Article Text

Abstract
Approximately 5% of individuals infected with hepatitis B virus (HBV) are coinfected with hepatitis D virus (HDV). Chronic HBV/HDV coinfection is associated with an unfavourable outcome, with many patients developing liver cirrhosis, liver failure and eventually hepatocellular carcinoma within 5–10 years. The identification of the HBV/HDV receptor and the development of novel in vitro and animal infection models allowed a more detailed study of the HDV life cycle in recent years, facilitating the development of specific antiviral drugs. The characterisation of HDV-specific CD4+ and CD8+T cell epitopes in untreated and treated patients also permitted a more precise understanding of HDV immunobiology and possibly paves the way for immunotherapeutic strategies to support upcoming specific therapies targeting viral or host factors. Pegylated interferon-α has been used for treating HDV patients for the last 30 years with only limited sustained responses. Here we describe novel treatment options with regard to their mode of action and their clinical effectiveness. Of those, the entry-inhibitor bulevirtide (formerly known as myrcludex B) received conditional marketing authorisation in the European Union (EU) in 2020 (Hepcludex). One additional drug, the prenylation inhibitor lonafarnib, is currently under investigation in phase III clinical trials. Other treatment strategies aim at targeting hepatitis B surface antigen, including the nucleic acid polymer REP2139Ca. These recent advances in HDV virology, immunology and treatment are important steps to make HDV a less difficult-to-treat virus and will be discussed.
- hepatitis D
- antiviral therapy
- immunology in hepatology
- chronic viral hepatitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
At least 12 million individuals infected with hepatitis B virus (HBV) are coinfected with hepatitis D virus (HDV) and have a high risk to develop liver cirrhosis and hepatocellular carcinoma within a few years.
Until 2020, there was no specific treatment option for the large majority of these patients; off-label use of pegylated interferon-α (pegIFNα) displays only approx. twenty per cent off-therapy virological response rates and is contraindicated in many patients.
The identification of sodium taurocholate cotransporting polypeptide (NTCP) as cell entry receptor for both, HBV and HDV, allows the development of novel cell culture models and contributes to the development of novel treatment strategies.
Beside de novo entry of virions via NTCP, cell division is an important mechanism of HDV spread; thus, combination therapies targeting these different mechanisms of viral spread are expected to show synergistic effects.
Characterisation of HDV-specific immune responses will contribute to a better understanding of the mechanisms of HDV clearance versus persistence. It may also lead the way to novel treatment concepts, combining compounds that target host and/or viral targets and immunotherapeutic interventions.
Three novel anti-HDV compounds target host factors: the entry inhibitor bulevirtide (BLV, Hepcludex), the prenylation inhibitor lonafarnib (LNF), and the nucleic acid polymer REP2139Ca.
BLV was approved for conditional marketing in Europe in 2020. BLV in combination with (off-label) pegIFNα for 48 weeks or as monotherapy for a longer duration may allow sustained virological responses in a substantial proportion of patients.
LNF and REP2139Ca also display encouraging response rates but additional data from phase III trials (ongoing for LNF) will be required prior to final assessment and possible approval by regulatory authorities.
Introduction
The human hepatitis D virus (HDV) is unique among animal viruses. Enveloped in the hepatitis B virus (HBV) surface proteins, HDV constitutes the smallest human virus with a diameter of 35–36 nm (figure 1A). HDV requires HBV as a helper for entry into hepatocyte, intrahepatic spread and dissemination between its hosts.1 2 Although recent in vitro findings indicate that HDV may propagate independent from HBV, using envelope glycoproteins from several virus genera such as vesiculovirus, flavivirus and hepacivirus including hepatitis C virus (HCV),3 clinical investigations confirm its strong association with HBV infection (hepatitis B surface antigen, HBsAg positivity).4–6 Some estimates suggest that up to 60 million individuals may be infected with HDV,7 8 however, another meta-analysis indicates that 12 million people are affected.9 HBV/HDV coinfection is associated with a more severe course of the diseases and an increased mortality compared with HBV monoinfection. Simultaneous infection with HBV and HDV of adults results in clearance of both viruses in the majority of individuals. In contrast, superinfection of an HBV-infected patient with HDV typically results in the development of persistent HBV/HDV coinfection which may lead to liver cirrhosis, liver failure and eventually hepatocellular carcinoma (HCC) within short time. Indeed, 50%–70% of patients with chronic HBV/HDV coinfection develop cirrhosis within 5–10 years after diagnosis, corresponding to a threefold increase compared with HBV-monoinfected patients.10 The risk for HCC development is increased compared with HBV monoinfection with an odds ratio (OR) of 1.28–2.77, depending on the selection of studies included in the meta-analysis.11 Due to this increased complication rate, HDV coinfected patients account for approx. 25% of HBsAg-positive liver transplant recipients in the European Liver Transplant Registry.10 Until recently, no approved antiviral treatment was available against HDV, thus, a more precise understanding of HDV virology and anti-HDV immune responses is essential to develop and establish novel therapeutic regimens.
