Article Text

Abstract
Objective Intratumor heterogeneity drives cancer progression and therapy resistance. However, it has yet to be determined whether and how subpopulations of cancer cells interact and how this interaction affects the tumour.
Design We have studied the spontaneous flow of extracellular vesicles (EVs) between subpopulations of cancer cells: cancer stem cells (CSC) and non-stem cancer cells (NSCC). To determine the biological significance of the most frequent communication route, we used pancreatic ductal adenocarcinoma (PDAC) orthotopic models, patient-derived xenografts (PDXs) and genetically engineered mouse models (GEMMs).
Results We demonstrate that PDAC tumours establish an organised communication network between subpopulations of cancer cells using EVs called the EVNet). The EVNet is plastic and reshapes in response to its environment. Communication within the EVNet occurs preferentially from CSC to NSCC. Inhibition of this communication route by impairing Rab27a function in orthotopic xenographs, GEMMs and PDXs is sufficient to hamper tumour growth and phenocopies the inhibition of communication in the whole tumour. Mechanistically, we provide evidence that CSC EVs use agrin protein to promote Yes1 associated transcriptional regulator (YAP) activation via LDL receptor related protein 4 (LRP-4). Ex vivo treatment of PDXs with antiagrin significantly impairs proliferation and decreases the levels of activated YAP.
Patients with high levels of agrin and low inactive YAP show worse disease-free survival. In addition, patients with a higher number of circulating agrin+ EVs show a significant increased risk of disease progression.
Conclusion PDAC tumours establish a cooperation network mediated by EVs that is led by CSC and agrin, which allows tumours to adapt and thrive. Targeting agrin could make targeted therapy possible for patients with PDAC and has a significant impact on CSC that feeds the tumour and is at the centre of therapy resistance.
- pancreatic cancer
- cell biology
- carcinogenesis
- molecular carcinogenesis
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE84,85 partner repository with the dataset identifier PXD023529 and 10.6019/PXD023529. All the codes to reproduce the data alignment and statistical analysis are publicly available at GitHub repository (GitHub, RRID:SCR_002630, https://github.com/fjcamlab/exonet_MS).
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Intratumor heterogeneity is a feature of pancreatic ductal adenocarcinoma (PDAC) tumours that contributes to their dismal prognosis.
Cooperation between subpopulations of cancer cells has been postulated to promote disease progression and therapy resistance.
Cancer cells communicate with cells of the tumour microenvironment and distant organs by means of EVs to support tumour progression.
What are the new findings?
EVs from subpopulations of PDAC cells establish an organised communication network, the Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network (EVNet), which confers plasticity to the tumour and supports tumour progression.
Communication from cancer stem cells (CSC) to non-stem cancer cells activates YAP in PDAC tumours and drives disease progression. Mechanistically, we found that agrin in CSC EVs acts through the LRP-4 receptor to activate YAP in PDAC tumours.
Targeting the EVNet and CSC EVs by means of Rab27a or agrin inhibition impairs PDAC tumour growth.
How might it impact on clinical practice in the foreseeable future?
Circulating agrin-positive EVs are potential biomarkers to determine the response to therapy and the risk of PDAC progression.
Our results suggest that antiagrin therapy could target CSC and therefore significantly impact tumour progression and therapy resistance, thus opening the possibility of targeted therapy for patients with PDAC.
Introduction
Intratumor heterogeneity is described as one of the main drivers of pancreatic ductal adenocarcinoma (PDAC) progression and therapy resistance.1 Genomic, non-genomic and functional cell state heterogeneity results in distinct cancer cell behaviours and represents one of the major hurdles for cancer treatment. Evidence of PDAC heterogeneity shows that the presence of distinct cell-surface molecules defines subpopulations of cancer cells with different tumorigenic capacity.2 3 In particular, pancreatic cancer stem cells (CSC) are characterised by the expression of specific surface markers and are associated with poor prognosis in PDAC.4 5 CD24, CD44, CD133 and EpCAM identify subpopulations with distinct tumorigenic capacity in pancreatic cancer.2 3 It has been postulated that cooperation between subpopulations of cancer cells is critical to maintain heterogeneity and potentiate tumor-promoting functions.6 However, how this cooperation takes place remains elusive.
Extracellular vesicles (EVs) are central mediators of intercellular communication.7 8 EVs are secreted by all cell types and can be found in all body fluids.9 EVs make use of their genetic and molecular cargo to reprogramme recipient cells.10 In particular, cancer EVs promote the differentiation of cancer-associated fibroblasts (CAFs),11 enhance angiogenesis,12 modulate the antitumour immune response,13 establish a premetastatic niche14 15 and confer metastatic properties to non-metastatic cancer cells.16 17 However, the exchange of EVs between subpopulations of cancer cells with distinct phenotypical and tumour-forming abilities has yet to be addressed.
For the first time, our report unravels an organised network of communication led by CSC EVs and demonstrates its significance in the biology of the tumour. We reveal that this communication is a fundamental process for cooperation within the tumour and contributes to its plastic nature and resistance to therapy.
Results
Subpopulations of PDAC cells use EVs to form an organised communication network, the EVNet and the preferential communication route occurs from CSC to NSCC
To determine whether and how cancer EVs flow between subpopulations of PDAC cells, we developed a tracking system using EVs markers tagged with distinct fluorescent reporters. A library of fluorescently labelled stable clones expressing markers highly represented in EVs (CD63, CD81, CD82 or Rab5) fused with reporter proteins (turboGFP, tdTomato, eYFP and mPlum, respectively) was established in a human PDAC cell line (MIA PaCa-2, figure 1A). Using imaging flow cytometry, we demonstrated that EVs isolated from human pancreatic cancer cell lines, including MIA PaCa-2, are positive for CD63, CD81, CD82 and Rab5 (online supplemental figure 1A).
Supplemental material

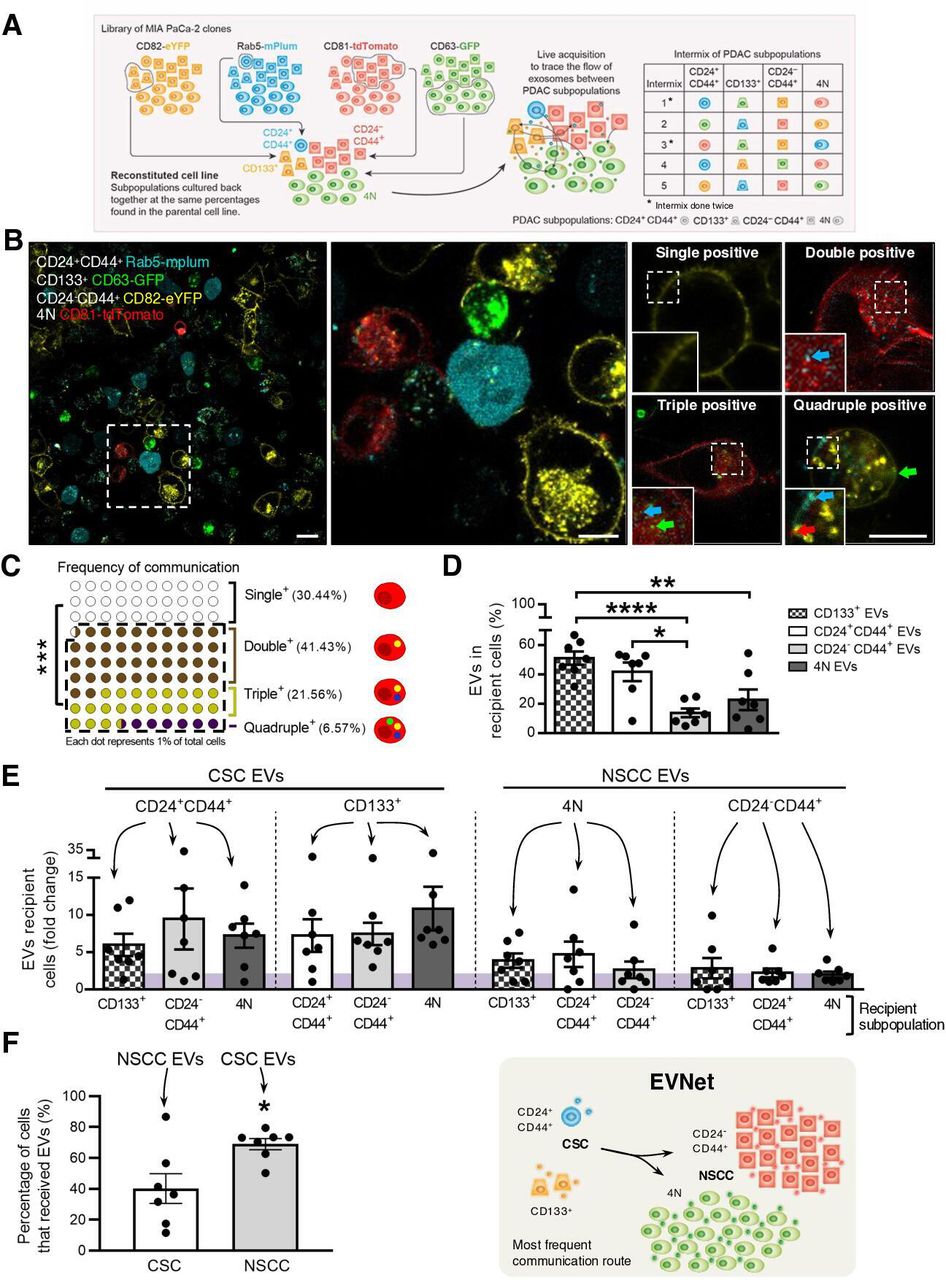
The most frequent communication route in the PDAC EVNet occurs from CSC to NSCC. (A) Schematics of the methodological approach: MIA PaCa-2 clones expressing different fluorescently labelled markers are cultured at the percentages found in the parental cell line and are analysed through flow cytometry in order to trace the flow of EVs among subpopulations. A total of seven intermix of colour-coded subpopulations was analysed, corresponding to five distinct intermixes. Subpopulations: CD24+CD44+ refers to CD24+CD44+CD133−EpCAM−; CD133 +refers to CD133+CD24−CD44−EpCAM−; CD24-CD44+ refers to CD24−CD44+CD133−EpCAM−; 4N refers to CD24−CD44−CD133−EpCAM−. (B) Representative confocal microscopy live images of 72-hour culture MIA PaCa-2 colour-coded subpopulations (CD133+ CD63-GFP, CD24+44+ Rab5-mPlum, CD24-CD44+ CD82-eYFP and 4N CD81-tdTomato). Right panel: representative images of a cell where EVs uptake was not detected (single positive), a cell that received EVs from one different subpopulation (double positive), a cell that received from two other subpopulations (triple positive) or from all four subpopulations (quadruple positive). Scale bars from left to right: 20, 10 and 10 µm. (C) Dot plot representing the percentage of single-positive, double-positive, triple-positive and quadruple-positive cells found in the cocultures (n=7, Mann-Whitney test ***p<0.001). (D) Quantification of the percentage of cells that received EVs from CD133+, CD24+CD44+, CD24−44+ and 4N subpopulations (n=7, one-way analysis of variance (ANOVA) *p<0.05, **p<0.01, ****p<0.0001). (E) Quantification of cells of different subpopulations that receive EVs from CSC or NSCC (highlighted on top). The fold change was calculated against the minimum percentage of communication found in each coculture (n=7). (F) Quantification of the percentage of cells that received EVs (left) and schematic representation of the most frequent communication route in the EVNet, from CSC to NSCC (right, comparison of CSC positive for NSCC EVs and NSCC positive for CSC EVs; n=7, Mann-Whitney test, *p=0.0379). Data are mean±SEM. CSC, cancer stem cell; EVs, extracellular vesicles; EVNet, Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network; NSCC, non-stem cancer cells; PDAC, pancreatic ductal adenocarcinoma.
Based on cell-surface markers, we have identified four subpopulations of cancer cells in PDAC cell lines (n=4), patient-derived xenografts (PDXs n=5) and a genetically engineered mouse model (GEMM, online supplemental figure 1B–D). CD133+ and CD24+CD44+ cells both identify two rare subpopulations characterised as CSC.2 3 The third and fourth subpopulations are CD24−CD44+ and 4N (CD133−CD24−CD44−EpCAM−), which represent the most abundant subpopulations and identify NSCC. We have confirmed their CSC and NSCC phenotypes using spheroid forming assays, which demonstrated that CSC have a significantly enhanced capacity to form spheres compared with NSCC (online supplemental figure 1E). Importantly, across the different models used, the proportions of CSC versus NSCC are consistent (MIA PaCa-2: CSC 3.81% and NSCC 93.08%, PDX: CSC 0.98% and NSCC 99.37%, and KPC (LSL-KrasG12D/+, LSL-Tp53R172H/+ and Pdx-1-Cre): CSC 2.21% and NSCC 97.49%; online supplemental figure 1BCD). In addition, the frequencies of the CSC and NSCC in the stable clones used to evaluate the flow of EVs between distinct subpopulations were not significantly different when compared with the parental cell line (online supplemental figure 1F).
We used fluorescence-activated cell sorting (FACS) to isolate one specific cell subpopulation (CD133+, CD24+CD44+, CD24-CD44+ or 4N) from each stable colour-coded clone (MIA PaCa-2 CD63-GFP, CD81-tdTomato, CD82-eYFP and Rab5-mPlum; figure 1A). Each subpopulation secretes EVs with a different colour (figure 1A). To mix the subpopulations and trace the flow of EVs between them, we cultured them back together at the same percentages found in the parental cell line in a total of seven experiments (figure 1A). Confocal imaging analysis after 72 hours of culture (figure 1B) showed that 69.56%±11.92% of cells were positive for EVs of other subpopulations, demonstrating that cancer EVs spontaneously flow between them (figure 1C). The majority of cells received EVs from one or two other subpopulations (double or triple positive cells, respectively; figure 1C and online supplemental figure 1G). Cells that received EVs from every other subpopulation (quadruple positive) were scarce (6.57%±7.82%, figure 1C and online supplemental figure 1G). The percentage of recipient cells that were positive for EVs from CD133+ and CD24+CD44+ subpopulations (CSC) was significantly higher than that of cells that were positive for EVs from CD24−CD44+ and 4N subpopulations (NSCC, figure 1D–F). This was true independently of the EVs marker or the fluorescent protein used to tag these subpopulations. Hence, the flow of EVs from CSC to NSCC constitutes the most frequent communication route between subpopulations of PDAC cells in comparison to communication to CSC and within both subpopulations (figure 1).
We next tested whether the secretion rate of EVs from each subpopulation could explain the directionality of the EVNet and the predominance of the communication route from CSC to NSCC. To this end, we quantified EVs isolated from each subpopulation by nanoparticle tracking analysis (NTA). We demonstrated that CSC secreted more EVs per cell than the 4N subpopulation (figure 2A). Although this could partially contribute to the communication route observed from CSC to NSCC, it is important to note that the difference in the numbers of EVs secreted per cell does not reach a 2.5-fold increase (figure 2A). In striking contrast, CSCcorrespond to about 3.81% of the cells, while NSCC are the most prevalent (93.08%, online supplemental figure 1B). Thus, the number of EVs secreted per cell per se does not explain the directionality of the flow of EVs from CSC to NSCC.

