,139 the diagnosis of NASH is still based on liver biopsy, an invasive procedure not suitable for the large proportion of general population affected by NAFLD. In 2007, the NAFLD fibrosis score (NFS) was introduced as a simple scoring system to distinguish NAFLD with and without advanced fibrosis.140 Subsequently, further fibrosis tests (table 1), including fibrosis-4 (FIB-4) index,141 fibrotest/fibrosure,142 enhanced liver fibrosis test143 and liver stiffness measurement by vibration-controlled transient elastography,144 have entered clinical practice. Most importantly for primary care, these NITs show excellent area under the receiver operating characteristic (AUROCs) for the diagnosis of advanced fibrosis and cirrhosis.145 The non-invasive diagnosis of advanced fibrosis has been further improved by the sequential combination of different NITs thereby refining the patient referral pathway between primary care or diabetologists and liver specialists.146 Longitudinal retrospective studies have demonstrated that NITs calibrated on liver fibrosis are themselves prognostic markers able to stratify the risk of liver-related outcomes and mortality in NAFLD.147 148 These data reinforce the relevance of using NITs instead of liver biopsy for the management of NAFLD patients in clinical practice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Timeline of major developments in the field of NAFLD over the past 200 years. Major landmarks in the field until 2000 are shown with solid lines and black font, while developments after 2000 that cannot yet be viewed in a historical context are shown in dashed lines and lighter font. NAFLD, non-alcoholic fatty liver disease.
Non-invasive tests for fibrosis assessment in NAFLD
NITs, such as the multiparametric NIS4 test,149 have not yet achieved sufficient accuracy and validation for the non-invasive diagnosis of NASH in routine practice. This, therefore, remains a significant challenge, and large consortia (LITMUS in Europe, NIMBLE in USA) are currently working to identify and validate new NASH biomarkers.
Pathological diagnosis in the near future will be supported by artificial intelligence. Future attempts to improve NASH diagnosis will apply machine learning to develop fully automated software applications for quantification of steatosis, inflammation, ballooning and fibrosis.150 Advances in microscopy techniques, such as second harmonic generation/two-photon excitation fluorescence imaging, could potentially improve reproducibility and standardisation of liver biopsy assessment.151
A great deal of our previously discussed knowledge has been derived from animal experiments under standardised conditions.152 Efforts are now focused on next-generation mouse models, which are particularly suitable for genetic manipulation but out of the focus of this work.153
What will the future bring in regard to NAFLD? As the global epidemic of obesity fuels metabolic conditions, the clinical and economic burden of NAFLD will become enormous.154 Models based on published estimates predict a growth of up to 30% in total NAFLD cases between 2016 and 2030.155 NASH prevalence will increase by 15%–56%, while advanced liver disease and liver-related mortality will more than double as a result of ageing Western populations. Obeticholic acid could represent an important milestone in the history of NASH if it becomes the first licensed treatment based on the favourable results of the interim analysis of the pivotal phase 3 trial.156 For those with morbid obesity, rates of bariatric procedures will further increase.157 Given the rapidly growing global burden of NAFLD/NASH, efforts to discover accurate, non-invasive diagnostic and prognostic biomarkers, to develop effective treatments for individuals with advanced NASH and to implement preventive methods must continue.4 The remark of Louis from 1843 on NAFLD as the most frequent and significant disease of the liver is still true today and guides our efforts in the future.15
Ethics statements
Acknowledgments
The authors thank Ms Alexandra Weisgram for her excellent secretarial assistance and Dr Camilla Graham for language editing and insightful comments.
References
Footnotes
Twitter @Dina Tiniakos
Correction notice This article has been corrected since it published Online First. The second author affiliation has been amended and the funding and competing interests statements added.
Collaborators None.
Contributors Concept and supervision: AG and MT; acquisition of data: AG, DT, HD and MT; analysis and interpretation of data: AG, DT, HD and MT; drafting of the manuscript: AG and MT; critical revision of the manuscript for important intellectual content: AG, DT, HD and MT; obtained funding: AG, DT and MT; material support: AG, DT and HD.
Funding This work has been supported by the Liver Investigation: Testing Marker Utility in Steatohepatitis (LITMUS) project. The LITMUS project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement no 777377. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.
Competing interests AG has declared receiving grants from Intercept, Novartis, Exalenz, Falk and Kibion and has received personal fees from Intercept, Novartis, Gilead, Pfizer, Falk, MSD, BMS, Ipsen, Sanofi-Aventis,Bayer, Eisai, CSL Behring, Sequana, Merz, Abbvie and Alexion. DT reports consultation fees from Intercept Pharmaceuticals Inc, Allergan plc, Cirius Therapeutics Inc, Alimentiv Inc, Clinnovate Health UK Ltd and an educational grant from Histoindex Pte. MT has received research grants from Albireo, Cymabay, Falk, Gilead, Intercept, MSD and Takeda and travel grants from Abbvie, Falk, Gilead and Intercept. He further has advised for Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, Genfit, Gilead, Intercept, Jannsen, MSD, Novartis, Phenex, Regulus and Shire and has served as speaker for Falk Foundation, Gilead, Intercept and MSD. He is also co-inventor of patents on the medical use of NorUDCA filed by the Medical Universities of Graz and Vienna.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.