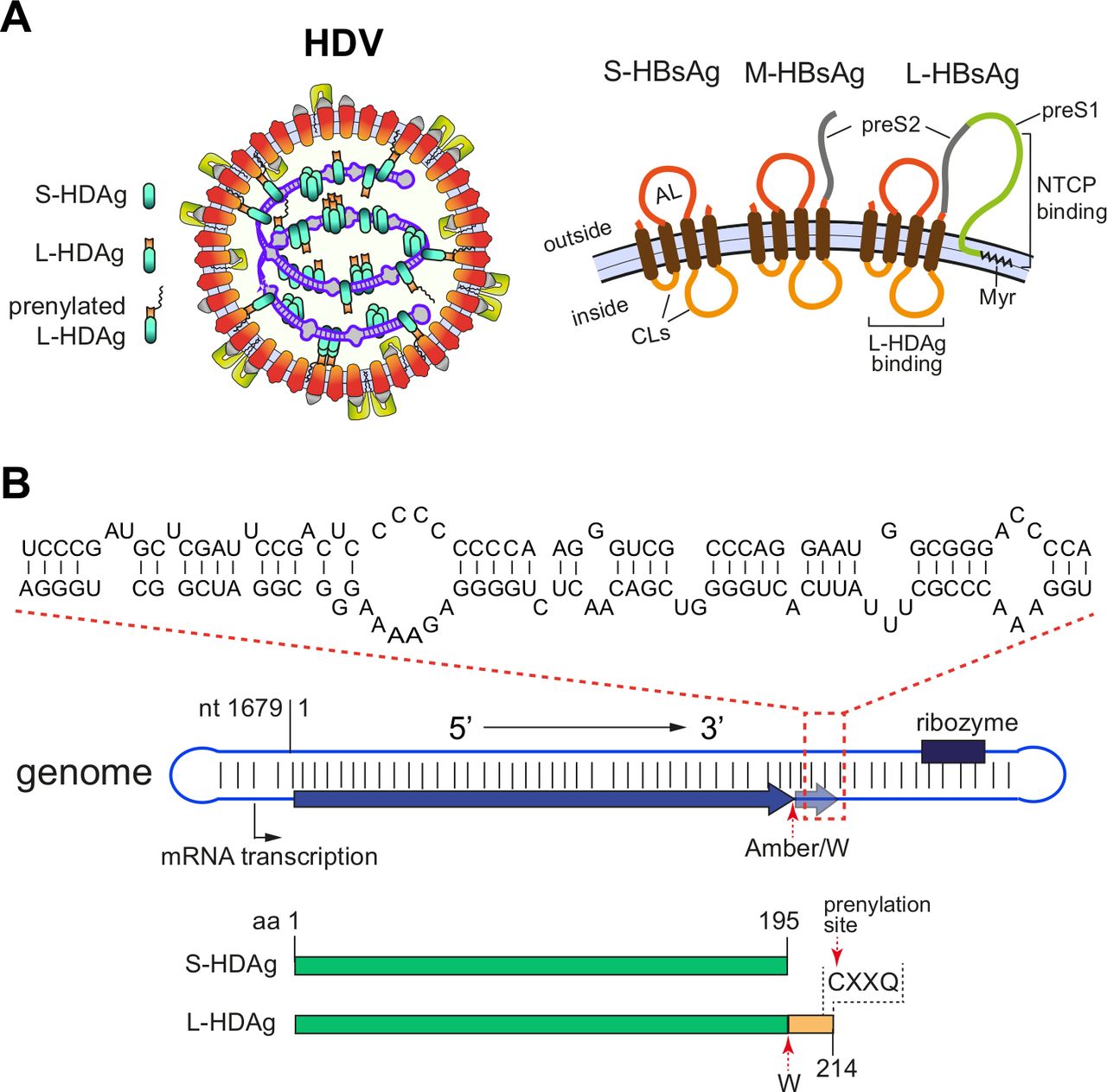
Structures of HDV virion and genome. (A) Schematic representation of HDV virion (left) and envelope proteins (right). HDV virion has a ribonucleoprotein (RNP) complex inside and an HBV derived envelope outside. The RNP consists of the HDV genome and two isoforms of hepatitis D antigen (HDAg), L-HDAg and S-HDAg. Prenylation of L-HDAg is essential for envelope acquisition. The envelope contains three HBV envelope proteins: small-HBsAg (S-HBsAg), medium-HBsAg (M-HBsAg) and large-HBsAg (L-HBsAg). M and L share the same sequence with S, however, contain N-terminal extensions: preS2 for M and preS1 plus preS2 for L. The preS1 domain of L is critical for binding of the receptor sodium taurocholate cotransporting polypeptide (NTCP), while the cytosolic loops (CLs) are important for encapsulation of HDV RNP through interaction with HDAg. (B) HDV genome structure and key elements. As a single-strand circular RNA, HDV genome forms an unbranched rod-like structure through high rate of intramolecular base-pairing. A representative region consisting of short stems and bulges is depicted on top. S-HDAg and L-HDAg are encoded by unedited and adenosine deaminases acting on RNA 1 (ADAR1)-edited (Amber stop codon to TGG (W)) genomic RNAs, respectively. The C terminal prenylation motif (CXXQ) is indicated. The numbering of nucleotide and protein sequences is based on a HDV genotype one strain (GenBank: M21012.1). HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HDV, hepatitis D virus; L-HDAg, large HDAg; S-HDAg, small HDAg.
HDV structure, replication, mechanisms of persistence and antiviral targets
Molecular biology and the burden of HDV infection
HDV genotypes and endemic hotspots
Due to the sequence variations found in HDV isolates, eight clades, termed genotypes 1–8, have been classified.12 They show remarkable differences in their replication efficacies.13 Genotype 1 is globally scattered while HDV genotypes 2–8 can be attributed to distinct geographic regions in the world. While the HDV median prevalence in HBsAg carriers is estimated to about 5%, it typically manifests in hotspots10 like Mongolia, the Middle East, Usbekistan or parts of South America where up to 80% of HBsAg carriers also display markers (anti-HDV antibodies, HDV RNA) of an HDV infection.10 Due to the large gaps of knowledge on reliable epidemiological data HDV prevalence may be profoundly underestimated and needs more attention in the future.7
HDV genome structure
The HDV genome consist of 1672–1697 ribonucleotides (genotype-dependent) and forms a single stranded covalently closed circular RNA molecule of negative polarity (defined in relation to the (+) stranded mRNA encoding the hepatitis D antigen (HDAg)). Both, genomic and antigenomic RNA is characterised by a high degree of self-complementarity (>70%) leading to recurrent back-folded stretches of base paired rods, that are interrupted by short loops.14 This peculiar structure resembles the structure of plant viroid RNA and mimics a DNA double helix (figure 1B). Different from plant viroids, HDV RNA associates with the viral HDAg but also the protein bromodomain adjacent to zinc finger domain 2B (BAZ2B), involved in chromatin remodelling.15 Such ‘molecular mimicry’ complexes of dsDNA enables the host DNA-dependent RNA-polymerase (Pol II) to accomplish RNA-dependent RNA synthesis. HDAg is encoded as two isoforms within a segment of the HDV genome, namely the small HDAg (S-HDAg, 195 aa, 24 kDa) and the large HDAg (L-HDAg, 214 aa, 27 kDa). While S-HDAg is necessary to initiate and maintain replication, L-HDAg negatively regulates replication and triggers envelopment of the virus into the HBV surface proteins. Both antigens are post-translationally modified in order to fulfil their distinct functions.1 2
HDV replication and envelopment of HDV ribonucleoproteins into HBsAg
Following entry and delivery of the genomic HDV ribonucleoprotein complex (RNP) to the nucleus of an infected hepatocyte, S-HDAg-encoding mRNA is transcribed and translated. S-HDAg expression is required for maintenance of nuclear RNA replication functioning as a ‘reprogramming factor’ to adopt Pol-II to an RNA substrate. During RNA synthesis, which proceeds via consecutive rolling circle mechanisms that switch between (−) and (+) strand synthesis (see reference 2 and figure 2A), the de novo synthesised genomic HDV RNA underlies editing by the cellular ADAR-1 enzyme.16 This editing results in a mutation in the UAG stop codon of the S-HDAg open reading frame to a UGG (Trp-codon). After transcription of the corresponding HDAg-mRNA, the ribosome introduces a Trp residue and further on a C-terminal extension of 19–20 aa (genotype dependent) leading to L-HDAg. Accordingly, both HDAgs share the N-terminal S-HDAg domain and are able to bind genomic and antigenomic HDV RNA to form RNPs (figure 1). In addition, L-HDAg is subjected to prenylation by the cellular farnesyltransferase (target of lonafarnib (LNF)) at a conserved Cys-residue (Cys-211) within the C-terminal extension. When HBsAg is expressed in the same cell, prenylated L-HDAg recognises a hydrophobic element within the cytosolic loop of the small HBV envelope protein (S-HBsAg). Since S-HBsAg alone triggers self-assembly and secretion of HBV subviral particles (SVPs), expression of HBsAg in RNP containing cells is sufficient for HDV secretion. Through incorporation of the large HBV envelope protein L-HBsAg, the particles gain infectivity and support transmission into sodium taurocholate cotransporting polypeptide (NTCP)-receptor expressing cells to disseminate within the liver (figure 2A) and between hosts17 (for more details see reference 1 2). The HBV M-protein is redundant for both, particle release and entry.18 19 Since in a natural infection HBsAg can be expressed from both HBV cccDNA (in HBV replicating cells) and chromosomally integrated HBV DNA,20 21 enveloped HDV serum RNA particles may arise from both sources.