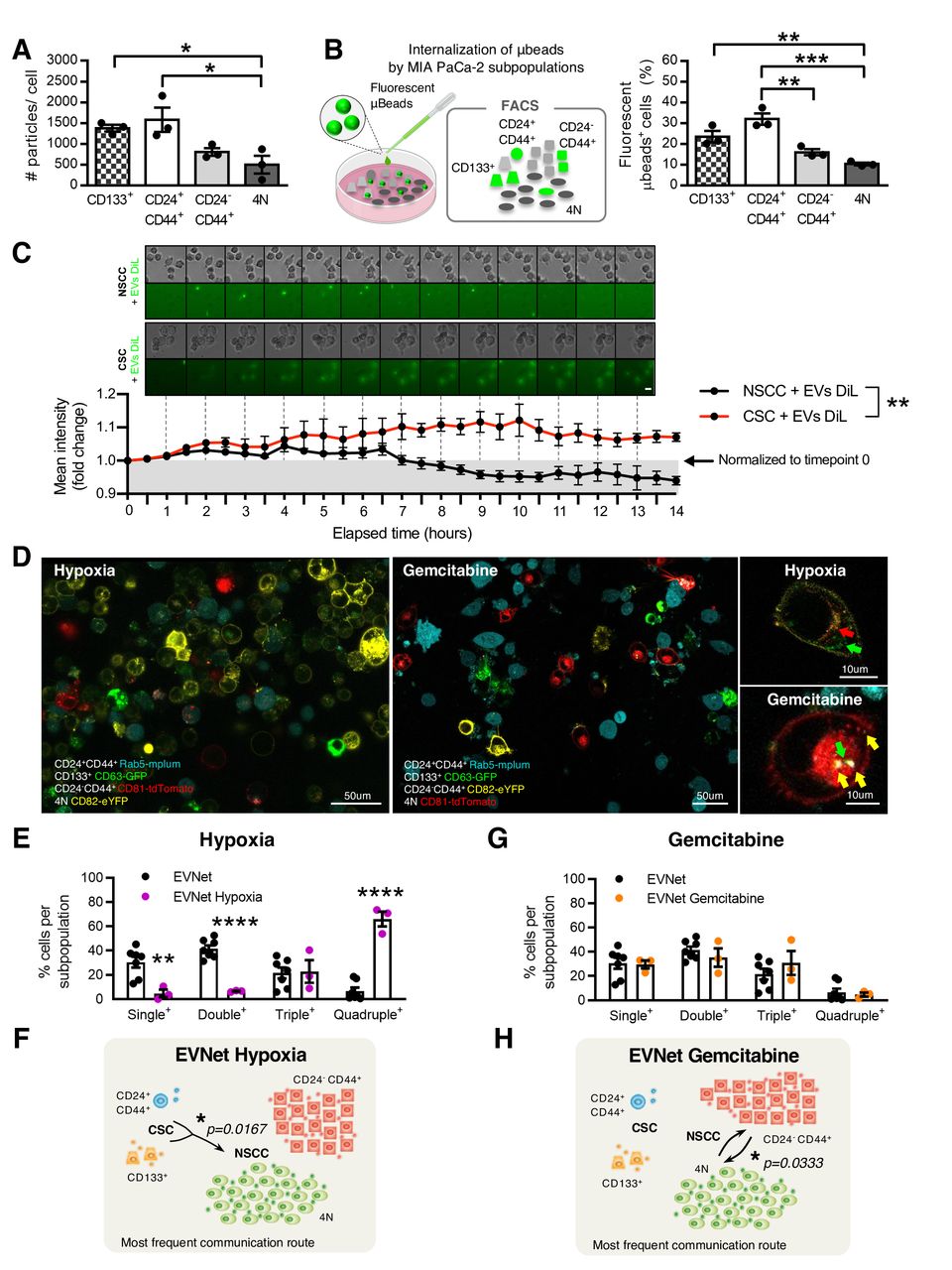
The EVNet is an organised and plastic communication network of EVs. (A) Quantification of the number of particles secreted by individual MIA PaCa-2 subpopulations measured by NTA (n=3, one-way ANOVA; *p<0.05). (B) Experimental set-up (left): MIA PaCa-2 subpopulations were treated with fluorescent microbeads and analysed through flow cytometry. Quantification of the percentage of subpopulations showing uptake of fluorescent microbeads (right, n=3, one-way ANOVA; **p<0.01, ***p<0.001). (C) Representative time-lapse images of CSC and NSCC treated with 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate (DiL)- labelled EVs (top). Graphic depicts fold change of mean fluorescence intensity relative to timepoint 0 (bottom, n=5 images per group, two-way ANOVA; **p=0.0037). (D) Representative confocal microscopy live images of the MIA PaCa-2 cells with colour-coded subpopulations cultured for 72 hours in hypoxia (1% O2, left) or treated with gemcitabine (1 µM, right). Scale bars on larger panels: 50 µm, and on smaller panels: 10 µm. (E) Quantification of the percentage of single-positive, double-positive, triple-positive and quadruple-positive cells in MIA PaCa-2 cultures in hypoxia (1% O2, n=3; one-way ANOVA; **p<0.01, ****p<0.0001). (F) Respective graphical representation of the communication network of communication established between subpopulations of cancer cells under hypoxic conditions. The most frequent communication route from CSC to NSCC is significant in comparison to the same route in the EVNet (Mann-Whitney test, *p=0.0167). (G) Quantification of the percentage of single-positive, double-positive, triple-positive and quadruple-positive cells in MIA PaCa-2 cultures treated with gemcitabine (1 µM, n=3) and (H) respective graphical representation of the communication network established between subpopulations of cancer cells under gemcitabine treatments. The most frequent communication routes occur between the two subpopulations of NSCC compared with the same routes in the EVNet (Mann-Whitney test, *p=0.0333). Data are mean±SEM. ANOVA, analysis of variance; CSC, cancer stem cell; EVs, extracellular vesicles; EVNet, Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network; FACS, fluorescence activated cell sorting; NSCC, non-stem cancer cell; NTA, nanoparticle tracking analysis.
We then sought to determine whether the observed routes of communication are dependent on the intrinsic ability of each subpopulation to endocytose EVs. We treated the MIA PaCa-2 cell line with fluorescent polystyrene carboxyl-functionalised microbeads (100 nm average diameter, figure 2B). Flow cytometry analysis showed that subpopulations of CSC have the highest rate of nanoparticle uptake compared with NSCC (figure 2B). This indicates that the EVNet observed is also not dependent on the endogenous endocytic capacity of each subpopulation. In addition, using time-lapse microscopy, we demonstrated that the degradation rate of EVs taken up by NSCC is significantly faster in comparison to EVs taken up by CSC (figure 2C). Thus, the observed communication routes are also not likely to be dependent on differences between the degradation rate of EVs by CSC and NSCC.
Taken together, our data indicate that distinct PDAC subpopulations use EVs to establish an organised communication network, the EVNet. In the EVNet, communication occurs more frequently from the rare subpopulations of CSC to NSCC. The routes of communication in the EVNet are not determined by the number of secreted EVs, their degradation rate in recipient cells or the endocytic capacity of each subpopulation of PDAC cells, thus, suggesting that this is a regulated process.
EVNet is plastic and supports adaptation of cancer cells to microenvironmental changes
Heterogeneity plays a key role in tumour plasticity, which is crucial in the response to microenvironmental changes and therapy. We sought to determine whether the EVNet could constitute a mechanism by which the tumour promptly and effectively adapts to changes in its environment, such as hypoxia (frequently found in PDAC18) and chemotherapy. We found that hypoxia significantly increased the communication between cancer cells (figure 2D,E). As such, there was a significant decrease in the single positive cells (cells that have not received EVs, 30.44±11.92% to 4.72±5.37%), as well as a significant increase in the amount of quadruple positive cells (6.57%±7.82% to 66.04±10.64%, figure 2E) in comparison to the EVNet. This increased communication could be due to enhanced secretion of EVs, which was demonstrated by the increased level of the EVs markers CD81, CD9 and syntenin-1 in EV preparations after exposure to hypoxia (online supplemental figure 2A).
Supplemental material
Along with the increase in overall communication, CSC to NSCC remains the most predominant communication axis, and there is a significant increase in the number of EVs that participated in this communication route (figure 2F). Conversely, although gemcitabine does not yield a significant alteration in the percentage of cancer cells that exchange EVs (figure 2D,G), the EVNet undergoes reshaping, with the most frequent communication routes now occurring between subpopulations of NSCC (CD24−CD44+ and 4N), which increases in frequency significantly in comparison to the same route in the EVNet (figure 2H). In addition, cells treated with gemcitabine take up more beads (online supplemental figure 2B), which suggests that cells are more permeable. Hence, the communication routes can be less specific.
To gain further insight into the role of the EVNet in adaptation to microenvironmental changes, we established a stable clone of the MIA PaCa-2 cell line expressing a doxycycline-inducible (Tet-On) short hairpin that targets Rab27a. Rab27a is a small GTPase involved in the exocytosis of vesicles of endosomal origin.19 Western blot validated downregulation of Rab27a using the Tet-On system (online supplemental figure 2C). Consistent with prior reports,20 21 NTA analysis showed that the number of EVs released was significantly decreased on downregulation of Rab27a (online supplemental figure 2D). To control for the effect of Rab27a downregulation in the secretion of components not related with vesicles in our model, we used a cytokine array to evaluate the secretion levels of 42 proteins. We observed that on Rab27a downregulation, there were no significant differences in the 42 proteins analysed (online supplemental figure 2E). In order to identify the possible role of EVs in the adaptation of cancer cells to hypoxia and gemcitabine treatments, we performed Rab27a knockdown in cells subjected to both conditions and examined cell death. We demonstrated that cancer cells with Rab27a knockdown and consequent impairment of EVs secretion become significantly more sensitive to both conditions, which is reflected in a significant increase in cell death. This suggests that EVs contribute to cancer cells’ adaptation and potentially for therapy resistance (online supplemental figure 2FG).
In sum, we have provided evidence of the EVNet in cancer and identified the communication route from CSC to NSCC as the most frequent one in PDAC. Most importantly, we demonstrated that the EVNet is plastic and changes the rate of communication or its routes when faced with distinct environmental challenges.
Inhibition of Rab27A impairs PDAC progression
We next aimed to understand the biological significance of the EVNet in PDAC. For this, MIA PaCa-2 Tet-On shRab27a cells were orthotopically implanted in the pancreas of immunodeficient mice (Rag2−/−Il2rg−/−), and doxycycline was administered in food pellets (figure 3A). Doxycycline-treated mice (n=8) showed a significant decrease in tumour growth, which was monitored by ultrasound, and there was a significant reduction in the number of liver macrometastases in comparison to the control group (n=7; figure 3B,C, and online supplemental figure 3). We did not observe any histological differences between tumours of both groups, and liver metastasis was histologically confirmed (figure 3D). Using immunohistochemistry (IHC), we demonstrated that tumours of doxycycline-fed MIA PaCa-2 Tet-On shRab27a mice had a significant downregulation of Rab27a (online supplemental figure 3B). To control for a potential effect of doxycycline in our analysis, we orthotopically implanted MIA PaCa-2 Tet-On shScramble cells in the pancreas of immunodeficient mice (control group n=6, doxycycline group n=6) and demonstrated that doxycycline did not affect tumour growth and metastatic capacity, thus confirming that our results are not associated with doxycycline (online supplemental figure 3CD).
Supplemental material
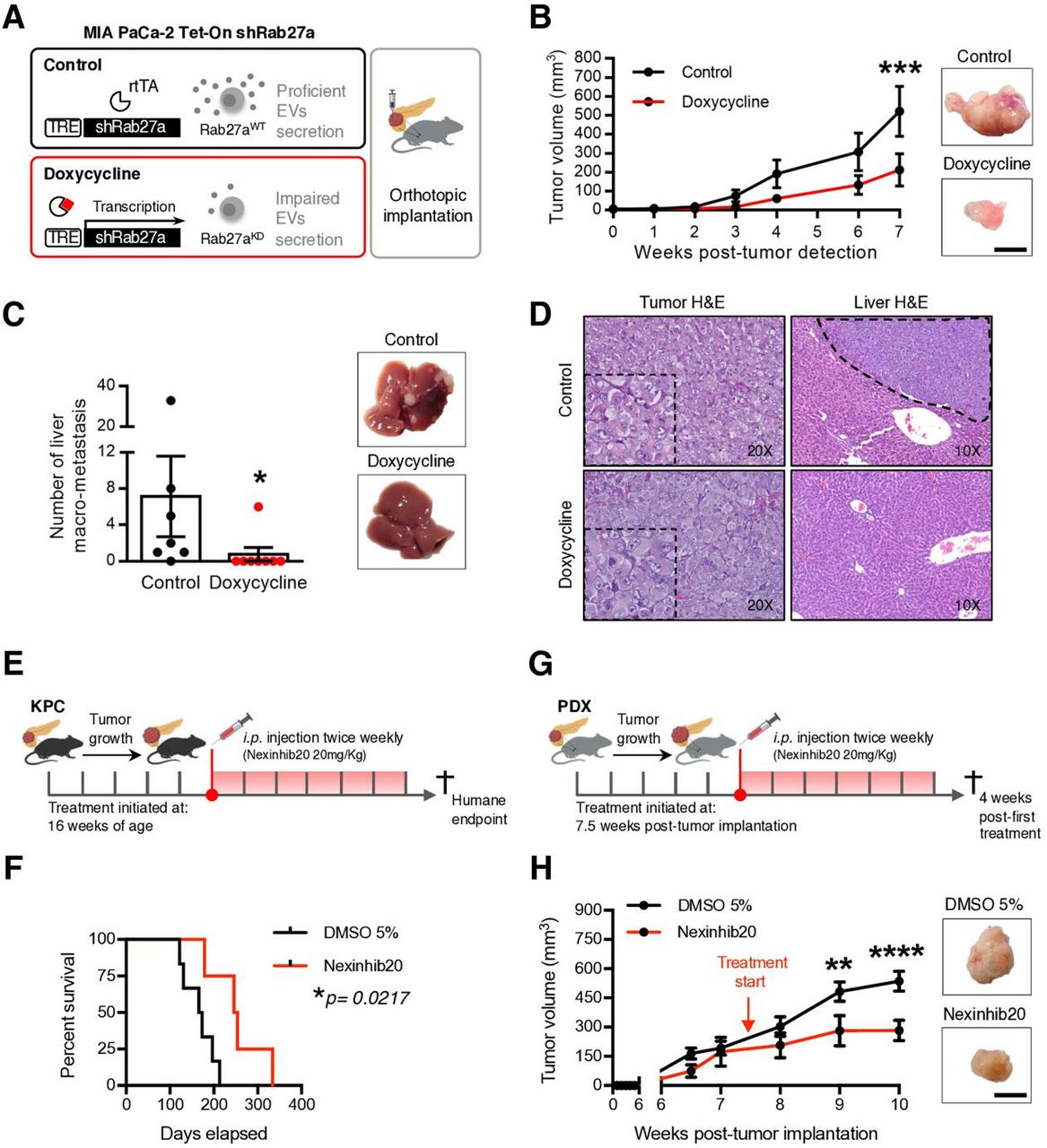
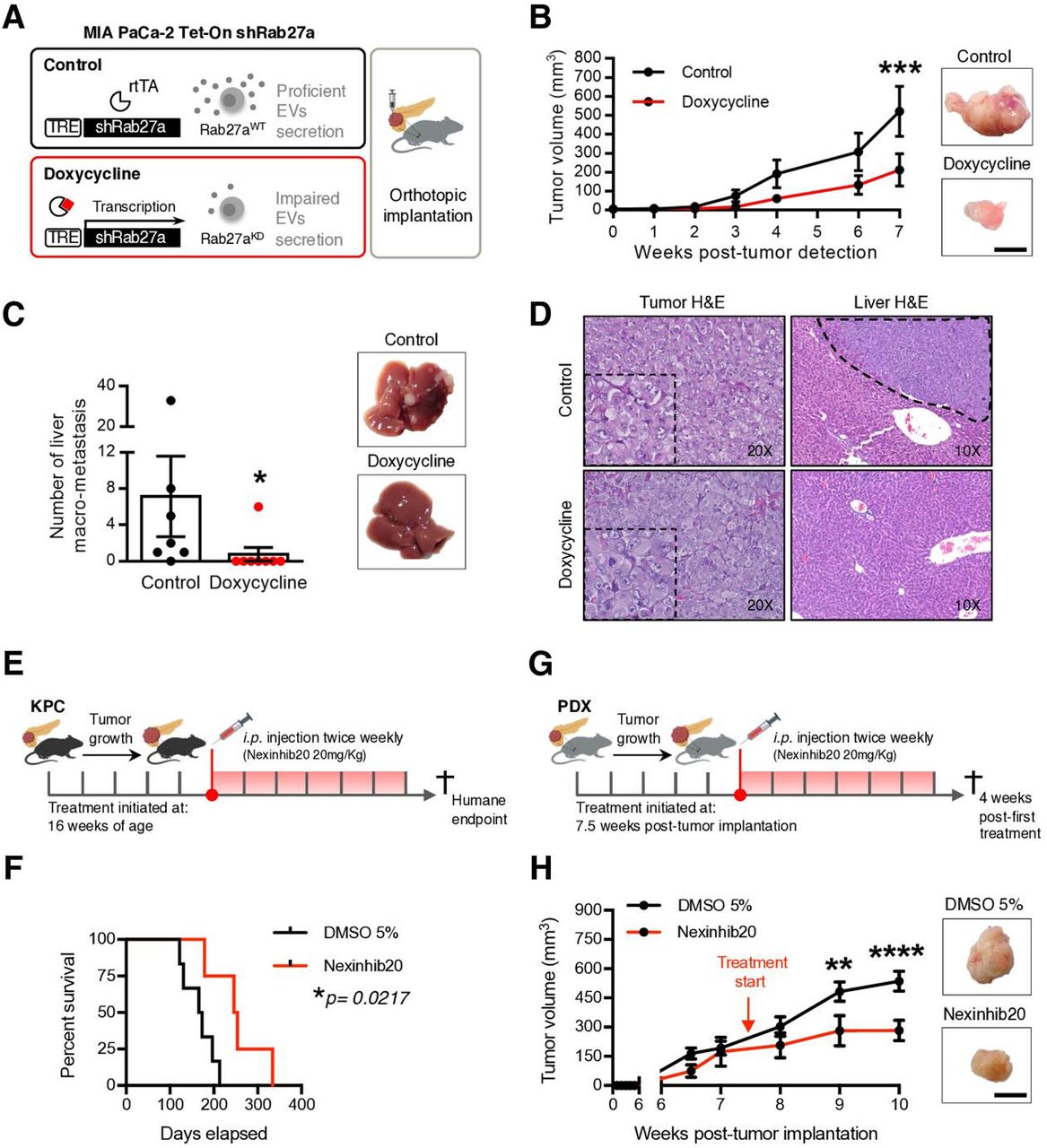
Impairment of the EVNet hampers tumour growth in PDAC orthotopic, GEMM and PDX models. (A) Experimental layout: in order to impair cancer EVs secretion, a doxycycline-inducible (Tet-On) shRNA against Rab27A was transfected into MIA PaCa-2 cells, which were then orthotopically implanted into the pancreas of Rag2−/−Il2rg−/− mice. (B) Tumour growth curve measured by ultrasound and representative photos of MIA PaCa-2 Tet-On shRab27a tumours treated with doxycycline (n=8) and control (non-treated, n=7, two-way ANOVA; ***p<0.001). Scale bar: 10 mm. (C) Liver macrometastasis quantification and representative photos of livers of the MIA PaCa-2 Tet-On shRab27a model treated with doxycycline (n=8) and control (n=7) (Mann-Whitney test, *p<0.05). (D) Representative H&E staining of orthotopic MIA PaCa-2 Tet-On shRab27a tumours (left, with zoom inset) and liver metastasis (right, dashed line) treated with doxycycline and control. (E) Experimental outline of KPC mice treated with Nexinhib20 (20 mg/kg) or dimethyl sulfoxide (DMSO) (5%) at 16 weeks of age when tumours are mature and are sacrificed at humane endpoint. Treatments were administered two times per week by intraperitoneal injection. (F) Kaplan-Meier curve of the overall survival of KPC mice treated with Nexinhib20 (n=4) vs DMSO 5% (n=6) (log-rank Mantel-Cox test, *p=0.0217). (G) Experimental layout of mice injected orthotopically in the pancreas with PDX treated with Nexinhib20 (20 mg/kg) or DMSO (5%) two times per week. Treatment was started 7.5 weeks post-tumour implantation, and mice were sacrificed 4 weeks late, at 11.5 weeks post-tumour implantation. (H) Tumour growth curve of PDX pancreas orthotopic tumours measured by ultrasound treated with Nexinhib20 (n=6) or DMSO 5% (n=6) and representative photos of tumours at the time of euthanasia (two-way ANOVA; **p<0.01, ****p<0.0001). Arrows depict timepoints where treatment was started (7.5 weeks). Scale bar: 10 mm. Data are mean±SEM. ANOVA, analysis of variance; EVs, extracellular vesicles; EVNet, Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network; GEMM, genetically engineered mouse model; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft; TRE, tetracycline response element.