HDV life cycle, spreading pathways and drug targets. HDV virions first attach to heparan sulfate proteoglycans (HSPGs) and then to the viral receptor NTCP to enter host cells. After membrane fusion, the ribonucleoprotein (RNP) is released and further transported to the nucleus to initiate RNA replication. The incoming genome (G) serves as the template for the first rolling circle amplification. The resulting antigenome (AG) multimers are cleaved in cis by the intrinsic ribozyme and ligated into circular monomers. After a second rolling cycle using the AG as the template, HDV G multimers are synthesised and further cleaved to produce monomers. The HDV AG might be edited by ADAR1, yielding an extended HDAg ORF that produces L-HDAg, some of that is further prenylated. S-HDAg and L-HDAg (intact and prenylated) are transported into the nucleus to regulate virus replication or bind to the HDV RNA to form RNP. The G-containing RNP can be exported to the cytoplasm and encapsulated into HBV envelope through the interaction between L-HDAg and S-HBsAg. HDV virions are released through the ER-Golgi secretory pathway. besides the HBV envelope-dependent de novo infection, HDV can also spread through division of infected cells in an HBV-independent manner (below). Bulevirtide (BLV) blocks de novo infection by efficient binding of the viral receptor NTCP. Lonafarnib (LNF) prevents the prenylation of L-HDAg by inhibiting the farnesyl transferase and consequently impairs HDV assembly and secretion. The target(s) of nucleic acid polymer (NAP) is unclear. It may inhibit assembly/release of HDV virions and/or HDV ribonucleoprotein assembly via direct interaction with the HDAg. IFNs, including MDA5-mediated HDV-induced IFNs and therapeutic IFNα and IFNλ, induce IFN stimulated genes (ISGs) which profoundly suppress HDV amplification during cell division. HBV, hepatitis B virus; HDV, hepatitis D virus; IFN, interferon; L-HDAg, large hepatitis D antigen; NTCP, sodium taurocholate cotransporting polypeptide; S-HBsAg, small hepatitis B surface antigen.
Receptor interaction and the consequence of HBV integration
Liver tropism and hepatic receptors of HBV and HDV
The liver tropism of HBV and HDV is primarily determined by a specific interaction of an extended receptor binding domain (RBD) (aa 1–75) in the preS-1-part of the HBV L-protein18 and the hepatic NTCP receptor.22 23 NTCP interaction of HBV and HDV requires prior attachment to heparan sulphate proteoglycans (HSPGs)1 (figure 2A). This mandatory step presumably triggers the release of the otherwise hidden preS-receptor binding site. HSPG-requirement explains how neutralising anti-HBsAg-specific antibodies, although they do not directly interfere with preS/NTCP-interaction, block entry and control infection.
NTCP exclusively locates at the basolateral/sinusoidal membrane of differentiated, polarised hepatocytes. NTCP-expression ceases when differentiated hepatocytes proliferate.24 NTCP is also downregulated in transformed cell lines of hepatic origin such as HepG2, HuH7 and Hep3B. Thus, proliferating normal hepatocytes, transformed hepatoma cells, and probably also tumour cells in HCC lack NTCP25 and do not support entry of HBV and HDV. Though, proliferating cells support spread of HDV RNA (depending on their interferon (IFN) activated state)26 27 but loose HBV cccDNA (figure 2B).28 A deeper understanding of these peculiar differences of HDV and its helper HBV will be crucial for understanding persistence and is important for the development of successful therapeutic interventions.
Studying HDV replication in vitro
Constitutive NTCP expression provides susceptibility to HDV infection of hepatic and even non-hepatic cells. Different NTCP-expressing cell lines have been developed in recent years and are used as HDV/HBV infection systems to study fundamental aspects of virus replication but also for the identification of novel drug candidates. Although NTCP-expression is sufficient to permit HDV entry and the onset of replication, assembly and secretion of viral particles cannot be achieved due to the lack of the HBV envelope proteins required for virus release. This can be overcome by cell lines that express both, the NTCP receptor and the HBV envelope proteins.29 30 Such cell lines support the complete replication cycle of HDV with entry, initiation of RNA replication, processing of L-HDAg and release of infectious progeny virus. Remarkably, coexpression of NTCP and its ligand (the L-protein) neither interferes with HDV particle secretion nor does it induce receptor downmodulation as described for many viruses (eg, HIV31 or even the duck HBV32). This has important clinical implications on intrahepatic persistence of HDV RNA and the envisaged results of treatment responses. Different in vivo models for HDV have been developed and are summarised in.33
Consequences of HBV integration and clonal expansion of integrants
One implication of HBV integration is that the replication space of HDV in an HBV-infected liver may not be restricted to cells that transcribe HBsAg from HBV cccDNA but also to hepatocytes that express HBsAg from integrated HBV DNA. Such integrates establish instantly when double stranded linear HBV DNA-containing particles, a ‘by-product’ of HBV replication, enter hepatocytes via NTCP.34 Early during acute HBV infection, the integration rate is low and restricted to single cells, however under circumstances like continuous inflammation accompanied by liver regeneration, or transformation, such cells clonally expand. Although integrated HBV DNA cannot function as a ‘provirus’ (like retroviruses), it characteristically encodes HBV envelope proteins which, when located in transcriptionally active sites of the chromosome and contribute to serum HBsAg in patients. If such clonally expanded ‘hepatocyte islands’ carrying identical integrations maintain NTCP expression, they constitute HBV cccDNA-independent replication space for HDV in the liver. Considering that cccDNA gets lost during cell division,28 35 it is reasonable to assume that especially in HBeAg-negative chronic carriers of HBV, who have been shown to produce the majority of HBsAg from integrates,36 HDV serum RNA may significantly originate from cells with HBV integrates. Thus, therapeutic approaches targeting cccDNA would only partially affect HDV.
Cell division mediated intrahepatic spread of HDV RNA
Besides spreading of enveloped HDV via an NTCP-receptor dependent de novo infection pathway, another mode of HDV RNA dissemination has been described recently.26 It is characterised by the direct transfer of replication competent HDV RNA between cells during mitosis. This process can proceed in the absence of HBsAg. To what extent it contributes to HDV persistence in the liver of infected patients is unknown and needs to be investigated in the future. The effectiveness of cell division-mediated RNA spread however profoundly depends on recognition of replicating HDV RNA by MDA537 and the degree of IFN induction (figure 2B).
Interfering with viral replication
Viral targets
In contrast to other RNA viruses that encode RNA-dependent RNA-polymerases (RdRP) for replication and mRNA synthesis, HDV recruits and reprogrammes the cellular Pol II to achieve these goals.2 Accordingly, an important viral drug target (the RdRP) is lacking. Nevertheless, crucial steps in the viral life cycle like the ribozyme-mediated self-cleavage of genomic and antigenomic RNA oligomers or the HDAg-dependent regulation of RNA replication and mRNA synthesis are attractive viral structures suitable for drug targeting. Inactivation of the S-HDAg could induce a selective shut down of RNA synthesis (eg, through abrogation of its RNA binding activity or inactivation of its cofactor function for Pol II). Alternatively, abolition of the interaction of the prenylated C-terminus of L-HDAg with the cytosolic loop in the HBV S-domain by small molecules would inhibit virus release similar to LNF, which targets the corresponding host enzyme (see below and figure 2A). No such drugs have been identified so far, however applying the new replication systems mentioned above will facilitate screening approaches and drug candidate identification in the future.
Cellular targets
At present all strategies that have been clinically developed address cellular targets. Host factor targeting bears the problem that the drug inactivates the cellular function of the target and thus, besides affecting the virus, also induces side effects. Conversely, host factor targeting profits by a higher barrier to develop drug resistance. The most advanced drug against HDV is bulevirtide (BLV), formerly called Myrcludex B. BLV addresses NTCP thereby blocking virus entry.17 Another drug, LNF, inactivates farnesyltransferase, thereby preventing envelopment of RNPs with HBsAg (figure 2A).38 While interference with prenylation results in a direct inhibition of virion release on target engagement, entry inhibition affects serum HDV-RNA levels through an indirect effect, namely the reduction of the pool of HDV producing cells by cell turnover through sustained inhibition of de novo infection. According to these differences in their mode of action, the kinetics and the degree of drug-mediated suppression of HDV serum RNA levels differ substantially. The third drug that is presently developed clinically are nucleic acid polymers (NAPs). These molecules have been associated with multiple modes of action including HBsAg secretion inhibition of SVPs and virions by targeting (a) host factor(s) and direct interaction with HDAg.39 40 In addition, it has been assumed that NAPs impact immunological mechanisms by yet ill understood mechanisms.41 Lastly, IFN-α (IFNα), an off-label drug used for chronic HBV/HDV coinfection since the 1980s, inhibits HDV replication, likely through induction of antiviral interferon stimulated genes (ISGs) and/or adaptive immunity (figure 2B). Notably, IFNs (IFNα and IFNλ) can profoundly suppress cell division-mediated HDV spread, which is not achievable by entry or release inhibition.27
Beside these well-characterised host factors additional approaches using siRNA or drug libraries42 in susceptible cell lines will allow to identify novel host factors in the future and it will be a challenging task to identify those that allow intervention and are tolerable regarding side effects.
Immune response to HDV
Innate immunity
Interferon
IFNs are the main mediators of early containment of viral replication: They bridge the gap until adaptive immunity is induced and play a crucial role in this induction process. While HBV as a ‘stealth’ virus undermines the IFN system by avoiding recognition in acute as well as chronic infection,43–45 HDV resembles HCV46 and induces an IFN response, but may have several methods to counteract or even take advantage of it. Indeed, induction of an IFN response has been demonstrated in cell culture models as well as in a mouse model of acute and chronic coinfection.37 47–49 Induction is limited to IFNβ and λ, but not α, and compared with other RNA viruses (eg, Sendai virus) rather weak.37 Recognition of RNA intermediates is sensed by the pattern recognition receptor melanoma differentiation antigen 5 (MDA5) (figure 2B), but not retinoic acid inducible gene I (RIG-I) or toll-like receptor 3 (TLR3).37 Since MDA5 is preferentially localised in the cytoplasm, the exact mechanisms of HDV sensing is not yet clear. The suppressive effect of IFNα on HDV replication in vitro is more profound during an early stage of infection (eg, the establishment of replicative intermediates) in vitro, while established infections in the absence of cell division are less affected.37 50 Importantly, IFN has a dominant effect on cell-division mediated HDV spread.27 This sensitivity of HDV to IFN during cell division is not yet understood on the molecular level but may be due to the resolution of the nuclei, exposure of viral RNA to induced ISGs, and the subsequent elimination/degradation of HDV RNA. Of note, this finding has implications for future combination therapies with innate immune modulators (eg, TLR agonists) but also IFN (eg, IFNλ).
HDV-induced IFNs may suppress HBV replication, partially explaining that patients coinfected with HDV usually display low HBV viral loads.47 In addition, HDV may take advantage of the IFN response: it leads to IFNβ/λ-mediated upregulation of HBV antigen presentation, resulting in increased T-cell induced HBV suppression and thus a further shift towards a dominance of HDV over HBV replication.51 How HDV counteracts the IFN response is so far only partially understood. There are conflicting results regarding the down-regulation of ISG transcription by HDV-mediated inhibition of phosphorylation and nuclear translocation of STAT1/2.37 48 52 Alternatively, HDV may hide from recognition as well as clearance by the IFN system by compartmentalisation to the nucleus as well as protecting its RNA within the RNP as well as HBsAg.49
Natural killer cells
Natural killer (NK) cells are increased in frequency in the peripheral blood in chronic viral hepatitis, irrespective of the exact viral pathogen, however, they display a less activated phenotype and are compromised in cytolytic function and cytokine production. Untreated HBV/HDV coinfected patients tend to have even higher peripheral NK cell frequencies compared with patients with other hepatitis virus infections, however, this difference is most likely due to the impact of disease activity and severity on NK cell frequency and function rather than the viral pathogen itself.53 These data also suggest, but do not proof, a role of NK cells in pathogenesis and disease progression. Of note, cytomegalovirus (CMV)-associated adaptive-like NK cell subsets are not affected by HDV or other hepatitis viruses.54 Early studies indicated a boost of NK cell activity during therapy in HBV/HDV coinfected patients that responded to IFNα treatment (clearance of intrahepatic delta antigen).55 More recent analysis demonstrated a change in NK cell differentiation during IFNα treatment, with selective loss of terminally differentiated NK cells, enrichment in immature NK cell subsets, and functional impairment.56 A high percentage of CD56dim NK cells at baseline was positively associated with treatment response,56 suggesting an important role of NK cells in IFN-induced viral control.