Next, we impaired EVs secretion in the PDAC GEMM KPC, which faithfully recapitulates the histopathology of the human disease, and in an orthotopic PDX model (figure 3E,G). Both models were treated with a specific small molecule inhibitor of Rab27a, Nexinhib20.22 Treatment of KPC mice with Nexinhib20 (n=4) significantly increased their survival in comparison to the controls (DMSO 5%, n=6; figure 3F). Importantly, treatment of the orthotopic PDXs with Nexinhib20 (n=6) also significantly reduced tumour growth in comparison to the control (DMSO 5%, n=6; figure 3H). Histological analysis and IHC of antihuman MUC1 on orthotopically implanted PDXs confirmed that they are PDAC tumours of human origin (online supplemental figure 4A, left panel). A tumour from the murine Panc02 pancreatic cancer cell line was used as a negative control (online supplemental figure 4A, right panel). Importantly, ex vivo treatment of cancer cells with Nexinhib20 isolated from five PDXs and two PDAC GEMMs (KPC and KPPC: LSL-KrasG12D/+, LSL-Tp53R172H/R172H, Pdx-1-Cre) led to a significant reduction in the number of secreted EVs (online supplemental figure 4B). Downregulation of secreted EVs in MIA PaCa-2 cells treated with Nexinhib20 was also demonstrated by reduction of the EVs marker CD6323 and β-actin (cytoskeletal protein abundant in EVs,23 online supplemental figure 4C,D). Cytochrome C and acetyl–cholinesterase were detected exclusively in cell lysates, demonstrating that our preparations of EVs were not contaminated (online supplemental figure 4E). The concentration of Nexinhib20 used was determined by a methylthiazolyldiphenyl–tetrazolium bromide (MTT) assay, which identified the maximum concentration that does not affect the viability of PDAC cells (online supplemental figure 4F).
Supplemental material
In summary, we demonstrated that Rab27a knockdown, which impairs communication mediated by EVs, in orthotopic, GEMM, and PDX models undermine disease progression.
Inhibition of Rab27A in CSC is sufficient to impair PDAC growth
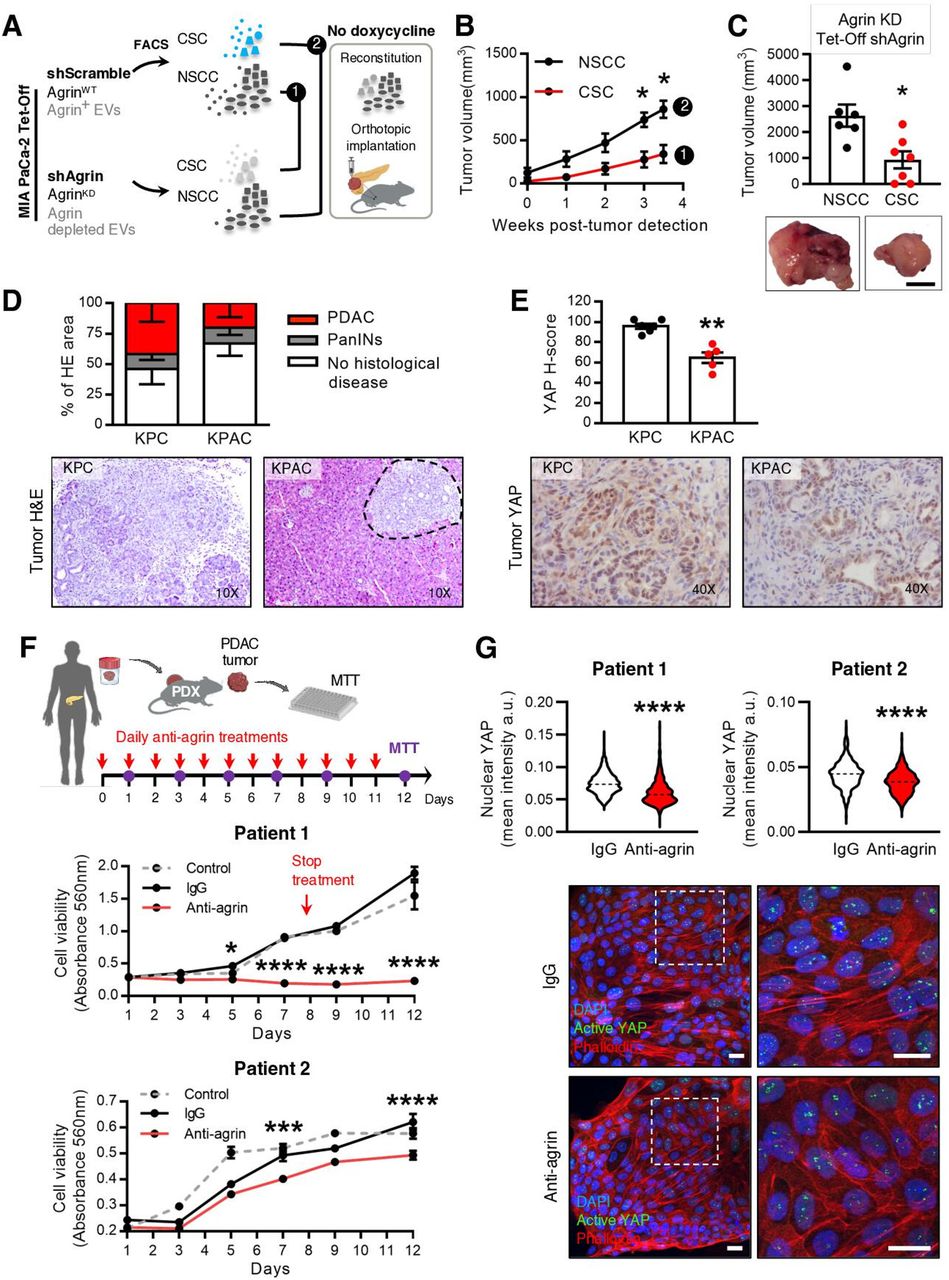
Since the most frequent communication route in the EVNet occurs from CSC to NSCC we set out to determine its role in the biology of the tumour. To specifically inhibit communication from CSC, we sorted CSC from MIA PaCa-2 Tet-On shRab27a and NSCC from MIA PaCa2 Tet-On shScramble cells. These were orthotopically implanted together, at the same percentages found in the parental cell line in immunodeficient mice (figure 4A, experiment 1). Doxycycline-fed mice (n=8) showed a significant delay in disease onset according to the number of weeks until tumour detection in comparison to the control (n=8, figure 4B). Moreover, the doxycycline group showed a significant decrease in tumour growth and weight in comparison to the control (figure 4C,D). In contrast, specific inhibition of communication originating from NSCC by knockdown of Rab27a (figure 4A, experiment 2) did not show any significant changes in disease onset (online supplemental figure 5A) or tumour growth and weight in comparison to the control (online supplemental figure 5BC). Tumours of both groups were histologically similar (figure 4D and online supplemental figure 5C). Hence, our data demonstrate that knockdown of Rab27a in CSC (which inhibits their communication ability) but not in NSCC is sufficient to impair tumour growth.
Supplemental material

Impairment of communication by EVs originated in CSC is sufficient to hamper tumour growth in PDAC orthotopic, GEMM and PDX models. (A) Experimental plan to impair specific routes of communication mediated by EVs in a PDAC orthotopic model using the MIA PaCa-2 Tet-On system. (B) Kaplan-Meier curve representing weeks to tumour detection of mice with CSC proficient (CSC Tet-On shRab27a control n=8) and CSC impaired EV secretion (CSC Tet-On shRab27a doxycycline n=8; paired t-test; *p<0.05). (C) Tumour growth curve measured by ultrasound of mice with CSC proficient (CSC Tet-On shRab27a control n=8) and impaired EV secretion (CSC Tet-On shRab27a doxycycline n=8; two-way analysis of variance; **p<0.01). (D) Tumour weight of groups described in (B,C). On the right, representative photos of tumours at time of euthanasia and respective H&Es (bottom, Wilcoxon test; **p<0.01). Scale bar: 10 mm. (E) Experimental layout to impair communication by CSC using a PDAC GEMM. Of note, as shown before (online supplemental figure 1D), the subpopulation positive for EpCAM (EpCAM+) was identified in the KPC and included in the NSCC. (F) Kaplan-Meier curve of the overall survival of mice with proficient CSC EV secretion (CSC were sorted from tumours of non-treated KPC iRab27aFrt/Frt mice, control n=7) and CSC with impaired EV secretion (CSC were sorted from tamoxifen-treated KPC iRab27aFrt/Frt mice, tamoxifen n=5, log-rank Mantel-Cox test; *p=0.0278). (G) Representative H&E staining of tumours in control and tamoxifen groups. (H) Experimental layout to impair communication by CSC EVs in a PDX model. (I) Tumour incidence in CSC proficient in EV secretion (CSC sorted from PDX tumour, treated ex vivo with DMSO 5% and injected with their NSCC counterparts), and CSC impaired EVs secretion (CSC were sorted from PDX tumour and treated ex vivo with Nexinhib20 (1 µM) before injection with their NSCC counterparts). DMSO 5% n=6, Nexinhib20 n=5 (Fisher’s exact test *p=0.0152). (J) Tumour volume and representative photos of tumours at time of euthanasia and respective H&ES of groups of mice described in (H,I). DMSO 5% n=6, Nexinhib20 n=5 (permutation test, *p=0.0123). Scale bar: 10 mm. (K) Representative flow cytometry analysis of viable cancer cells derived from PDX cells non-treated, DMSO 5% treated or treated with Nexinhib20 ex-vivo. Data are mean±SEM. CSC, cancer stem cell; EVs, extracellular vesicles; GEMM, genetically engineered mouse model; NSCC, non-stem cancer cell; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft.
To further confirm the role of CSC EVs in the progression of PDAC, we developed a GEMM that allows the inducible and conditional knockout of Rab27a mediated by flippase recombination (Rab27aFrt/Frt). We crossed Rab27aFrt/Frt and R26LSL-FLPOERT2/+ alleles24 with the KPC. The final model spontaneously develops PDAC (LSL-KrasG12D/+, LSL-Tp53R172H/+, Pdx-1-Cre, R26LSL-FLPoERT2/+, Rab27aFrt/Frt, hereafter referred to as KPC iRab27aFrt/Frt). Recombination of the Rab27a allele in the pancreas on tamoxifen treatment at birth via lactation was confirmed by PCR (online supplemental figure 5D). KPC iRab27aFrt/Frt treated with tamoxifen at birth showed a tendency for increased overall survival (online supplemental figure 5E,F). Mice also developed metastasis in the liver and lungs as the control group did (online supplemental figure 5G,H). We have used this GEMM to sort CSC from tumours of KPC iRab27aFrt/Frt tamoxifen-treated mice (Rab27a knockout), and NSCC were obtained from control mice (KPC iRab27aFrt/Frt non-treated or KPC Rab27aFrt/Frt without the R26LSL-FLPOERT2/+ allele treated with tamoxifen). The cells were orthotopically implanted together in wild-type (C57BL/6) mice (n=23) at the same percentages found in the original tumours (figure 4E). The control group was mice orthotopically implanted with CSC and NSCC from Rab27aWT tumours (n=23, figure 4E). The overall survival of mice bearing tumours with impaired secretion of CSC EVs was significantly increased compared with the control group (figure 4F). No major histological differences were noted in tumours of both groups (figure 4G).
We further validated our findings in a PDX model. CSC were isolated from a PDX tumour and treated with Nexinhib20 or vehicle (DMSO 5%) for 4 hours ex vivo. These cells were orthotopically implanted in immunodeficient mice (Rag2−/−IL2rg−/−) together with NSCC isolated from the same PDX at the same percentages found in the PDX tumour (figure 4H). Inhibition of the function of Rab27a and consequent impairment of the secretion of CSC EVs abrogated the incidence of tumours and metastasis (figure 4I,J, and online supplemental figure 6AB). Tumours and metastasis were histologically confirmed in control animals (figure 4J and online supplemental figure 6AB). Importantly, we found no differences in cancer cell viability after treatment with Nexinhib20 in comparison to the controls (DMSO 5% or untreated, figure 4K).
Supplemental material
We provide evidence that the inhibition of CSC EVs by Rab27a knockdown is sufficient to impair PDAC growth. Taking this into consideration, we next set out to identify the cargo of CSC EVs and investigate the underlying causes of the phenotype observed.