Mucosa-associated invariant T cells
Mucosa-associated invariant T (MAIT) cells occur in high frequencies in the liver.57 In patients with HBV/HDV coinfection, MAIT cells are activated, most likely due to increased interleukin 12 (IL-12) and IL-18 secretion by activated monocytes, leading to functional impairment and subsequent progressive loss of peripheral as well as intrahepatic MAIT cells with progressive disease.58
Adaptive immune response
Antibody response
Anti-HDV antibodies are detectable in rather low titres in acute-resolving HDV infection, but at higher titres in persistent infection.59 In patients with active hepatitis, anti-HDV IgM often persist at high titres. Thus, anti-HDV antibodies contribute little to viral control and clearance, most likely due to the lack of neutralising activity.
T cell response
Virus-specific T cells are the drivers of elimination in acute-resolving HBV as well as HCV infection. The important contribution of cellular adaptive immunity in HBV and HCV clearance have been demonstrated by (1) longitudinal studies demonstrating temporal association between the onset of virus-specific T-cell responses and viral elimination, (2) HLA association studies revealing protective class I and II alleles, and (3) direct antibody-mediated depletion of CD4+ and CD8+ T cells in the chimpanzee model.60 For HBV/HDV coinfection, the role of T cells has not been well defined, since adequate animal models are lacking and few HDV-specific T cell epitopes with defined HLA restriction were fine-mapped so far.
Indeed, the HDV-specific CD4+ T cell epitope repertoire has been analysed in two studies at a single-epitope resolution.61 62 Approx. 30%–40% of untreated HDV-infected patients displayed HDV-specific CD4+ T cell responses usually targeting 1–3 distinct epitopes. These responses were weak and only detectable after antigen-specific culture. CD4+ T cells targeted epitopes throughout the L-HDAg, with some preference for the N-terminal region (figure 3). In addition, CD4+ T cell epitopes displayed promiscuous HLA restriction. The two studies led to conflicting results regarding the association between detectable HDV-specific CD4+ T cell responses and clinical parameters. Nisini et al were able to detect HDV-specific CD4+ T cell responses only in patients with normal alanine aminotransferase (ALT; HDV-RNA data are not available for this early study).62 Landahl et al, in contrast, did not observe an association between HDV-specific CD4+ T cell responses and HDV-RNA status (18 untreated patients with detectable RNA vs 13 untreated patients with undetectable RNA), HDV viral load, transaminases or HBsAg levels. They found, however, an association between HDV-specific CD4+ T cell responses and low HBV viral load,61 paralleling the association between HBV-specific T-cell responses and low HBV viral load found in inactive HBsAg carriers and nucleoside/nucleotide analogue-treated patients in chronic HBV monoinfection.63

HDV regions targeted by HDV-specific CD4+ and CD8+ T cell epitopes. (A) Dominantly targeted CD8+ and CD4+ epitope regions (identified in olp studies) are indicated in blue and green colour, respectively, with intensity representing frequency of recognition. Fine-mapped CD8+ and CD4+ T cell epitopes are indicated by blue and green bars, respectively. HLA class I associated HDV polymorphisms (‘HLA footprints’) that correlate to fine-mapped epitopes are depicted by red arrowheads. (B) HLA restriction of fine-mapped HDV-specific CD8+ T cell epitopes, demonstrating a clear dominance of HLA-B restriction. HDV, hepatitis D virus; L-HDAg, large hepatitis D antigen; S-HDAg, small HDAg.
HDV-specific CD8+ T cell responses were comprehensively analysed only recently.61 64 65 Indeed, after peptide-specific culture using overlapping L-HDAg peptides, approx. 40% and 70% of untreated and LNF-treated patients, respectively, with chronic HBV/HDV coinfection displayed HDV-specific CD8+ T cell responses.61 65 HDV-specific CD8+ T cell epitopes clustered in the C-terminal part of HDAg that is unique to its large isoform (L-HDAg) and were dominantly restricted by HLA-B alleles61 65 (figure 3). Like HDV-specific CD4+ T cells, HDV-specific CD8+ T cells display low ex vivo frequencies similar to HBV- and HCV-specific CD8+ T cell frequencies, but substantially lower compared with Eppstein Barr Virus (EBV) -specific, CMV-specific and influenza-specific CD8+ T cells.64 65 Earlier studies in a small number of patients found positive HDV-specific CD8+ T cell responses in patients with resolved HDV infection only.66 67 In the more recent and comprehensive analyses, however, no difference was observed for the detection rate of proliferative HDV-specific CD8+ T cell responses between patients with resolved HDV infection and patients with chronic HBV/HDV coinfection.61 64 65 In comparison to previous findings in HBV and HCV infection, where patients with spontaneously resolved infection usually display substantially stronger T-cell responses on peptide stimulation, this finding is unexpected. It may be explained by the fact that many patients with resolved HDV infection are still chronically infected with HBV, leading to continued T-cell exhaustion, or long periods of HDV viraemia in many patients possibly leaving a functional scar (eg, partial exhaustion) on HDV-specific CD8+ T cells even after viral clearance similar to the findings in HCV-infected patients after DAA-mediated cure.68 In line with these considerations, IFNγ responsiveness of HDV-specific CD8+ T cells correlated negatively with HDV viral load in untreated patients in one study,65 but with HBV viral load in the other study.61
The mechanisms of HDV-specific CD8+ T cell failure in chronic HBV/HDV coinfection have been addressed recently.64 65 67 69 There is evidence that the two main mechanisms known for other viral infections also apply to HDV: mutational viral escape and CD8+ T cell exhaustion (figure 4). A large international collaborative study analysed HLA class I-associated viral sequence polymorphisms in 104 untreated patients with chronic HBV/HDV coinfection. Several HDV-specific CD8+ T cell epitopes were identified and viral sequence variations in these epitopes were confirmed to mediate viral escape by functional analyses.64 67 Of note, several of the newly identified HDV-specific CD8+ T cell epitopes corresponded to HDV-specific CD8+ T cell epitopes identified by using overlapping peptides (figure 3),61 65 indicating that the repertoire of HDV-specific CD8+ T cell epitopes is limited. Importantly, both approaches and all three studies consistently observed a dominance of HLA-B alleles in restricting HDV-specific CD8+ T cell responses.61 64 65 Strikingly, the majority of CD8+ T cell epitopes as well as HLA-associated sequence polymorphisms was linked to quite rare HLA class I alleles, while most common HLA class I alleles such as HLA-A*02 seem to contribute little to the HDV-specific CD8+ T cell epitope repertoire.64 Thus, viral escape may have led to the extinction of HDV-specific CD8+ T cell epitopes restricted by common HLA class I alleles at the population level.