Extracellular vesicles from CSC have unique protein cargo and are enriched in agrin
To determine the protein composition of the identified subpopulations of cancer cells and their respective EVs, we used four human PDAC cell lines (BxPC-3, PANC-1, T3M4 and MIA PaCa-2) and segregated the identified subpopulations by FACS. From each subpopulation, EVs were isolated by differential ultracentrifugation method25 (figure 5A). Biophysical characterisation of purified vesicles using NTA showed that isolated vesicles are within the expected size range for small EVs (124.1±4.5 nm; online supplemental figure 7A). Using transmission electron microscopy (TEM), we verified that the EVs exhibited a cup-shaped morphology and lipid bilayer (online supplemental figure 7B). We performed liquid chromatography–electrospray ionisation–tandem mass spectrometry (LC/ESI–MS/MS) to characterise the protein content of EVs and of the subpopulations of cells from which they originate (figure 5A). Overall, the alignment and filtering of peptides yielded a total of 6185 proteins (online supplemental figure 7C and online supplemental table 1). Unsupervised hierarchical clustering revealed two protein clusters that grouped proteins mostly detected in EVs or in cells, and a cluster of proteins detected in both fractions (figure 5B, online supplemental figure 7D and online supplemental table 2). Gene ontology analysis using the reactome pathway database revealed that each cluster of proteins is involved in distinct biological functions (figure 5C and online supplemental table 3). This suggests that the cargo of EVs is enriched in specific biological pathways, rather than mimicking the function of the cells’ proteome. The cluster of proteins found mostly in EVs is significantly enriched in the extracellular matrix (ECM)-proteoglycans and integrin cell-surface interaction pathways (figure 5C). This could indicate the involvement of these proteins in the specific uptake of EVs and thus in the establishment of specific communication routes. As expected, subcellular location of the proteins identified in EVs corresponded mainly to the cytosol, plasma membrane, vesicles and endoplasmic reticulum (online supplemental figure 7E). We identified 545 proteins in CSC EVs not detected in NSCC EVs (figure 5D). Functional enrichment analysis demonstrated that proteins detected only in CSC EVs versus the ones detected only in NSCC EVs belong to distinct biological pathways and thus have distinct phenotypical outcomes (figure 5E). The axon guidance pathway stood out as a highly abundant and significant pathway in only CSC EVs (p value=1.46815E-27; figure 5E and online supplemental table 4). The axon guidance pathway has been previously described aberrantly mutated in PDAC and to potentiate pancreatic carcinogenesis.26 To define whether there were significant global differences in protein concentration of certain biological categories, we performed a gene set enrichment analysis (GSEA) based on normalised protein count differences between CSC and NSCC EVs. Similarly, GSEA showed a significant enrichment of the axon guidance pathway in CSC EVs corresponding to 79 proteins (adjusted p value=0.001, figure 5F and online supplemental table 5). Most importantly, GSEA analysis confirmed that the proteins in the axon guidance pathway could discriminate CSC EVs from NSCC EVs (figure 5F). Out of these 79 proteins, we found that 14 of them were present in CSC EVs across all four cell lines (figure 5G). Agrin stood out due to its previously described role in cancer and specifically in PDAC.27–29 Agrin was found to be upregulated in PDAC and its overexpression promotes epithelial-to-mesenchymal transition contributing to metastasis, as well as poor overall patient survival.27 In addition, a previous proteomic analysis of PDAC extracellular matrix revealed that agrin present in tumours is not of stromal origin; instead, it is specifically derived from cancer cells.30 We have validated the enrichment of agrin in MIA PaCa-2 CSC EVs by FACS, demonstrating that the membrane anchored isoform of agrin is present in EVs (figure 5H). In addition, we performed size exclusion chromatography in MIA PaCa-2 EVs and demonstrated by western blot that agrin is present in the EVs fractions (SEC F7-10) as well as in the non-vesicular fractions (SEC F11-25, online supplemental figure 7F) as expected, since agrin has membrane-bound and secreted isoforms.31 EV fractions were confirmed by the presence of alix and syntenin-1 by western blot, and the presence of CD9, CD63 and CD81 was confirmed by flow cytometry (online supplemental figure 7FG).23 25 32 In a similar fashion, we performed Optiprep gradients and demonstrated that agrin is present in EVs and non-vesicular fractions (F1–6 and F7–12, respectively; online supplemental figure 7H). EVs fractions were characterised by the expression of CD81, syntenin-1 and alix and the non-vesicular fractions by the presence of HSP90 and histone H3 (online supplemental figure 7H).23 25 32
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material

Agrin is enriched in CSC EVs. (A) Experimental approach to perform LC/ESI–MS/MS in subpopulations of cells and respective EVs (CD24+CD44+, CD133+, CD24−CD44+, 4N and EpCAM+) in four human PDAC cell lines (BxPC3, PANC-1, T3M4 and MIA PaCa-2) for a total of 38 samples (MIA PaCa-2 cells are EpCAM−). (B) Heatmap depicting protein clusters present in PDAC subpopulations and respective EVs in four cell lines. Dendogram displays unsupervised hierarchical clustering showing separation of three protein clusters: EVs, cells and cells+EVs. The full heatmap is shown in online supplemental figure 7D. (C) Functional enrichment analysis of protein clusters. Dot plot representing the top 6 reactome-enriched pathways per cluster (adjusted p value <0.05).Gene ratio corresponds to the relative size of every pathway in each protein cluster. (D) Venn diagram of total proteins detected in CSC and NSCC EVs isolated from four PDAC cell lines. Edges represent the number of proteins detected only in CSC or NSCC EVs, and intersection represents the number of proteins common to both subpopulations. (E) Top 5 enriched reactome pathways in CSC, NSCC and in both subpopulations of EVs (adjusted p value <0.05). (F) Gene set enrichment analysis demonstrates that proteins in the axon guidance pathway separate CSC EVs from NSCC EVs. (G) Venn diagram depicting the intersection of the proteins found in CSC EVs across the four PDAC cell lines. Out of the 233 proteins common across all CSC EVs, 79 correspond to proteins of the axon guidance pathway. Out of these, 14 were present in CSC EVs across all four cell lines. These 14 were ranked by average DESeq2 normalised peptide counts (table on the right). (H) FACS analysis and representative histogram plots of agrin-positive EVs derived from MIA PaCa-2 NSCC and CSC subpopulations (n=4, Mann-Whitney test, *p<0.05). Data are mean±SEM. CSC, cancer stem cells; EVs, extracellular vesicles; GTP, Guanosine-5'-triphosphate; HSC, hematopoietic stem cells; IGFBP, Insulin-like growth factor binding protein; LC/ESI–MS/MS, liquid chromatography–electrospray ionisation–tandem mass spectrometry; NSCC, non-stem cancer cell; PDAC, pancreatic ductal adenocarcinoma; PKN, protein kinase N; RHO, ras homologous; ROBO, roundabout guidance receptor; SLIT, slit guidance ligand; SRP, signal recognition particle.
These results demonstrate that CSC EVs have a distinct protein cargo from that of NSCC EVs. Most importantly, we showed across four human PDAC cell lines, that a significant number of proteins exclusively detected in CSC EVs belong to biological processes involved in PDAC, such as the axon guidance pathway.26 Within this pathway, we verified that in CSC EVs, agrin is one of the most enriched proteins.
Agrin-Positive CSC EVs promote YAP activation
Agrin is associated with PDAC progression and is a marker of poor prognosis in PDAC patients.27 Agrin was also described to promote liver carcinogenesis by activation of the YAP transcription factor by binding to the LRP-4 receptor.28 YAP is a central player in the Hippo pathway, which is frequently dysregulated in cancer.33 The YAP translocation to the nucleus alters the transcriptional programme of cells, thus promoting proliferation and survival.34 Since agrin is enriched in CSC EVs, we tested the possibility of CSC agrin+ EVs modulating YAP activity and fuelling PDAC growth. We sorted CSC from MIA PaCa-2 Tet-On shRab27a and cultured them with NSCC sorted from MIA PaCa-2 Tet-On shScramble at the same proportions found in the parental cells (figure 6A). On doxycycline treatment, we prevented CSC from secreting EVs through Rab27a knockdown, inhibiting the most frequent communication route in the EVNet. We demonstrated that on downregulation of Rab27a in CSC, there was a significant decrease in the levels of active YAP (nuclear YAP) in comparison to the control (figure 6A). The same results were obtained using an antibody that detects total YAP (online supplemental figure 8A). We next confirmed the role of agrin in CSC EVs in the activity of YAP. We developed a Tet-Off system in the MIA PaCa-2 cell line to control the expression of agrin by means of a short-hairpin RNA. CSC were sorted from MIA PaCa-2 Tet-Off shAgrin and cultured with MIA PaCa-2 Tet-Off shScramble NSCC at the same proportions found in the parental cells (figure 6B). We demonstrated that knockdown of agrin in CSC alone (no doxycycline) is sufficient to significantly decrease YAP nuclear levels, in a similar fashion to what is seen when communication from CSC is impaired by Rab27a knockdown (figure 6B). We confirmed the agrin downregulation in cells and in CD133+ (CSC) EVs derived from the MIA PaCa-2 Tet-Off shAgrin by western-blot and FACS, respectively (online supplemental figure 8B). We also demonstrated that this mechanism is specific to CSC because coculture of NSCC from MIA PaCa-2 Tet-Off shAgrin with CSC from MIA PaCa-2 Tet-Off shScramble (knockdown of agrin in NSCC) does not decrease YAP activation (online supplemental figure 8C).
Supplemental material

Agrin-positive CSC EVs promote YAP nuclear location. (A) Experimental layout (top, left): CSC and NSCC were sorted from MIA PaCa-2 Tet-On shRab27a or shScramble, respectively, and cultured at the same proportions found in parental cells in order to assess YAP activity in these conditions. Representative confocal microscopy pictures of CSC from MIA PaCa-2 Tet-On shRab27a cultured with NSCC from MIA PaCa-2 Tet-On shScramble (cultured at the same percentages found in the MIA PaCa-2 Tet-On shRab27a). Active YAP (green), phalloidin (red) and nuclei (blue) (right). Quantification of YAP nuclear levels (mean intensity per cell) (n=1, six images per group, unpaired t-test; **p<0.01) (bottom, left). Data are min to max. Scale bar: 10µm. Dashed lines in violin plot represent median values. (B) Experimental layout (top, left): in order to assess the role of agrin in CSC EVs, a doxycycline-inducible (Tet-Off) shRNA against agrin was transfected into MIA PaCa-2 cells. CSC were sorted from Tet-Off shAgrin MIA PaCa-2 cells and cultured at the same proportions found in parental cells with MIA PaCa-2 Tet-Off ShScramble NSCC. Representative confocal microscopy pictures of CSC from MIA PaCa-2 Tet-Off shAgrin cultured with NSCC from MIA PaCa-2 Tet-Off shScramble (cultured at the same percentages found in the MIA PaCa-2 -Tet-Off shAgrin). Active YAP (green), phalloidin (red) and nuclei (blue) (right). Graph depicts quantification of YAP nuclear levels (mean intensity per cell; n=2, six images per group, unpaired t-test; ****p<0.0001) (bottom, left). Data are min to max. Scale bar: 10 µm. Dashed lines in violin plots represent median values. (C) Representative immunohistochemistry photos and quantification of per cent nuclear YAP-positive epithelial cells in MIA PaCa-2 Tet-On shRab27a tumors. Control n=7, doxycycline n=8 (unpaired t-test, **p<0,01). Data are mean±SEM. (D) Representative immunofluorescence of LRP-4 (purple) in MIA PaCa-2 NSCC treated with CSC EVs isolated from MIA PaCa-2 CD63-turboGFP. Representative orthogonal view YZ (middle panel). Arrows identify colocalisation between CD63-turboGFP CSC EVs and LRP-4. Scale bars 10µm. (E) Fold change of AREG, CXCL5, STAT3, CYR61, ALX and VIM gene expression in MIA PaCa-2 NSCC transfected with siSCR and treated with CSC EVs (red), MIA PaCa-2 NSCC transfected with siLRP-4 (grey) and MIA PaCa-2 NSCC transfected with siLRP-4 and treated with CSC EVs (blue) in comparison with NSCC transfected with siSCR (baseline, 1) analysed by qPCR. AREG CT levels were undetermined in MIA PaCa-2 NSCC transfected with siLRP-4 condition. β-actin was used as endogenous control for comparative CT method. CSC EV treatment (10 µg) was performed 72 hours after transfection with siRNA, and gene expression was evaluated 24 hours after treatment. (F) Cell viability measured by absorbance at 570nm (MTT assay) at days 1, 4 and 6 after the beginning of the experiment in MIA PaCa-2 NSCC (dark blue), NSCC treated with CSC EVs (light blue), NSCC treated with DMSO and CSC EVs (light red) and NSCC treated with verteporfin and CSC EVs (dark red). DMSO and verteporfin (10 µg/mL) treatments were performed at days 1 and 4. CSC EVs treatments (1 µg) were performed at days 1 and 4 two-way ANOVA; *p<0.05, ****p<0.0001. CSC, cancer stem cells; DAPI,4′,6-diamidino-2-phenylindole; EVs, extracellular vesicles; NSCC, non-stem cancer cells. rtTA, reverse tetracycline-controlled transactivator; VIM, vimentin.
In addition, we showed that doxycycline treatments are not the underlying cause of the observed YAP phenotype using cultures of MIA PaCa-2 CSC Tet-Off shScramble and parental NSCC (online supplemental figure 8D). Most importantly, active YAP is also significantly decreased in cancer cells of MIA PaCa-2 Tet-On shRab27a tumours with impaired EV secretion, thus confirming that EVs are involved in YAP activation (figure 6C). Altogether, these results demonstrate that agrin knockdown in CSC phenocopies inhibition of CSC EVs by Rab27a knockdown, suggesting the involvement of EVs-associated agrin in YAP activation.
Finally, we evaluated whether the LRP-4 receptor could be the mediator of YAP activation by agrin as previously described.35 We demonstrate that LRP-4 is enriched in NSCC compared with CSC, which indicates a potential role of the agrin-LRP-4 axis in supporting the most frequent route of communication of the EVNet (online supplemental figure 9A). Moreover, we treated NSCC with CSC EVs (tagged with CD63-turboGFP) and showed that CSC EVs colocalise with LRP-4 and are internalised by NSCC (figure 6D). Finally, since agrin has a membrane anchored isoform that we can detect in EVs by FACS analysis, we also found that agrin colocalises with the LRP-4 receptor in MIA PaCa-2 cells (online supplemental figure 9B).
Supplemental material
To validate that CSC agrin+ EVs promote YAP activation through LRP-4, we treated NSCC with CSC EVs and modulated LRP-4 expression. First, we demonstrated that the YAP-regulated genes AREG,36 CXCL5,37 STAT3,38 CYR61,39 ALX40 and VIM41 are overexpressed on treatments with CSC EVs (figure 6E; fold change of black bar in reference to baseline NSCC siScramble, 1 . Second, we validated that NSCC transfected with siLRP-4 show downregulation of the evaluated genes, indicating the crucial role of LRP-4 in regulating YAP activation (figure 6E; fold change of red bar in reference to baseline NSCC siScramble, 1). Finally, we demonstrated that NSCC with LRP-4 downregulation do not recover the expression of YAP-regulated genes on treatment with CSC EVs in comparison to NSCC siScramble (figure 6E, blue bar).
Genes regulated by YAP are involved in proliferation and invasion in PDAC.42 43 We sought to evaluate the role of YAP in mediating the phenotype observed on modulation of the EVNet, which occurs mainly through CSC agrin+ EVs signalling. To this end, we performed an MTT assay using NSCC treated with CSC EVs in the presence or absence of verteporfin, a YAP inhibitor.44 45 First, we demonstrated that treatments of NSCC with CSC EVs promote cell proliferation (figure 6F). Second, we showed that this effect is highly dependent on YAP since NSCC treated with verteporfin and CSC EVs do not show any effect on cell proliferation (figure 6F).
Our findings demonstrate that CSC EVs and agrin promote YAP activation and, thus, dysregulate the Hippo pathway through LRP-4, which could explain their contribution to tumour growth.
Agrin is a potential therapeutic target for CSC in PDAC
Inhibition of secretion of EVs in CSC through knockdown of Rab27a is sufficient to impair tumour growth in orthotopic, GEMM and PDX models. We next sought to evaluate whether the same impact on disease progression results from downregulation of agrin specifically in CSC EVs, while maintaining the communication originating from this subpopulation of cancer cells. To answer this, CSC were sorted from MIA PaCa-2 Tet-Off shAgrin, NSCC from MIA PaCa-2 Tet-Off shScramble, and they were orthotopically implanted together in immunodeficient mice at the same proportions found in the parental cells (figure 7A, experiment 1). In the same manner, NSCC were sorted from MIA PaCa-2 Tet-Off shAgrin, CSC were sorted from MIA PaCa-2 Tet-Off shScramble cells, and they were orthotopically implanted in immunodeficient mice (figure 7A, experiment 2). In both groups, in the absence of doxycycline treatment, agrin was specifically downregulated in either CSC or NSCC, respectively (figure 7A). We found that downregulation of agrin in CSC and their respective EVs (n=7) led to a significantly slower tumour growth in comparison to the downregulation of agrin in NSCC (n=6, figure 7B). This was also reflected in a significant decrease in tumour volume at euthanasia (figure 7C). In addition, when the same groups were treated with doxycycline (normal levels of agrin expression), we observed no differences in tumour growth (online supplemental figure 10A,B). We also demonstrated that the relative tumour growth in the CSC group is decreased on knockdown of agrin, which is not true in the NSCC group (online supplemental figure 10C). Together, these results demonstrate the significant impact of agrin-positive CSC and respective EVs on PDAC growth.
Supplemental material

Agrin in CSC promotes PDAC progression and blocking agrin in PDX cells impairs their proliferation. (A) Experimental layout: to impair agrin expression in specific cancer cell subpopulations, CSC and NSCC were sorted from the MIA PaCa-2 Tet-Off shAgrin and shScramble clones, respectively, and then orthotopically implanted into the pancreas of Rag2−/−Il2rg−/− mice. (B) Tumour growth curve measured by ultrasound of untreated (agrin kD) tumours CSC from MIA PaCa-2 Tet-Off shAgrin plus NSCC from MIA PaCa-2 Tet-Off shScramble (injected at the same percentages found in the cells of origin, n=7) and NSCC from MIA PaCa-2 Tet-Off shAgrin with CSC from MIA PaCa-2 Tet-Off shScramble (injected at the same percentages found in the cells of origin, n=6, two-way ANOVA; *p<0.05). (C) Quantification of tumour volume at euthanasia and representative photos of pancreas tumours (Mann-Whitney test, *p<0.05). Scale bar: 10 mm. (D) Histological evaluation of the percentage of the pancreas that showed no histological disease, PanINs and PDAC area in KPC and KPAC mice euthanised at 14 weeks of age and corresponding H&E pictures (KPC n=6, KPAC n=5). (E) YAP H-score and representative immunohistochemistry photos in KPC and KPAC (KPC n=6, KPAC n=5) (Mann-Whitney test,**p<0.01). (F) Schematic representation of PDX ex vivo treatment with human antiagrin neutralising antibody. Cell viability was measured by absorbance at 560 nm (MTT assay). PDX cells were treated ex vivo either from day 0 to day 8, every day (patient 1), or from day 0 to day 11, every day (patient 2), with antiagrin blocking antibody (Mab5204 10 μg/mL), IgG (10 μg/mL) or untreated (control). Comparison was performed with PDX ex vivo treated with IgG (two-way ANOVA; *p<0.05, ***p<0.001, ****p<0.0001). Arrow indicates timepoint that treatment was stopped in patient 1. (G) Quantification of YAP nuclear levels in PDX cells treated ex vivo from day 0 until day 5, every day, with antiagrin blocking antibody (Mab5204 10 μg/mL) or IgG (10 μg/mL) (top) (mean intensity per cell, n=1, six images per group, unpaired t-test; ****p<0.0001). Data are min to max. Dashed lines in violin plots represent median values. Representative confocal microscopy photos of treated PDX cells. Scale bar: 20 µm. Active YAP (green), phalloidin (red) and nuclei (blue) (bottom). Data are mean±SEM. ANOVA, analysis of variance; KD, knockdown; KPAC, agrin knockout KPC; CSC, cancer stem cells; EVs, extracellular vesicles; FACS, fluorescence activated cell sorting; MTT, methylthiazolyldiphenyl–tetrazolium bromide; NSCC, non-stem cancer cell; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft.
To further confirm the role of agrin in PDAC progression, we crossed the KPC model with an agrin-floxed mouse to generate the KPC Agrin knockout (KPAC: LSL-KrasG12D/+, LSL-Tp53R172H/+, Agrinfl/fl, Pdx-1-Cre). KPAC spontaneously develops PDAC and recombination of the agrin allele in PDAC tumours was confirmed by PCR (online supplemental figure 10D). KPAC mice showed delayed disease progression compared with KPC mice, as demonstrated by the histopathological analysis of the pancreas of both models in a cross-sectional study (figure 7D). Most importantly, we found that KPAC tumours have a significant downregulation of active YAP expression in cancer cells (figure 7E).
Taking this in consideration, we investigated the impact of targeting agrin in two PDXs using a neutralising antihuman agrin antibody (figure 7F).28 Antiagrin, IgG or no treatments were given to PDX cancer cells, and cell viability was assessed by MTT assay (figure 7F). We determined that antihuman agrin treatments significantly decreased cell growth in comparison to treatments with IgG or no treatment in two different PDXs (figure 7F and online supplemental figure 10E). Accordingly, antihuman agrin treatments significantly decreased the levels of active YAP in PDX cells (figure 7G).
Overall, our results demonstrate that CSC EVs are enriched in agrin that has a tumor-promoting role. This further validates the biological significance of the CSC axis of communication in the EVNet. Most importantly, we have identified agrin as a novel target for therapeutic intervention in pancreatic cancer.
Circulating agrin-positive EVs are a prognostic marker for disease progression
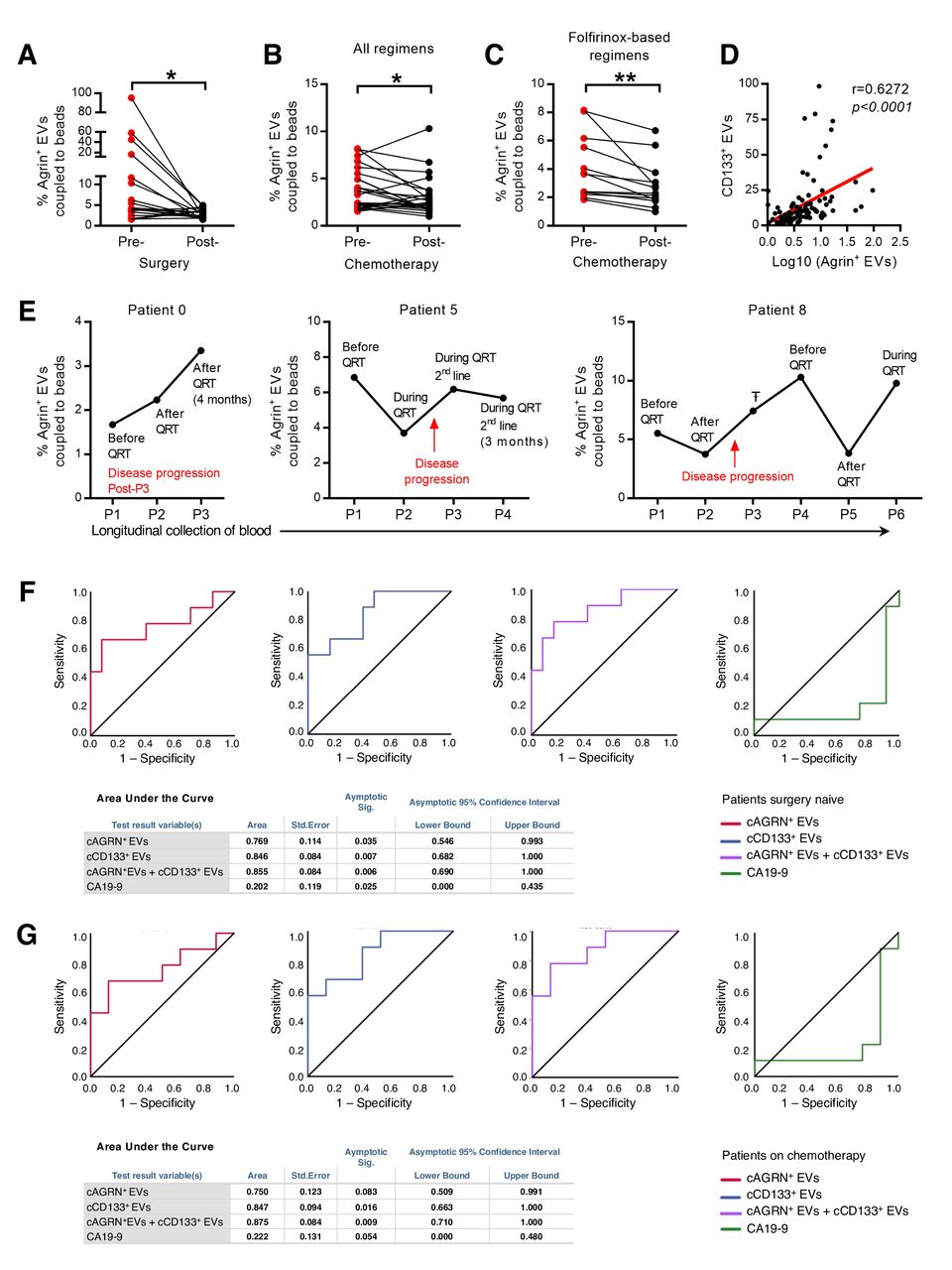
To validate our findings, we gathered a cohort of 110 serum samples that were longitudinally collected from 44 patients with PDAC (online supplemental table 6). Circulating agrin-positive EVs coupled to beads (cAGRN+EVs) were identified in the serum of patients with PDAC by imaging flow cytometry (online supplemental figure 11A). The analysis of cAGRN+EVs by FACS before and after surgery (n=19, patients eligible for surgery) revealed a significant decrease on tumour resection (figure 8A). In addition, we also found a significant reduction in the percentage of cAGRN+EVs in patients with PDAC after chemotherapy treatments (n=24, figure 8B). This was only true for patients who had gone through folfirinox-based treatment regimens (figure 8C and online supplemental figure 11B). This is in accordance with the therapeutic benefit associated with folfirinox-based therapies in PDAC.46 Thus, we demonstrated that the percentage of cAGRN+EVs is directly proportional to tumour burden.
Supplemental material
Supplemental material

Circulating agrin-positive EVs are a biomarker of disease progression and predict response to treatment in patients with PDAC. (A) Presurgery and postsurgery analysis of the percentage of circulating agrin-positive EVs coupled to beads in the serum of PDAC patients (n=19, paired t-test; *p<0.05). (B,C) Prechemotherapy and postchemotherapy analyses of the percentage of circulating agrin-positive EVs coupled to beads in the serum of patients with PDAC treated with all regimens (B) (n=24, paired t-test; *p<0.05) and folfirinox-based regimens (C) (n=13, paired t-test; **p<0.01). (D) Correlation between the percentage of circulating CD133-positive EVs coupled to beads and the log10 of the percentage of agrin-positive EVs coupled to beads in the serum of patients with PDAC (n=106, Spearman r=0.6272). (E) Analysis of the percentage of circulating agrin-positive EVs coupled to beads in the serum of three patients with PDAC throughout time. QRT, chemotherapy, ŦAborted surgery due to non-resectable tumour. (F) Receiver operating curve analysis for the percentage of agrin-positive (red), CD133-positive (blue), and combination of agrin-positive and CD133-positive (purple) EVs coupled to beads and CA19-9 (green) in the serum of patients with PDAC not submitted to surgery (n=22 in circulating agrin-positive and CD133-positive EVs coupled to beads analysis and n=20 in CA19-9 analysis). (G) Receiver operating curve analysis for the percentage of agrin-positive (red), CD133-positive (blue), and combination of agrin-positive and CD133-positive (purple) EVs coupled to beads and CA19-9 (green) in the serum of patients with PDAC not submitted to surgery and treated with chemotherapy (n=17). In tables: a means under the non-parametric assumption and b means null hypothesis: true area=0.5. EVs, extracellular vesicles; PDAC, pancreatic ductal adenocarcinoma.
Using serum samples from a cohort of 106 patients with PDAC (online supplemental table 7), we found a significant correlation between the percentage of cAGRN+EVs and cCD133+EVs (circulating EVs in part originated from CSC, r=0.6272, p<0.0001; figure 8D). These data provide further evidence in support of the CSC origin of the majority of the cAGRN+EVs. Longitudinal evaluation of cAGRN+EVs in patients with PDAC demonstrated that an increase in cAGRN+EVs occurred concomitantly or even before imaging detection of disease progression during chemotherapy or after it was ceased (figure 8E). Most importantly, we showed that in patients that did not have surgery, a 1% increment of cAGRN+EVs presented a 2.94-fold increased risk for disease progression [OR (CI)=2.94(1.03–8.43), p=0.045]. Interestingly, a 1% increment in cCD133+EVs also resulted in significantly increased risk for PDAC progression [OR(CI)=1.77(1.09–2.88), p=0.022].
Supplemental material
Finally, we investigated the performance of cAGRN+EVs as a prognostic biomarker for disease progression by receiver operating characteristic curve (ROC) analysis. Circulating agrin-positive EVs behaved as a specific and sensitive biomarker for disease progression in patients that did not have surgery (AUC=0.77; p=0.035; figure 8F). Similar behaviour was observed for cCD133+EVs (AUC=0.85, p=0.007; figure 8F). Additionally, combination of both markers increases the performance in predicting disease progression (AUC=0.86, p=0.006). In comparison, in the same cohort of patients, we confirmed that CA19-9 was not a good biomarker to predict disease progression (AUC=0.2, p=0.025; figure 8F). Additionally, in patients who did not have surgery but did have chemotherapy, the percentage of cAGRN+EVs together with cCD133+EVs was also a significant predictor of disease progression (AUC=0.875, p=0.009; figure 8G). Altogether, our results demonstrate that cAGRN+EVs, alone or in combination with cCD133+EVs, constitute prognostic biomarkers associated with increased risk for disease progression.
Furthermore, we analysed the expression levels of agrin and YAP protein in patients with PDAC using The Cancer Genome Atlas (TCGA). As expected, higher expression of YAP protein was associated with higher risk of recurrence (OR=4.76, CI 1.91 to 12.60; p=0.0007). In addition, patients exhibiting both high levels of YAP protein and agrin expression showed significantly increased risk for recurrence (OR=6.15, CI 1.45 to 26.11; p=0.01) and had a worse prognosis in comparison to patients with low levels of YAP and agrin (online supplemental figure 11C). Furthermore, we found that concomitantly high levels of agrin and low levels of non-active YAP (phosphoS127 YAP) are associated with worse prognosis in comparison with low levels of agrin and high levels of non-active YAP (online supplemental figure 11D). Most importantly, low levels of non-active YAP alone do not correlate with a worse prognosis (online supplemental figure 11E), thus reflecting the role of agrin in YAP activation and consequent tumor-promoting function in PDAC.
Therefore, we conclude that the EVNet, through agrin in CSC EVs, is associated with increased risk of disease progression and could be a novel target for CSC therapeutic intervention in PDAC (figure 9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic of the intercellular communication mediated by EVs between subpopulations of pancreatic cancer cells, the EVNet and the role of CSC agrin-positive EVs in PDAC. Our work demonstrates that subpopulations of PDAC cells establish an organised and plastic communication network, the EVNet, in which the preferential communication route is from CSC to NSCC, by means of CSC agrin-enriched EVs. Specific inhibition of this route is sufficient to impair the growth of PDAC tumours in orthotopic, GEMM and PDX models. In PDAC human samples, we have confirmed that cAGRN+EVs are a prognostic biomarker for disease progression and are associated with therapy response. CSC, cancer stem cell; EVs, extracellular vesicles; EVNet, Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network; GEMM, genetically engineered mouse model; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft.
Discussion
Tumours are complex entities composed of different cancer cell subpopulations with distinct cellular behaviour and survival capacity, dictated by both cell and non-cell autonomous changes.2 3 47 48 It is postulated that cancer cell subpopulations with distinct capacities interact with each other and cells from the tumour microenvironment, and that this communication is crucial for cancer cells to overcome microenvironmental changes and thrive.49 50 EVs are known to mediate intercellular communication51 and have been described to carry cargo that modulate recipient cells, both nearby and in distant organs, contributing to tumour survival, progression and the formation of the pre-metastatic niche.11 13–15 Here, we demonstrate that cancer EVs are exchanged locally between subpopulations of cancer cells establishing a non-random communication network, the EVNet. Although CSC represent a rare subpopulation, we provide evidence that EVs act as mediators of a frequent communication route in the EVNet from CSC to NSCC. Importantly, we demonstrate that the EVNet reshapes when challenged with hypoxia or gemcitabine, thus supporting a role in the cooperation between subpopulations within the tumour. These findings suggest that the EVNet could be a mechanism used by cancer cells to adapt to the tumour milieu, allowing them to survive in harsh environments and resist therapy, ultimately supporting disease progression. Although our experimental model allows us to assess intercellular communication between distinct subpopulations of cells, we cannot discard the possibility of the existence of intrasubpopulation communication. Nevertheless, the biological significance of such phenomenon, in which cancer cells exchange the same set of information they already contain, would be difficult to interpret and beyond the scope of the current study. We also need to consider that, although we identified the EVNet established among four specific subpopulations of cancer cells, there are certainly other subpopulations defined by distinct sets of cell-surface markers, which might also communicate by means of EVs and that have not been assessed in our model.
Studies show that malignant cells released EVs that are taken up by less malignant cancer cells.17 This transfer of information leads to enhanced migratory and metastatic capacity,17 and the so-called ‘amoeboid’ phenotype16. We demonstrated that the transfer of information present in CSC EVs to other subpopulations of cancer cells is crucial for tumour growth, indicating that CSC are at the centre of the biological impact of the EVNet on PDAC. Transcriptomic studies have identified two main classes of PDAC cells: ‘classical’ and ‘basal-like’.52 53 These cancer cell subtypes coexist in tumours and dictate the tumour differentiation state, as well as how it responds to therapy.54 This highlights the plasticity of PDAC, which is driven not only by genetic and epigenetic cues, but also by extracellular signals from the tumour microenvironment.54 Considering our data, in which CSC(more tumorigenic) communicate with NSCC (less tumorigenic), one could speculate that EVs could also dictate cellular subtype and PDAC fate.
To study the biological significance of the CSC to NSCC communication axis, we limited the release of EVs by targeting Rab27a, a GTPase known to be involved in the late stages of EVs biogenesis.19 21 55 Besides its role in EVs release, Rab27a has also been described to modulate the secretion of non-vesicular components, which could represent a limitation of our study.20 21 However, our data show that in our model, Rab27a downregulation impairs EVs release without interfering with exocytosis of 42 investigated secreted proteins. Most importantly, treatments using CSC EVs could reproduce our phenotype, namely, YAP activation, overexpression of YAP target genes and increased proliferation of NSCC Therefore, we could demonstrate a role of CSC EVs in PDAC biology. In agreement with our findings, previous studies suggest that high expression of Rab27a predicts poor survival of PDAC patients,56 and its downregulation in a murine PDAC cell line compromises metastasis.57
We found that agrin is significantly enriched in CSC EVs and in non-vesicular fractions, which was expected because of its membrane-anchored and secreted isoforms.58 Agrin plays an oncogenic role in hepatocellular and pancreatic carcinoma and is associated with poor prognosis in PDAC.27 This proteoglycan was also identified as a surfaceome protein overexpressed in PDAC in a Kirsten ras oncogene homolog (KRAS)-dependent manner,29 as well as a matrisome protein upregulated in PDAC cancer cells that promotes epithelial-to-mesenchymal transition.27 We demonstrate that agrin+ EVs modulate YAP activity. Since we also observe agrin in non-vesicular fractions, we cannot exclude the possibility of both forms of agrin contributing to the phenotypes observed.