Mechanisms of HDV-specific CD8+ T cell failure. HDV-specific CD8+ T cells targeting viral epitopes with wild-type sequence display a chronically activated phenotype and are functionally partially exhausted (left), HDV-specific CD8+ T cells targeting viral epitopes with sequence variations (viral escape) do not recognise the antigen anymore and display a memory-like phenotype (right). HDV, hepatitis D virus.
HDV-specific CD8+ T cells targeting viral epitopes with mutated sequences displayed a memory-like phenotype (CD127+, programmed cell death protein 1 [=(PD-1)+, T cell factor 1 (TCF-1)+) and low expression of activation markers such as CD38,64 65 consistent with the phenotype of virus-specific CD8+ T cells lacking antigen stimulation due to viral escape in chronic HCV infection.70 HDV-specific CD8+ T cells that targeted viral epitopes without evidence of viral escape, however, displayed a different phenotype with higher expression of CD38 and lower expression of CD127 and TCF-1.65 These cells were, however, not terminally exhausted, since they displayed low levels of CD57 and—mostly strikingly—expressed multiple inhibitory/exhaustion markers to a lesser degree compared with HBV-specific, CMV-specific and EBV-specific CD8+ T cells.65 While it is at first contraintuitive that virus-specific CD8+ T cells in the setting of the ‘most severe’ viral hepatitis display lower exhaustion markers compared with other persistent viral infections, this observation may partially explain the similar strength of HDV-specific CD8+ T cell responses in resolved versus chronic infection. It is also in agreement with the previous report that HDV-specific T cells are functionally restored by the third signal cytokine IL-12 rather than by checkpoint inhibitors such as anti-PD-L1 or anti-CTLA4.69 CD38+ HDV-specific CD8+ T cells, and thus HDV-specific CD8+ T cell targeting non-mutated HDV epitopes, were associated with increased aspartate aminotransferase (AST) levels, indicating, but by far not proving, a causative role of HDV-specific CD8+ T cells in immunopathology of chronic HBV/HDV coinfection.65
Some of the most important questions in HDV-specific immunity remain to be solved. For example, regarding the natural course of persistent infection, the precise mechanism of immunopathology leading to unfavourable outcomes in the majority of patients remains elusive. From an immunologist’s perspective, the interplay between HBV-specific and HDV-specific immunity is a clear focus for further investigation, especially since current studies still ‘ignore’ the dual role of HBsAg as surface antigen of both, HBV as well as HDV. Lastly, and most important from a translational perspective: Are HDV-specific immune responses (at least partially) restored during therapy with novel antiviral strategies, similar as observed for HBV during NUC treatment and HCV during DAA therapy60? HDV-specific T-cell responses show little recovery during the treatment with pegylated IFNα (similar to what has been observed for HBV- and HCV-specific T-cell responses, likely due to the T-cell-suppressive effects of IFNα), but do they restore and play a role in viral clearance during BLV therapy? First analyses that were limited to three cases with cirrhosis could not readily identify such a T-cell restoration,71 however, further studies need to address this in larger patient cohorts also including less advanced disease. Of note, restoration of HDV-specific immunity by new antiviral treatments targeting host and/or viral targets may also pave the way for therapeutic vaccination that may be an attractive intervention in the setting of an otherwise ‘suppressive’ and not ‘curative’ treatment strategy.
Anti-hepatitis D treatment
Endpoints of therapy
The ideal endpoint for any anti-HDV therapy would be HBsAg loss with anti-HBs seroconversion. Elimination of replicating HDV RNA from the liver in HBsAg positive patients would be an alternative, however, it would require biopsies from patients and is not applicable in clinical practice. A more practical primary endpoint outcome is serum or plasma HDV RNA (as a surrogate marker of liver HDV-RNA levels) below the limit of detection by a sensitive and specific PCR assay during therapy, at the end of treatment (EOT) and off-therapy, at least 24 weeks after treatment discontinuation.72 However, given the high risk of late post-treatment virological relapses described after IFN–based therapies, a sustained off-therapy response should be confirmed over time, well beyond 24 weeks after treatment discontinuation. The proportion of patients with a≥2 log IU/ml decline of HDV RNA coupled with normal ALT have also recently been suggested as reasonable secondary endpoints for clinical trials.73 To comply with these stringent virological endpoints, it is of paramount importance to rely on commercially available, validated, WHO standardised, sensitive and specific HDV RNA assays that may allow to compare viral kinetics within as well as across studies.73
Current anti-HDV treatment
IFNα
Although not FDA or EMA approved, standard and pegIFNα treatments have been widely used as anti-HDV strategy in the last 20–30 years. PegIFNα is the only treatment regimen currently recommend by international guidelines.74–76 A 48-week course of weekly subcutaneous injections of pegIFNα suppresses HDV replication in approximately 20%–30% of the patients 24 weeks off therapy, yet with significant side effects. Continuous administration of IFN for more than 48 weeks may lead to a lower likelihood of disease progression, with HBsAg loss occurring in about 10% of these patients during long-term follow-up.77 Although the long term, off-treatment virological/biochemical response induced by IFN treatment has been associated with improved outcomes,72 73 IFN has limited use in clinical practice given the fact that this drug is contraindicated in elderly people or in those with autoimmune disease stigmata or with advanced or decompensated liver disease. Moreover, many patients have been already unsuccessfully exposed to standard or pegIFNα in the past. Combination of pegIFNα-2a with adefovir for 48 weeks78 or with tenofovir disoproxil fumarate (TDF) for 96 weeks did not significantly improve the off-treatment virological responses.79 Of note, in approximately 50% of week 24 off-treatment responders, a late virological relapse was observed, further challenging the long-term effectiveness of this treatment.80
The general failure of IFN treatment to induce a long-term (>24 weeks) sustained virological response on HDV may be due to the persistence of HDV in the liver even at very low HBsAg levels. This concept is supported by the observation that after liver transplantation, HDV can persist in the liver for many months even in the absence of liver HBV DNA/cccDNA and serum HBsAg and HDV RNA.81
New anti-HDV strategies: completed phase II studies
NAPs with pegIFNα
Phosphorothioate NAPs are oligonucleotides with a broad spectrum of inhibitory activity against several viruses, whose exact mechanism in HDV is still unknown (see previous section on virology).