YAP is a known oncogenic driver in PDAC, although tumours with genetic aberrations in the Hippo/YAP signalling pathway are rare,33 59 suggesting that YAP activation is driven by non-genetic mechanisms, which include inhibition of the Hippo kinases.60 Binding of agrin to LRP-4 and integrins at the cell surface inactivates merlin and LATS1/2,35 which directly phosphorylate YAP, leading to its cytoplasmic retention.45 Limiting YAP transcriptional function through agrin inhibition is expected to suppress tumour progression. In support of this hypothesis, we observed that downregulation of agrin in CSC and respective EVs slows tumour growth in comparison to NSCC, and most importantly antiagrin treatments impair PDX cancer cells proliferation. These results are dependent on LRP-4 and YAP activation, since downregulation of LRP-4 or inhibition of YAP hampers phenotypes mediated by agrin present in CSC EVs. Moreover, we have also identified two other receptors for agrin—β1 integrin and DAG1—in NSCC. These have been implicated in the regulation of YAP and could present alternative mechanisms for the effect of agrin on YAP activity.61 62 Overall, our results demonstrate the impact of the agrin–YAP signalling in PDAC, promoted, at least in part, by CSC EVs. YAP activation is responsible not only for cell proliferation but also for its role in the conversion of fibroblasts into CAFs63 and suppression of T-cell function.64 Since we show that CSC agrin+ EVs modulate YAP activity in NSCC, there is also a possibility of these vesicles being involved in the reprogramming of the tumour microenvironment, which is also a role previously attributed to cancer EVs.13 65 66 Although our data support a role for agrin and the hippo pathway as modulators and effectors of EV-mediated communication from CSC to NSCC in PDAC pathogenesis, it can always be argued that there are additional pleiotropic effects of these molecules in CSC biology. In support of the specificity of our results, we have demonstrated LRP-4-dependent activation of YAP and its downstream targets by CSC EVs, resulting in increased proliferation of NSCC, without modulating the expression of Rab27a or agrin.
Finally, several biomarkers were described to be useful in diagnosing and monitoring PDAC, but there is a lack of biomarkers to inform treatment and phenotype characteristics of PDAC tumours.67–69 We showed that cAGRN+EVs correlate with the levels of circulating EVs that are positive for the CSC marker CD133. This further confirms that agrin present in EVs is mainly derived from CSC, which is also supported by its significant decrease on surgery. The significant decrease of cAGRN+EVs in patients with folfirinox-based therapy, but not other regimens, is consistent with the therapeutic benefit of folfirinox in PDAC in comparison to other regimens.70 71 In addition, we observed that elevated levels of cAGRN+EVs signifies a threefold increased risk for disease progression. Our results highlight the potential of cAGRN+EVs as an indirect measure of the amount of CSC present in a patient’s tumour, thus correlating with poor prognosis and therapy resistance. This is further supported by the observation that an increase in the amount of cAGRN+EVs precedes imagiological detection of progression. Finally, our data suggest that antiagrin therapy could target CSC in PDAC, which holds great potential to overcome therapy resistance and prevent cancer progression.
In conclusion, we describe for the first time the EVNet. We show that this organised communication network is plastic and allows cancer cells to adapt to harsh conditions, and that the communication route from CSC to NSCC is crucial for tumour progression. We identify agrin as a critical cargo of CSC EVs, promoting YAP activity in recipient cells and, consequent PDAC progression. Finally, we provide evidence that cAGRN+EVs can be used as a prognostic biomarker in PDAC. Our results could have profound clinical implications because we demonstrate that agrin is a potential target to impair CSC function in PDAC.
Methods
The file ‘Source Data’ contains all the source data related to the manuscript and is deposited in the Figshare Data Repository.72
Cell culture
The following cell lines were used in our study: human PDAC cell lines: MIA PaCa-2 (ATCC Cat# CRL-1420, RRID:CVCL_0428); PANC-1 (ATCC Cat# CRL-1469, RRID:CVCL_0480), BxPC-3 (ATCC Cat# CRL-1687, RRID:CVCL_0186); and T3M4 (RCB Cat# RCB1021, RRID:CVCL_4056 kindly provided by Dr Christoph Kahlert, Universitatsklinikum Carl Gustav Carus an der Technischen Universitat Dresden, Germany), and the 293T (ATCC Cat# CRL-3216, RRID:CVCL_0063) cells. All cells were tested for mycoplasma during our study, and all cells were STR profiled.73 All cells were cultured in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco). All stable clones and subpopulations derived from any of the aforementioned cell lines were cultured in the same conditions. Primary cultures derived from PDXs of human PDAC tumours were cultured in RPMI-1640 medium supplemented with 20% (v/v) FBS 100 U/mL penicillin and 100 µg/mL streptomycin. All cell lines were kept at 5% CO2 and 37°C in a humidified atmosphere.
Transfection and lentiviral transduction
In order to develop the stable clones of the MIA PaCa-2 cell line expressing colour-coded EVs, the following plasmids were inserted in the pLVX Puro backbone (RRID:Addgene_66604) through EcoRI-XhoI site via PCR method: CD63-turboGFP (Origene RG217238), YFP-CD82 (RRID:Addgene_1819) and tdTomato-CD81-10 (RRID:Addgene_58078). Rab5-mPlum plasmid was generated by cloning Rab5B coding region (Molecular Cloud OHu20962) into pmPlum plasmid (RRID:Addgene_54629) through NheI-BamHI site and, ultimately, through the insertion of the resulting Rab5B-mPlum sequence into the pLVX Puro backbone using EcoRI-XhoI site via PCR method.
To transduce MIA PaCa-2, lentiviral particles were produced by transfecting 293 T cells with the expression plasmids mentioned previously plus psPAX2 packaging (RRID:Addgene_12260) and VSV/G envelope (RRID:Addgene_8454) plasmids using Lipofectamine 2000 (ThermoFisher 11 668–019). Culture medium was collected 72 hours post-transfection and filtered using a 0.2 µm filter (Whatman) prior to MIA PaCa-2 cell transduction. Polybrene was added to increase virus infection efficiency (10 µg/mL). After infection, puromycin (1 μg/mL Sigma-Aldrich P8833) was used and fluorescence activated cell sorting (FACS) based on the expression of fluorescent proteins was performed to obtain stable clones.
For downregulation experiments, Rab27A shRNA (5′-CCCAGTGTACTTTACCAATATA-3′) and scramble shRNA (5′-CAACAAGATGAAGAGCACCAA-3′) (Mission shRNA Sigma-Aldrich) were cloned into pLKO-Tet-ON (RRID:Addgene_21915) through AgeI-EcoRI site via PCR method and GenScript ClonEZ method, respectively. Additionally, Agrin shRNA (5′-CGACGUGUGCUGUGAAGAATT-′’) and scramble shRNA, mentioned previously, were cloned into pCW57.1-MAT2a (RRID:Addgene_100521) through NheI/XcmI site via PCR method. Lentiviral particles were produced as previously described. Agrin shRNA clones were selected with Blasticidin (VWR A3784.0010).
Flow cytometry
Single-cell suspensions of PDAC cell lines were blocked for 15 min on ice with blocking buffer (FBS 10% in phosphate-buffered saline (PBS) 1×) prior to staining. Next, cells were centrifuged at 1200 revolutions per minute (RPM) for 5 min and incubated for 30 min on ice with an antibody mix in staining buffer (FBS 2% in PBS 1×) to identify the cancer subpopulations used in this study: CD24+44+EPCAM−CD133−, CD24−44+EPCAM−CD133−, CD133+CD24−44−EPCAM−, EPCAM+CD133−CD24−44− and CD24−44−CD133−EPCAM− (4N). The antibodies used were CD24-PE 1:8 (BD Biosciences Cat# 555428, RRID:AB_395822), CD44-APC 1:40 (BD Biosciences Cat# 560890, RRID:AB_2033959), CD133-PE Vio770 1:4 (Miltenyi Biotec Cat# 130-102-891, RRID:AB_2660071), CD133-PE-Cy7 1:62.5 (BioLegend Cat# 372810, RRID:AB_2686968), EPCAM-Fluorescein isothiocyanate (FITC) 1:16 (Sigma-Aldrich Cat# SAB4700424, RRID:AB_10896600), EPCAM-FITC 1:8 (Abcam, Cat# ab8666, RRID:AB_306701) and EPCAM-PerCP-Cy5 1:32 (BioLegend Cat# 369803, RRID:AB_2650899). Afterwards, cells were washed two times with PBS 1× and filtered through a 35 µm cell strainer prior to cell sorting on BDFACS Aria II Cell Sorter (BD Biosciences).
Tumours derived from PDAC GEMMs and PDXs were minced and digested in digestion buffer (0012% Dispase II Sigma-Aldrich D4693, 0012% Collagenase Sigma-Aldrich C7657 in HBSS 1×) for 20 min at 37°C in slow agitation. Afterwards, blocking buffer (FBS 10% in HBSS 1×) was added to stop digestion and cells were centrifuged at 600 g for 5 min. Then, cells are filtered through a 70 µm strainer (Falcon) and incubated with red blood cell lysis buffer for 5 min at room temperature. HBSS 1× is added in excess to stop the reaction and cells are centrifuged as previously described to obtain a single-cell suspension. Staining protocol for the different PDAC subpopulations and consequent cell sorting was as previously described for PDAC cell lines. Antibodies used for isolation of mouse PDAC subpopulations were CD133-APC 1:400 (Thermo Fisher Scientific Cat# 17-1331-81, RRID:AB_823120), CD24-FITC 1:3000 (BD Biosciences Cat# 553261, RRID:AB_394740), CD44-PerCP-Cy5.5 1:500 (BD Biosciences Cat# 560570, RRID:AB_1727486) and EPCAM-PE 1:350 (BD Biosciences Cat# 563477, RRID:AB_2738233). In addition, a viable dye was used to exclude dead cells (Fixable Viability Dye eFluor 780 1:100 000 eBioscience 65-0865-14).
In order to evaluate the presence of Agrin EVs by flow cytometry, 5×109 EVs were incubated with aldehyde/sulfate 4 µm beads for 45 min in rotation. After, 1 M glycine was added and incubated in rotation at room temperature for 1 hour. After centrifugation at 12 000 RPM for 2 min, supernatant was discarded and beads are resuspended in bovine serum albumin (BSA) 10% and incubated for 45 min in rotation. Next, beads were resuspended in 20 µL of BSA 2%. Ten microlitres was used for incubation with primary antibody and secondary antibody, and the other 10 µL was used for incubation with secondary antibody only (control). Incubation with antiagrin antibody was performed overnight at 4°C and in rotation (1:25 Santa Cruz Biotechnology Cat# sc-374117, RRID:AB_10947251). On the following day, washing steps were performed using BSA 2%. Next, incubation with secondary antibody was performed for 30 min at room temperature (Donkey antimouse IgG Alexa 488 1:100 Molecular Probes Cat# A-21202, RRID:AB_141607). Washing steps were performed with BSA 2% prior to FACS analysis in BDFACS Canto II (BD Biosciences). As a gating strategy, first beads were identified in the foward scatter - height (FSC-H) versus side scatter - height (SSC-H) plot. Finally, single beads were analysed for FITC signal. Gate is selected at 103 for the control sample and laser power is adjusted until 1% positivity is detected in the control. The exact same sample but containing the antibody of interest (experimental sample) is rune with the exact same settings. For each sample, this procedure is repeated. Analysis of cAGRN+EVs and cCD133+EVs in serum patient samples was performed blinded.
Spheroid forming assay
Sorted MIA PaCa-2 subpopulations (gating strategy online supplemental figure 1B) were cultured in DMEM-F12 (Corning Cat# 10–090 CV) supplemented with 1 µg/mL hydrocortisone, 4 ng/mL heparin, 10 ng/mL EGF and 20 ng/mL FGF and 1% penincilin/strepmycin (Gibco) in ultralow-adherence sixwell plates (Corning 3471). Spheres were imaged by light microscopy (BZ-X710, Keyence, 4× objective) at day 23, and their area was measured in FIJI (Fiji, RRID:SCR_002285).
Viability assay
MIA PaCa-2 Tet-On shRab27a cells (untreated and treated with doxycycline 200 ng/mL) were exposed to normoxia (21% O2)/hypoxia (1% O2), or to gemcitabine (Sigma Cat# G6423, 1 uM)/vehicle (cell culture grade water, Corning Cat# 25–055 CV) for 72 hours.
Cells were trypsinised and viability/apoptosis was assessed with the FITC annexin V and 7-AAD apoptosis detection kit (BioLegend Cat# 640922), according to manual instructions. As positive control, viable cells (not exposed to gemcitabine or hypoxia) were boiled at 95°C for 5 m and spiked into viable cells. Stained cells were analysed with a BD LSRFortessa X-20 system (RRID:SCR_019600).Live cells were considered as 7-aminoactinomycin D (7AAD) and annexin V negative, early apoptosis as annexin V positive, late apoptosis as annexin V and 7AAD positive and dead/necrosis as 7AAD positive.
Immunofluorescence assay
After culture, media was removed and coverslips were washed with PBS. Cells were fixed using 4% paraformaldehyde in PBS 1× for 10 min at room temperature and washed three times with PBS 1×. Cells were permeabilised with 0.03% Triton-X100 in PBS 1× for 15 min. Coverslips were rinsed three times with PBS 1× and then blocked for 1 hour at room temperature in 10% FBS 0.01% Triton-X100 in PBS 1×.
Coverslips were incubated with primary antibody in 2% FBS 0.01% Triton-X100 at 4°C overnight. Next, coverslips were washed four times with PBS 1× and were incubated with secondary antibodies (1:400) and phalloidin (1:500) for 1.5 hour. Coverslips were washed with PBS 1×, four times. To label nuclei coverslips were incubated with Hoescht (Invitrogen) 1:5000 solution in 0.01% Triton-X100 in PBS 1×. Coverslips were mounted in glass slides with ProLong Diamond Antifade Mountant (Invitrogen).
For the experiments on figure 6A and B (two replicates), (online supplemental figure 5C) (two replicates) anti-active YAP (Abcam Cat# ab205270, RRID:AB_2813833) was used in a concentration of 1:200 and an anti-rabbit Alexa 488-conjugated secondary antibody 1:400 (Abcam Cat# ab150077, RRID:AB_2630356). Alexa 568 conjugated phalloidin (Invitrogen A12380) was used in 1:500 dilution. For experiments on online supplemental figure 5A) (two replicates) an anti-total YAP 1:200 (Santa Cruz Biotechnology Cat# sc-376830, RRID:AB_2750899) was used. For the experiment on figure 5D and online supplemental figure 5D) anti-LRP-4 (Thermo Fisher Scientific Cat# MA5-27674, RRID:AB_2735260) was used in 1:100 dilution and anti-Agrin (Abcam Cat# ab85174, RRID:AB_1860988) was used in 1:200 dilution. For figure 6D, an anti-mouse Alexa488 conjugated secondary antibody (Molecular Probes Cat# A-21202, RRID:AB_141607) 1:200 was used to label LRP-4 and an antirabbit Alexa 488-conjugated secondary antibody 1:400 (Abcam Cat# ab150077, RRID:AB_2630356) was used to label Agrin. In online supplemental figure 5D, an Alexa594 secondary antibody (Thermo Fisher Scientific Cat# A-21203, RRID:AB_141633) was used to label LRP-4.
Microscopy imaging and analysis
MIA PaCa-2 colour-coded cultures were plated on Ibidi multiple well plates with coverslip bottom and were cultured for 72 hours in a 37°C incubator with 5% CO2 (seven replicates were performed). Before imaging, cultures cell media was changed to phenol-free complete RPMI media (Gibco).
A Leica TCS SP5 inverted confocal system was used to image live cells at 37°C using an HCL PL APO CS 40×/ N.A. 1.3 oil objective. Sequential acquisition was used to acquire each fluorescent protein (turboGFP, eYFP, tdtomato and mPlum) laser lines used were 488, 514, 561 and 594 nm, respectively. Images were acquired in tile scan. Prior to image analysis channel cross talk was corrected using the Channel dye separation tool in LAS AF software (Leica Application Suite X, RRID:SCR_013673), where a reference for each channel was manually selected. Individual cells were manually segmented in FIJI (Fiji, RRID:SCR_002285), and single-cell 256 bin histograms were obtained using a script that applied getHistogram function to single-cell regions of interest (ROIs) added to ROI manager. For hypoxia condition an automated segmentation was used based on edge detection and Sobel filter applied to the bright field images.
Background levels were manually determined for each channel (or fluorescent reporter) by measuring pixel intensities in aROI without cells present. The following analysis was performed in MATLAB V.R2016A (MATLAB, RRID:SCR_001622): the original cell channel, including the corresponding subpopulation, was determined by summing pixel frequencies above background level for each channel, where the maximum sum value was considered as the original channel. Communication rates were determined by the presence or absence of pixels with intensities above a threshold level for each channel. Thresholds were determined by measuring intensity range of positive cells for each one the channels/fluorescent reporters. A cell containing pixels above threshold was classified as ‘1’, while a cell without positive pixels was classified as ‘0’. The classification was performed for each channel and the percentage of receiving cells for each reporter was calculated. Presented images were cropped and contrast was optimised in FIJI (Fiji, RRID:SCR_002285).
Tet-On and Tet-Off cocultures were plated into 10 mm coverslips placed in a 24-well plate and cultured for 72 hours in a 37°C incubator with 5% CO2. Images of YAP-1 stained cultures (secondary antibody Alexa 488) were acquired with Leica TCS SP5 inverted confocal system using a HCL PL APO CS 63 x/N.A. 1.40 oil objective. Six images with five z-stacks were acquired for each condition. Quantification was performed in z-maximum projection images obtained in FIJI (Fiji, RRID:SCR_002285). Hoescht signal was used to segment cell nuclei and YAP-1 single-cell nuclear intensity was measured in Cell Profiler V.3.1.9 (CellProfiler Image Analysis Software, RRID:SCR_007358).
Z-stack images of LRP-4, Agrin and LRP-4 stained NSCC treated with CSC-CD63-turboGFP EVs were acquired in Leica TCS SP5 inverted confocal system using a HCL PL APO CS 63 x/N.A. 1.40 oil objective and optical zoom. Presented images were max z-projected, cropped and contrast was optimised in FIJI (Fiji, RRID:SCR_002285).
Fluorescent microbeads uptake
MIA PaCa-2 cells were incubated with green fluorescent polystyrene microbeads with -COOH functional group (100 nm EPRUI Nanoparticles & Microspheres Co., EPRUI-GF-100C) in a ratio of 10 000 microbeads per cell for 24 hours (experiment with three replicates). On the next day, single-cell suspension was obtained and staining protocol, as previously described, was performed to identify cancer cell subpopulations CD24+44+, CD24−44+, CD133+ and 4N. Antibodies used were CD24-PE 1:8 (BD Biosciences Cat# 555428, RRID:AB_395822), CD44-APC 1:40 (BD Biosciences Cat# 560890, RRID:AB_2033959), CD133-PE-Cy7 1:62.5 (BioLegend Cat# 372810, RRID:AB_2686968) and EPCAM-PerCP-Cy5 1:32 (BioLegend Cat# 369803, RRID:AB_2650899). Afterwards, FACS analysis was performed in in BDFACS Canto II (BD Biosciences).
MIA PaCa-2 cells treated for 3 days with gemcitabine (Sigma Cat# G6423, 1 uM) or vehicle (cell culture grade water, Corning Cat# 25–055 CV) were incubated with green polystyrene microbeads, as described earlier. On the next day, cells were analysed for the percentage of Alexa 488 positive cells in BD LSRFortessa X-20 system RRID:SCR_019600).
Mice
C57BL/6 (IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664) and Rag2−/−Ilrg2−/− (IMSR Cat# JAX:014593, RRID:IMSR_JAX:014593) mice were orthotopically injected in the pancreas with cells derived from PDAC cell lines, PDAC GEMMs or PDAC PDXs. PDAC PDXs were established by subcutaneous implantation of pancreas biopsies material in sex-matched Rag2−/−Ilrg2−/− (IMSR Cat# JAX:014593, RRID:IMSR_JAX:014593) mice. For pancreas, orthotopic injections cells from digested tumours were used. The number of cells used varies according to the experiment. Briefly, after acclimatisation for about 3 day,s mice were anaesthetised by intraperitoneal administration of a ketamine/xylazine solution (12.5 mg/kg, 12.5 mg/kg) followed by the subcutaneous administration of a buprenorphine solution (0.08 mg/kg). A small incision was made in the left abdominal flank and the pancreas was exposed and placed on a sterile gauze embedded in NaCl 0,9%. Using a Hamilton syringe, we injected cells slowly in the mouse pancreas. Then, peritoneal and skin layers were sequentially closed with PGA sutures (Surgicryl PGA 6–0). Afterwards, the anaesthetic effect was reversed through subcutaneous administration of an atipamezole solution (2.5 mg/kg). Mice used in experiments related with Mia PaCa-2 Tet-ON shRab27A or Mia PaCa-2 Tet-OFF shAgrin clones were fed with doxycycline supplemented diet (+625 mg/kg doxycycline 720 ppm doxycycline hyclate, Ssniff) or standard diet, as a control. Tumour growth was monitored by ultrasound (Micro Ultrasound Vevo 2100, RRID:SCR_015816). Mice were euthanised when the tumour reached 1500 mm3 or when presented with severe symptoms.
Rag2−/− mice were a kind gift from Dr Nuno Alves, i3S, Porto, Portugal, and Ilrg2−/− mice were a kind gift from Dr James Di Santo, Institute Pasteur, Paris, France.74 C57BL/6 mice were obtained from the i3S animal facility.
KPC (Pdx1-Cre, LSL-KrasG12D/+, LSL-Trp53R172H/+) alleles were purchased from Jackson Laboratory: B6.FVB-Tg(Pdx1-cre)6Tuv/J (IMSR Cat# JAX:014647, RRID:IMSR_JAX:014647); B6.129S4-Krastm4Tyj/J (IMSR Cat# JAX:008179, RRID:IMSR_JAX:008179); 129S-Trp53tm2Tyj/J (IMSR Cat# JAX:008652, RRID:IMSR_JAX:008652), as well as Agrin floxed allele B6;129-Agrntm1Rwb/J (IMSR Cat# JAX:031788, RRID:IMSR_JAX:031788).
Rab27AFrt/Frt allele was developed in collaboration with Cyagen. Exon 4 of the Rab27A gene was selected as conditional knockout (cKO) region. cKO region was first flanked at the 3′ end by a validated Rox-neomycin-Rox-Frt3 cassette and by a single Frt3 site at the 5′ end. Homology arms and cKO region were generated by PCR using BAC clone RP23-10I10 and RP23-336J19 from the C57BL/6J library as template. The 3 prime was cloned into Basic Vector by In-Fusion Enzymes, and the Basic Vector came from digested products of AflII/HindIII. It was subsequently confirmed to be the correct targeting vector by diagnostic PCR, restriction digests and sequencing. The 5 prime and cKO regions was then cloned by In-Fusion Enzymes, and final vector came from digested products of PmeI/AsisI. It was subsequently confirmed to be the correct targeting vector by diagnostic PCR, restriction digests and sequencing. Then, this plasmid was linearised with NotI and used for electroporation of ES cells (C57BL/6). Neomycin drug selection was performed and 366 drug-resistant clones were obtained. PCR screening confirmed six potentially targeted clones and all of them were confirmed correct by Southern blotting. Afterwards, selected ES cells were used for blastocyst injection, followed by chimaera production. Neo cassette flanked by Rox sites was self-deleted during germline transmission and is not present in the final construct.
The R26LSL-FLPoERT2/+ was kindly provided by Dr David Goodrich, Roswell Park Cancer Institute, Buffalo, USA.
KPC iRab27AFrt/Frt (Pdx1-Cre; LSL-KrasG12D/+, LSL-Trp53R172H/+, R26LSL-FLPoERT2/+, Rab27AFrt/Frt) developed spontaneous PDAC tumours in a similar way to the KPC mice. Tamoxifen treatments to induce recombination were given to pups via lactation through oral gavage of the mother with 6 mg of tamoxifen diluted in corn oil (20 mg/mL Sigma-Aldrich T5648) at days 0, 1, 2 and 4 postbirth.
Treatments with Nexinhib20 (Sigma-Aldrich SML1919) were performed in PDAC GEMMs and PDAC PDXs by multiple researchers (injections were not performed unblinded). KPC mice were treated two times per week (intraperitoneal injection, 20 mg/kg) starting at 16 weeks of age. DMSO 5%, vehicle, was used as control. Mice were euthanised when presented severe symptoms such as low body composition, jaundice and ascites. Rag2−/−Ilrg2−/− mice injected with PDAC PDX cells were randomly assigned using randomisation function implemented in IBM SPSS V.25 and V.26 to treatment groups, Nexinhib20 or DMSO 5%, on detection of tumour mass (average of 183 mm3) by ultrasound in all animals of the experiment. Mice were treated two times per week (intraperitoneal injection, 20 mg/kg) for 4 weeks and euthanised after the last treatment.
Ex vivo treatments with Nexinhib20 were also performed in PDAC PDXs. CSC (CD133+ and CD24+44+) isolated from PDAC PDXs were treated with 1 µM Nexinhib20 for 4 hour and NSCC (CD24−44+ and 4N) were not treated. The control group corresponds to CSC treated with DMSO (vehicle) and NSCC not treated. Afterwards, CSC and NSCC were orthotopically injected in the pancreas (at the same percentages found in the PDX tumour), as previously described.
For the in vivo presented in figures 4 and 7, subpopulations of MIA PaCa-2 Tet-On shRab27A/Scramble and Tet-Off shAgrin/Scramble were sorted, mixed and injected in pairs (control and doxycycline). For each replicate (pair), an independent sorting was performed.
Prior to the study, sample sizes were estimated by power analysis (0.05 significance and power 80%).
Patient serum and tissue collection
Serum samples from patients with PDAC were obtained from Hospital Beatriz Ângelo, Lisboa, Portugal, as well as from PDAC patients from Centro Hospitalar de São João (CHSJ), Porto, Portugal. Informed consent was obtained from all patients included in this study in both Hospitals. A cross-sectional study including 62 patients with PDAC was performed at CHSJ from October 2016 to January 2019. Endoscopic ultrasound (EUS)-guided tissue acquisition was performed in all patients with PDAC followed by blood sample collection. Clinical information was age, sex and tumour stage. Exclusion criteria included patients younger than 18 years of age, pregnancy/breast feeding, unable to perform endoscopic procedures or contrast administration, contraindications to CT or MRI, coagulopathy (prothrombin time >50% of control, activated partial thromboplastin time (aPTT) >50 s or international normalized ratio (INR) >1.5), patients on chronic anticoagulant therapy and platelet count below 50x109/L. Collected tissue from patients with PDAC was subcutaneously implanted in immunodeficient mice Rag2−/−Ilrg2−/− for establishment of PDXs. The successful implantations where then passed up to a maximum of six passages in mice, and experiments were performed orthotopically in the pancreas.
A longitudinal study including 44 patients with PDAC was performed at Hospital Beatriz Ângelo from October 2017 and October 2019. All patients were submitted to blood sample collection at different timepoints during follow-up. Clinical information was age, sex, tumour stage, date of surgery, treatment regimen and respective timeline and CA19.9 levels. Patients included in this cohort were followed up either at Hospital Beatriz Ângelo or Hospital da Luz and were not submitted to surgery and/or chemotherapy treatment before this study.
Blood samples collected were centrifuged at 2000 g for 10 min at 4°C and supernatant (serum) was collected and snap freezed. Serum samples were stored at −20°C.
Blinding is not relevant for our study.
EVs isolation
EVs derived from PDAC cells were obtained from cell supernatant. Cells were cultured in RPMI medium supplemented with EVs-depleted FBS for 72 hours. Next, the medium was centrifuged at 2500 RPM for 10 min, followed by a centrifugation at 4000 RPM for 5 min to remove cell debris. Then, medium was filtered using a 200 nm filter (GE Healthcare) and ultracentrifuged at 100 000 g overnight at 4°C. On the following day, supernatant was discarded and EVs pellet was resuspended in a solution of 8M/2.5% sodium dodecyl sulfate (SDS)–urea (Sigma Aldrich) for LC-MS/MS or 100 µL of PBS 1× for FACS, TEM or Image Stream. NanoSight NS300 (NTA, RRID:SCR_014239) analysis was performed for each sample.
For ex vivo evaluation of the number of EVs secreted by cells derived from tumours of PDAC GEMMs or PDXs, single-cell suspension was obtained as previously described and the same number of cells was plated for the treated and control condition. Then, cells were treated with 1 µM Nexinhib20 for 72 hours in RPMI medium supplemented with EV-depleted FBS. DMSO was used as control in the corresponding volume. After 72 hours, cell culture medium was collected and EVs were isolated using the Total Exosome Isolation Reagent (from cell culture media) (Thermo Fisher Scientific, 4478359).
EV isolation was performed from human serum samples. Serum (200 µL) was centrifuged at 10 000 RPM for 2 min and was diluted afterwards with PBS 1× in 400 µL and filtered through a 200 nm filter. Next, samples were ultracentrifuged at 100 000 g overnight at 4°C. On the following day, EV pellet was ressuspended in 300 µL of PBS 1× for NanoSight NS300 (NTA, RRID:SCR_014239) analysis.
Size-exclusion chromatography
MIA PaCa-2 EVs isolated via ultracentrifugation (as previously described) were further purified according to the method described in Kugeratski et al.75 Briefly, a qEV original column 35 nm (Izon SP5) was used to separate 500 μL of EVs preparation in 25 fractions of 500 μL (fractions 1–6 were discarded). For downstream analysis, fractions 7–10 and fractions 11–25 were pooled together and spun down by ultracentrifugation (100 000 g for 3 hours). Fractions were analysed by western blot using precasted polyacrylamide gradient gels (Bolt 4%–12% Bis Tris Plus, Invitrogen) and transferred with a semidry system (Trans-Bolt turbo, Biorad) into methanol activated polyvinylidene difluoride (PDVF Transblot turbo, Biorad) membrane. Expression of EVs markers syntenin-1 (1:5000, Abcam Cat# ab133267,RRID:AB_11160262) and Alix (1:1000 CST Cat# 2171, RRID:AB_2299455) was detected using antirabbit-horseradish peroxidase (HRP) (1:5000 CST Cat# 7074S RRID:AB_2099233) and antimouse-HRP (1:2000 R&D Cat# HAF007 RRID:AB_357234) secondary antibodies, respectively.
The expressions of transmembrane EV markers CD9, CD81 and CD63 were performed by coupling EVs to aldehyde/sulfate beads (Invitrogen, Cat# A37304) according to the protocol described in Kugeratski et al.75 Anti-CD9 (Sigma-Aldrich, Cat# SAB4700092 RRID:AB_10900636), anti-CD63 (BD, Cat# 556019 RRID:AB_396297) and anti-CD81 (BD, Cat# 555675 RRID:AB_396297), together with the secondary antibody anti-mouse Alexa 488 (Invitrogen Cat# A21202, RRID:AB_141607) were used to detect these EV markers.
Optiprep density gradient
MIA PaCa-2 EV preparation (isolated via ultracentrifugation, as previously described) was bottom loaded into an Optiprep density gradient as described in Kugeratski et al.75 Briefly, 500 μL of EVs preparation was mixed into Optiprep (Sigma, Cat# D1556) at 36% concentration (final volume 2.5 mL) and loaded into an ultracentrifuge tube. Next, 2.5 mL of 30%, 24%, 18% and 12% Optiprep concentrations (2.5 mL) ws sequentially layered on top. Gradients were centrifuged for 15 hours at 120 000 g (no break, rotor SW41, Beckman coulter). Afterwards, 12 fractions of 1 mL were collected, resuspended in 11 mL of PBS 1× and centrifuged at 120 000 g for 4 hours. Each fraction pellet was resuspended in 40 μL of lysis buffer (8 M urea, 2.5% SDS, cComplete 1× (Roche) and PhosphoSTOP 1× (Roche).
Fractions 1–12 were analysed by western blot, as described previously, for CD81 (1;5000 Santa Cruz Cat# sc-166029, RRID:AB_2275892) syntenin-1 (1:5000, Abcam Cat# ab133267, RRID:AB_11160262), Alix (1:1000 CST Cat# 2171, RRID:AB_2299455), and non-EV markers HSP90 (1:1000 CST Cat# 4877S, RRID:AB_2233307) and histone H3 (1:1000, Abcam Cat# ab201456, RRID:AB_2650560).
For agrin detection (1:200 Abcam Cat# ab85174, RRID:AB_1860988), transfer was performed in a wet transfer system (Biorad) into a 0.45 µm PDVF methanol-activated membrane (Immobilion Cat# IPVH00010, Merck).
Antirabbit-HRP (1:2000, Abcam Cat# ab16284, RRID:AB_955387) was used to detect Alix, HSP90, histone H3 and agrin; antirabbit-HRP (1:5000 CST Cat# 7074S RRID:AB_2099233) was used to detect syntenin-1; and anti-mouse-HRP (1:2000 R&D Cat# HAF007 RRID:AB_357234) was used to detect CD81.