In the non-randomised, open-label phase 2 REP301 study, 12 treatment-naïve non-cirrhotic patients with chronic HBV/HDV coinfection from Moldova received 500 mg of REP2139Ca intravenously once per week for 15 weeks, followed by 15 weeks of 250 mg REP2139Ca+pegIFNα, then followed by 33 weeks of pegIFNα monotherapy (overall 63 weeks of therapy).82 At 24-week off-treatment follow-up, 5 (42%) patients were HBsAg negative, 5 (42%) anti-HBs positive, 7 (58%) with HBV DNA <10 IU/mL and 7 (58%) with negative HDV RNA by Robogene assay. A marked increase in ALT levels was observed after the introduction of pegIFNα in five patients but all remained asymptomatic. All patients experienced at least one adverse event, mostly pegIFNα-related. The virological responses observed at week 24 off-therapy were confirmed when the off-treatment follow-up was extended up to 3 years.83
LNF with or without pegIFNα
LNF is an orally administered inhibitor of farnesyl-transferase that blocks the prenylation of L-HDAg, showing an intracellular accumulation of RNPs and a dose-dependent reduction of serum HDV RNA. To optimise the risk-to-benefit ratio of this regimen, LNF was combined with ritonavir (RTV) to enable achieving higher (fourfold to fivefold) systemic exposure while improving its gastrointestinal tolerability, and with pegIFNα to achieve a more profound inhibition of viral replication and HBsAg levels.
In the phase II LOWR HDV-2, 3 and 4 studies,84–86 the efficacy and safety of different doses of LNF+RTV in monotherapy or combined with pegIFNα administered for 12 or for 24 weeks were assessed. In summary, an all oral antiviral strategy based on LNF 50 mg two times per day+RTV 100 mg two times per day led to HDV RNA decline ≥2 log or below the lower limit of quantification (LLoQ) in 39% of patients (7 of 18) and ALT normalisation in 60% at week 24 (EOT) (tables 1 and 2). A combined therapy based on LNF+RTV+pegIFNα increased the EOT responses to 89% (8 of 9) and 78%, respectively (tables 1 and 2). To date, the off-treatment virological or biochemical response rates of these regimens are not available.
Virological response in HDV patients treated with pegIFNα, BLV with or without pegIFNα, pegIFNλ, and LNF+RTV with or without pegIFNα
Biochemical response in HDV patients treated with pegIFNα, BLV with or without pegIFNα, pegIFNλ, and LNF+RTV with or without pegIFNα
PegIFNλ with or without LNF+RTV
Lambda IFN binds a unique receptor vs type I IFN, highly expressed on hepatocytes, which may lead to a better safety profile of this compound compared with pegIFNα.
The phase II LIMT HDV study evaluated the safety, tolerability, and efficacy of subcutaneous pegIFNλ monotherapy administered at the dose of 120 vs 180 μg QW in addition to TDF/entecavir (ETV) in 30 patients with chronic HBV/HDV coinfection87 (tables 1 and 2). At week 72, by per-protocol (PP) analysis, the proportion of patients with undetectable HDV RNA, ALT normalisation and combined endpoints (ALT normalisation +≥2 log IU/mL HDV RNA decline vs BSL) was 16% vs 36%, 26% vs 36% and 11% vs 29%. Adverse events typically seen with pegIFNα were fewer, but 10% of patients experienced hyperbilirubinaemia and ALT/AST increases, that were reversible with dose reduction and without any signs of decompensation.
In the open-label phase II single arm LIFT HDV study, 26 HDV patients received LNF+RTV+pegIFNλ 180 μg QW for 24 weeks88 (tables 1 and 2). At week 24 (EOT), HDV-RNA decline was 3.2 (2.5–4.0) log IU/ml, 25/26 (96%) patients had a >2 log RNA decline and 11/26 (42%) patients had HDV RNA undetectable or <LLoQ. Dose reductions were required in 3/26 (11%) patients and treatment discontinuation in 4/26 (15%). At week 48, 24 weeks off-therapy, the virological response defined as HDV RNA <40 IU/mL by Quest Diagnostics assay was 19% (5 of 26 patients) by intention-to-treat (ITT) analysis and 23% (5 of 22) by PP analysis.89
BLV with or without pegIFNα
BLV, previously named Myrcludex-B, and approved in 2020 in Europe under the branded name of Hepcludex, is a subcutaneously delivered lipopeptide that mimics the NTCP RBD of the L-HBsAg, inhibiting the HBV/HDV entry into the hepatocytes (see previous section on virology).
Short-term therapy
In the multicentre phase IIb MYR202 study,90 120 TDF-treated patients with chronic HBV/HDV coinfection were randomised to different BLV doses (2, 5 or 10 mg/day) or TDF monotherapy for 24 weeks. A 2-log decline or undetectable HDV RNA at EOT (week 24) was reached by 46%, 47% and 77% of the patients treated with increasing doses of BLV but only in 3% of those on TDF monotherapy. ALT normalised in 43%, 50%, 40%, and 6%, while HBsAg levels were not affected. BLV was well tolerated, and elevation of glycine-conjugated and taurine-conjugated bile salts had no clinical consequences. An HDV-RNA relapse occurred in 60%, 80% and 83% of EOT HDV-RNA responders and was associated with a moderate increase in ALT levels.
In the phase II MYR203 study91 treatment with BLV, at different doses and with or without peg-IFN, was extended to 48 weeks (tables 1 and 2). 90 patients with chronic HBV/HDV coinfection were randomised into six treatment arms. The primary efficacy endpoint, HDV RNA below the LLoD (10 IU/mL) at week 72 (24 weeks off-therapy), was achieved by 0%, 53.3%, 26.7%, 6.7%, 6.7% and 33.3% of patients randomised to pegIFNα 180 QW, 2 mg BLV+pegIFNα, 5 mg BLV+pegIFNα, 5 mg BLV, 10 mg BLV+pegIFNa and 10 mg BLV, respectively. The corresponding ALT normalisation rates were 10%, 53.8%, 33.3%, 23.1%, 35.7% and 35.7%. HBsAg loss or >1 log IU/ decline at week 72 was observed only in patients treated with BLV combined with pegIFNα: 40% for BLV 2 mg, 13.3% for 5 mg and 13.3% for 10 mg. BLV was generally well tolerated; an asymptomatic dose-related increase of total bile acids was observed. Most adverse events were observed in patients treated with pegIFNα, without any difference between patients treated as a monotherapy or in combination with BLV.
Long-term therapy
Two patients with compensated HDV-related cirrhosis, one with oesophageal varices, were treated with BLV monotherapy 10 mg for up to 3 years.71 Both cases normalised ALT levels before week 28 and achieved undetectable HDV RNA (<6 IU/mL by Robogene assay) before week 52. Biochemical and virological responses were maintained over 3 years of BLV administration without relapse or breakthrough, even after dose reduction of BLV from 10 to 5 and 2 mg/day.92 In the patient with more severe liver disease, virological response was associated with an excellent clinical response: oesophageal varices disappeared, histological/laboratory features of autoimmune hepatitis secondary to HDV infection resolved, AFP levels normalised and liver stiffness, platelets and albumin levels significantly improved. As far as safety is concerned, only asymptomatic, dose-related increase of total bile acids was observed. The optimal duration of long-term therapy with BLV monotherapy is at present unknown although application of an HCV-based kinetics model to HDV-infected patients suggests that after 3 years of continuous suppression of HDV replication by BLV more than 50% of the patients might achieve a long-term off-therapy virological response, despite persistence of HBsAg 93 and unpublished data).
New anti-HDV strategies: ongoing phase III studies
Two multicentre international phase III registration studies are ongoing (figure 5). In the D-LIVR study (EIG-LNF-011), 400 patients with chronic HBV/HDV coinfection will be randomised to four treatment arms (figure 5A) for 48 weeks. The primary endpoint is the proportion of patients achieving a≥2 log10 IU/ml reduction in serum HDV-RNA level and ALT normalisation at week 48 (EOT). In the MYR301 study, 150 patients with chronic HBV/HDV coinfection have been randomised to three different arms (figure 5B). The primary objective is to evaluate the safety and efficacy of different doses of BLV monotherapy administered up to 144 weeks. The total duration of the study is 240 weeks.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Study design of the two ongoing phase III studies assessing the efficacy and safety of new therapeutic regimens against HDV. (A) D-LIVR study. LNF +RTV: LNF 50 mg two times per day+RTV 100 mg two times per day. Primary endpoint: ≥2 log10 IU/mL decline in HDV RNA and ALT normalisation in week 48. All patients will be maintained on background HBV nucleoside/nucleotide analogue therapy. (B) MYR301 study. primary endpoint: undetectable HDV RNA or decrease by ≥2log10 IU/mL and ALT normalisation in week 48. If indicated treatment with nucleoside/nucleotide analogues according to European Association for the Study of the Liver (EASL)/American Association for the Study of the Liver (AASLD) guidelines. ALT, alanine aminotransferase; BLV, bulevirtide; HBV, hepatitis B virus; HDV, hepatitis D virus; LNF, lonafarnib; pegIFNα, pegylated interferon-α; RTV, ritonavir;
New therapeutic approaches targeting HBsAg
Apart from the above-mentioned therapies, any new therapeutic that leads to functional cure in HBV monoinfected patients could be helpful in HBV/HDV coinfected ones.72 RNA interference and antisense oligonucleotides showed substantial declines of HBsAg in few weeks of administration in the absence of peg-IFN, suggesting a potential role also in the treatment of coinfected patients.
Current treatment options
BLV at a dose of 2 mg sc every day was approved by EMA in 2020 and is beside off-label use of pegIFNαthe only treatment option, at least in the EU. FDA approval is pending. BLV could be used either in combination with pegIFNα (without formal EMA approval) or as monotherapy. A ‘curative’ strategy based on short-term (48 week) administration of sc daily injections of 2 mg BLV combined with pegIFNα may result in HBsAg response and sustained off-therapy HDV-RNA negativity in some patients and may be used preferentially in those patients with well-compensated disease. For the many HDV patients who cannot be treated with pegIFNα for different reasons, administration of BLV monotherapy may be a promising ‘suppressive’ strategy. However, the current data indicate that the approved low dose (2 mg every day) is suboptimal and the optimal duration of high-dose therapy and its long-term safety profile is currently investigated (MYR301 trial). Studies on the safety in patients with decompensated cirrhosis are required and a more convenient mode of administration or less frequent administrations would be desirable.
Conclusions
Forty years after the discovery of HDV, the first anti-HDV therapeutic has been approved in Europe, indicating the beginning of a new era for these difficult to treat/cure patients. Nevertheless, many questions related to HDV virology and immunology that affect sustained treatment responses to novel drugs remain to be solved (see box 1 ‘Open questions and future directions’), and a deeper understanding of virological and immunological mechanisms contributing to viral control will help to advance on the road to HDV eradication.
Open questions and future directions
Can hepatitis D virus (HDV) establish transcriptionally silenced but reactivatable episomes in hepatocytes as an additional mechanism of persistence?
To what extent do the eight HDV genotypes differ in replication efficacy and sensitivity against the upcoming novel treatments?
Do HDV-targeted therapies lead to a restoration of HDV-specific immunity?
Are these restored immune responses required for treatment response? Are they required for prevention of viral relapse? Of note, there may be differences in treatment regimens that are associated with alanine aminotransferase (ALT) flares (eg, REP2139Ca+pegylated interferon-α (IFN α) combination therapy) compared with treatment regimens without ALT flares (eg, BLV monotherapy).
How can a synergistic potential of antiviral drugs and immune-modulators be translated into curative regimens?
Are there baseline or on-treatment predictors (eg, immune responses, kinetics of virological response) to sustained HDV virological response for the different treatment strategies?
Is a sustained HDV virological response without hepatitis B surface antigen (HBsAg) loss a realistic and achievable aim for treatment regimens without IFNs?
Are drugs aiming at HBsAg loss effective and safe in patients with chronic hepatitis B virus/HDV coinfection?
Last but not least, since HDV is prevalent in low-income countries and migrant populations, it will be important to establish new concepts to foster diagnosis and access to care.
Ethics statements
Acknowledgments
We are grateful to Dr Zhenfeng Zhang for help with figures 1 and 2.
References
Footnotes
SU and CN-H contributed equally.
Contributors All authors contributed to the writing of the manuscript and design of figures, and approved the final version.
Funding This work received funding by German Center for Infection Research (DZIF), TTU Hepatitis, project 5.704 (to SU) and 5.822 (to SU and CN-H) and the Deutsche Forschungsgemeinschaft TRR179 (project no. 272983813; TP15 and TP9 to SU, TP2 to CN-H).
Competing interests SU: Advisory Board/Speaker Bureau for: GILEAD SCIENCES, MYR, VIRBIO, ASSEMBLY, JANSSEN, ENYO, PEPPERPRINT, ALIGOS. CN-H: Speaker Bureau for: ABBVIE, Falk Foundation, Novartis, MSD. PL: Advisory Board/Speaker Bureau for: BMS, ROCHE, GILEAD SCIENCES, GSK, ABBVIE, MSD, ARROWHEAD, ALNYLAM, JANSSEN, SBRING BANK, MYR, EIGER.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.