EV characterisation derived from cells exposed to hypoxia or gemcitabine
MIA PaCa-2 cells (triplicates) were exposed to hypoxia (1%O2)/normoxia (21% O2) or to gemcitabine (1 µM)/vehicle (cell culture grade water) for 72 hours and cultured in RPMI medium supplemented with EVs-depleted FBS. EVs were collected as previously described and pellets were resuspended in 100 µL of PBS 1×. For western blot, 20 µL of EV preparation from each condition was loaded into the gel. CD81 (1:5000 Santa Cruz Cat# sc-166029, RRID:AB_2275892), syntenin-1 (1:5000, Abcam Cat# ab133267, RRID:AB_11160262) and CD9 (1:1000, Abcam Cat# ab263019) were used to detect EVs.
Antirabbit-HRP 1:5000 (CST Cat# 7074S, RRID:AB_2099233) was used for syntenin-1, antirabbit-HRP 1:2000 (Abcam, Cat# ab16284, RRID:AB_955387) was for CD9 and anti-mouse-HRP (1:2000 R&D Cat# HAF007, RRID:AB_357234) for CD81.
EV characterisation derived from cells exposed to Nexinhib20
MIA PaCa-2 cells were cultured in in RPMI medium supplemented with EVs-depleted FBS for 72 hours in the presence of DMSO (5%) or Nexinhib20 (1 µM). Afterwards, EVs were isolated according to Crescitelli et al.76
Briefly, cells debris were eliminated through a centrifugation at 300 g for 10 min and a centrifugation at 2000 g for 20 min. Afterwards, supernatants were centrifuged at 16 500 g for 20 min to remove large vesicles and followed by 118 000 g centrifugation for 2.5 hour to collect small vesicles. All centrifugations were performed at 4°C.
For western blot, 20 µL of EV preparation from each condition was loaded into the gel. CD63 (1:500, BD Biosciences Cat# 556019, RRID:AB_396297) and β-actin (Sigma-Aldrich Cat# A3854, RRID:AB_262011) were used to detect EVs. Acetyl–cholinesterase (1:500, Abcam Cat# ab31276, RRID:AB_722529) and cytochrome C (1:200, Santa Cruz Biotechnology Cat# sc-7159, RRID:AB_2090474) were used to confirm purity of the small EVs isolated.
Transmission electron microscopy
EV characterisation by TEM was performed as previously described in Théry et al.77
Image stream
EVs were collected from 50 mL of MIA PaCa-2 cell supernatant cultured for 72 hours in RPMI medium supplemented with EV-depleted FBS. EV isolation was performed as previously described. Then, EV pellet was resuspended in 100 µL of PBS 1× and divided into four conditions for the analysis of CD63, CD81, CD82 and Rab5. A similar protocol to the one performed for the analysis of Evs by flow cytometry was done until detection in ImageStreamx MarkII Imaging Flow Citometry (Luminex Amnis Image Stream Multispectral Imaging Flow Cytometer, RRID:SCR_018589). Primary antibodies used were anti-CD63 1:400 (Santa Cruz Biotechnology Cat# sc-15363, RRID:AB_648179), anti-CD81 1:400 (Abcam Cat# ab109201, RRID:AB_10866464), anti-CD82 1:400 (Abcam Cat# ab59509, RRID:AB_2076398) and anti-Rab5 1:400 (Cell Signalling Technology Cat# 46449, RRID:AB_2799303). Secondary antibodies used were Donkey antimouse IgG Alexa Fluor 488 1:500 (Molecular Probes Cat# A-21202, RRID:AB_141607) and goat antirabbit IgG Alexa Fluor 488 1:500 (Thermo Fisher Scientific Cat# A-11034, RRID:AB_2576217). For each sample, control refers to EVs incubated with secondary antibody only.
Liquid chromatography–electrospray ionisation–tandem mass spectrometry
Protein digestion
Samples were dissolved with chaotropic lysis buffer containing 8.4 M urea (USB Corporation, Cleveland, Ohio, USA), 2.4 M thiourea (Sigma-Aldrich), 2.5% SDS (Sigma-Aldrich), 5 mM tris(2-carboxyethyl)phosphine (TCEP) (Sigma-Aldrich) and a protease inhibitor cocktail (Sigma-Aldrich), and incubated for 15 min on ice. Homogenisation of the pellet was achieved by ultrasonication for 5 min on ultrasonic bath Branson 2510 (Marshall Scientific, New Hampshire, USA). The homogenate was centrifuged at 20 000×g for 10 min at 4°C, and the supernatant containing the solubilised proteins was used for further analysis. Then, 20 µg of protein was precipitated by methanol/chloroform method and resuspended in 20 µL of multichaotropic sample solution UTT buffer (7 M urea, 2 M thiourea, 100 mM TEAB (Sigma-Aldrich).
The resuspended sample was reduced with 2 µL of 50 mM TCEP, pH 8.0, at 37°C for 60 min, followed by addition of 1 µL of 200 mM cysteine-blocking reagent MMTS (SCIEX, Foster City, California, USA) for 10 min at room temperature. Sample was diluted to 140 µL to reduce the urea concentration with 25 mM triethylamonium bicarbonate (TEAB). Finally, digestion was initiated by adding 1 µg of Pierce MS-grade trypsin (Thermo-Fisher Scientific) to each sample in a ratio 1:20 (w/w), and then incubated at 37°C overnight on a shaker. Sample digestion was evaporated to dryness in a vacuum concentrator.
Liquid chromatography and mass spectrometer analysis
The digested sample was cleaned up/desalted using Stage-Tips with Empore 3M C18 disks (Sigma-Aldrich). A 1 µg aliquot of each digested sample was subjected to 1D-nano LC ESI-MSMS analysis using a nano liquid chromatography system (Eksigent Technologies nanoLC Ultra 1D Plus, SCIEX) coupled to high-speed Triple TOF 5600 mass spectrometer (SCIEX TripleTOF 5600+Mass Spectrometer System, RRID:SCR_018053) with a Nanospray III source. The analytical column used was a silica-based reversed phase Acquity UPLC M-Class Peptide BEH C18 Column, 75 µm×150 mm, 1.7 µm particle size and 130 Å pore size (Waters). The trap column was a C18 Acclaim PepMapTM 100 (Thermo Scientific), 100 µm×2 cm, 5 µm particle diameter, 100 Å pore size, switched online with the analytical column. The loading pump delivered a solution of 0.1% formic acid in water at 2 µL/min. The nanopump provided a flow rate of 250 nL/min and was operated under gradient elution conditions. Peptides were separated using a 250 min gradient ranging from 2% to 90% mobile phase B (mobile phase A: 2% acetonitrile, 0.1% formic acid; mobile phase B: 100% acetonitrile, 0.1% formic acid). Injection volume was 5 µL.
Data acquisition was performed with a TripleTOF 5600 System (SCIEX). Data were acquired using an ionspray voltage floating of 2300 V, curtain gas of 35, interface heater temperature of 150C, ion source gas 1 of 25C, declustering potential (DP) 100 V. All data was acquired using information-dependent acquisition (IDA) mode with Analyst TF 1.7 software (SCIEX, AnalystTF Software, RRID:SCR_015785). For IDA parameters, 0.25 s MS survey scan in the mass range of 350–1250 Da were followed by 35 MS/MS scans of 100 ms in the mass range of 100–1800 (total cycle time: 4 s). Switching criteria were set to ions greater than mass to charge ratio (m/z) 350 and smaller than m/z 1250 with charge state of 2–5 and an abundance threshold of more than 90 counts (cps). Former target ions were excluded for 15 s. IDA rolling collision energy (CE) parameters script was used for automatically controlling the CE.
Raw data alignment and filters
The mass spectrometry data from obtained were processed using PeakView V.2.2 Software (SCIEX, PeakView Software, RRID:SCR_015786). Raw data file conversion tools generated Mascot General Format (.mgf) files, which were searched against a custom database composed of (1) Homo sapiens protein database from EMBL-EBI Reference Proteomes repository (release 2020_02 based on UniProt Release 2020_02, Ensembl release 98 and Ensembl Genome release 45), containing 20 596 protein coding genes; and (2) sequences of 116 contaminant proteins obtained from cRAP repository (version dated 2019_03_04, https://www.thegpm.org/crap/) (cRAP protein sequences, RRID:SCR_018187). MS-GF+ software1 (release v2020_03_14) (Innovative Research Cat# IR-MS-GF, RRID:AB_1501658) was used as a search engine. Search parameters were set as follows: carbamidomethyl c, as fixed modification and Gln to pyro-Glu (N-term Q), Glu to pyro-Glu (N-term E) and oxidation (M) as variable modifications. Peptide mass tolerance was set to ±25 ppm and two missed cleavages were allowed. Corresponding mzIdentML (.mzid) files were generated and loaded into the R environment for further statistical analyses.
Mass measurement error ±10 ppm and MS-GF +spectrum level E-value <1e-10 thresholds were defined for every sample.78 Then, Nelder-Mead optimisation was applied to maximise the number of identifications under false discovery rate (FDR) ≤1% at peptide level (mSnID R package). Proteins matching contaminants (cRAP repository), keratins and serum albumin were also removed, keeping a total of 6185 proteins and 38 biological samples. Each subpopulation was replicated in the four cell lines, with the exception of EpCAM+ that was not detected in MIA PaCa-2 cell line.
Data analysis
To cluster sets of proteins and associate them to group of samples, hierarchical clustering (Ward’s minimum variance method) and k-means clustering, both implemented from ComplexHeatmap R package, were applied on the binary matrix (1=detected protein, 0=non-detected protein) of 6185 proteins. Twelve initial protein clusters were grouped into four major clusters (‘not defined’, ‘cells’, ‘EVs’ and ‘cells & EVs’), depending on which group of samples the proteins were associated with.
For the differential analysis between group of samples (CSC vs NSCC), peptide spectrum matches PSM levels of all samples were summarised and quantified to protein counts (mSnbase R package,79 ‘sum’ method). Peptide counts of every protein were input to DESeq2 R package,80 and Wald’s test was applied to find and rank differential protein abundances between CSC and NSCC. FDR 5% threshold was defined for significant differentially expressed proteins. Furthermore, GSEA using reactome (ReactomePA R package81) as database was applied using protein DESeq2 logFCs as input.
Subcellular localisation data were retrieved from Human Protein Atlas82 database (dated May 2020) (HPA, RRID:SCR_006710) and assigned to detected proteins (only ‘main location’ field was considered). Enrichment of subcellular compartments in cells and EVs samples was calculated by simple linear model regression.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE83 84 partner repository with the dataset identifier PXD023529 and 10.6019/PXD023529.85
All the code to reproduce the data alignment and statistical analysis is publicly available at GitHub repository (GitHub, RRID:SCR_002630, https://github.com/fjcamlab/EVNet_MS).
IHC and histological analysis
Tissue samples were fixed in 10% formalin for at least 24 hours prior to paraffin embedding. Then, 4 µm sections were cut using a Microm HM335E microtome (Thermo Fisher Scientific). For IHC, sections were deparaffinised and hydrated prior to heat-mediated antigen retrieval for 40 min using a citrate buffer pH 6 solution (Vector Laboratories) for Rab27A staining or using a TRIS-EDTA pH 9 solution for YAP staining. After, incubation with 0,3% hydrogen peroxide solution (Sigma-Aldrich) diluted in methanol was performed for 15 min at room temperature. To inhibit unspecific staining, sections were blocked with protein block (DAKO) for 30 min at room temperature. Incubation with primary antibody was performed overnight at 4°C (anti-Rab27A 1:200 Sigma-Aldrich Cat# HPA001333, RRID:AB_1079730, anti-YAP 1:2000 Abcam Cat# ab205270, RRID:AB_2813833). On the next day, slides were incubated with HRP rabbit/mouse secondary antibody (DAKO, RRID:AB_2888627) for 30 min at room temperature and visualised using the DAB chromogen (DAKO). Rab27A H-score was performed as described previously.86 Briefly, for H-score evaluation, five fields were randomly selected at ×400 magnification. Cytoplasm staining intensity was determined for Rab27A. Score 0, 1, 2 or 3 corresponds to the presence of negative, weak, intermediate or strong staining. The number of cells for each staining intensity and total number of cells in each field was determined. H-score was calculated according to the following formula:
H-score=(% of cells stained at intensity 1×1)+(% of cells stained at intensity 2×2)+(% of cells stained at intensity 3×3). A maximum score of 300 is obtained when 100% of cells are stained at staining category 3 (strong).
YAP staining was quantified using the average of the percentage of nuclear YAP+ cells in five random fields (×400).
For histological analysis, sections were deparaffinised and hydrated and incubated with H&E for 1 min each.
Histopathological analysis
H&E-stained pancreas sections were evaluated for the presence of each disease stage: no histological disease, PanIN and PDAC. Pancreas sections were evaluated at ×40 amplification, and the percentage of each stage for each mouse was determined.
Cell proliferation assay
For Nexinhib20 toxicity evaluation, Mia PaCa-2 cells were treated with increasing concentrations of Nexihib20 for an MTT (Sigma-Aldrich M5655) assay. Five thousand cells were plated in quadruplicates for each condition in a 96-well plate. Nexinhib20 concentrations used were0.01, 0.05, 0.1, 0.5, 1.0 and 5.0 µM. Treatments were performed for 72 hours and vehicle (DMSO in corresponding volume) was used as a control. After 72 hours, 10 µL of MTT (5 mg/mL in NaCl 0,9%) was added to each well and incubated at 37°C for 3 hours. Afterwards, the medium was removed and 100 µL of DMSO was added per well followed by incubation with agitation at room temperature for 5 min. Finally, absorbance is read at 560 nm after a 5 s shake in a microplate reader (Model 680 Bio-Rad).
MTT assay was performed on PDX cells treated with antihuman agrin neutralising antibody. Seven thousand cells were plated in quadruplicates for each condition in 96-well plates. Antihuman agrin neutralising antibody (10 µg/mL, Millipore Cat# MAB5204, RRID:AB_2225272), IgG (10 µg/mL, Jackson ImmunoResearch Labs Cat# 115-005-003, RRID:AB_2338447) or no treatments were given daily for 7 days, after which treatments ceased and cells were kept for 5 more days without treatment. MTT evaluation was performed at days 1, 3, 5, 7, 9 and 12, as previously described. Also, MTT assay was used to access proliferation of sorted MIA PaCa-2 NSCC (CD24−CD44+ and 4N) treated with CSC EVs (1 μg), and YAP inhibitor verteporfin (Sigma, Cat# SML0534 10 μg/mL, diluted from a 1 mg/mL stock) or DMSO (1:100 (v/v)). Sorted NSCC (3000 cells) were plated in triplicates in 96-well plates and treated (with EVs and verteporfin/DMSO) at days 1 and 4 postsorting. For this experiment, absorbance was read at 570 nm in a microplate reader (FLUOstar Omega, BMG Labtech)
EV degradation assay
EVs from MIA PaCa-2 cells obtained as previously described (1×1012 measured by NTA) were resuspended in 1 mL of PBS 1×, mixed with 4 μL of CM-Dil (Invitrogen, Cat# C7001, diluted 1:8 in DMSO) and incubated at 37°C for 1 hour. Total exosomes isolation kit (from cell culture media, Invitrogen) was used to collect stained EVs according to manual instructions. Pellet was resuspended in 200 μL of RPMI complete media. Previously plated CSC (CD24+CD44+ and CD133+) and NSCC (CD24-CD44+ and 4N) in 48 wells (5000 cells) were treated with 100 μL of DiL-stained EVs. For each condition, five fields were acquired with a ×20 objective and imaged for 24 hours, every 30 m (Olympus IX81 RRID:SCR_020341 and Slidebook 42 software, DiL was detected in tetramethylrhodamine (TRITC) channel). Mean grey value of TRITC channel for each field overtime was quantified in FIJI (RRID:SCR_002285).
PCR
Pancreas fragment was digested at 56°C for 2 hours on a thermal-shaker with lysis buffer (10 mM Tris (pH 7.5), 400 mM NaCl, 2 mM EDTA (pH 8.0)) (pH 7.3–7.5), 20% SDS (Merck 428018) and 20 µL of 20 mg/mL proteinase (Ambion RNA by Life Technologies AM2548). Then, 6 M NaCl was added to the extraction mixture; samples were mixed thoroughly by vortexing for 10 s, followed by centrifugation at 14 000 RPM for 15 min to precipitate the residual cellular debris. The supernatant was transferred to a clean eppendorf tube and 100% ethanol was added to each sample, mixed thoroughly by vortexing for 10 s, and centrifuged at 14 000 RPM for 5 min to pellet the DNA. The DNA pellets were washed with 70% ethanol, followed by centrifugation at 14 000 RPM for 5 min. The pellets were completely air dried and resuspended in sterile nuclease-free water.
For conventional PCR, we used a commercial master mix 2× My Taq HS Mix (Bioline Bio-25046). PCR amplifications were carried out in the T-100 Thermal Cycler (Biorad). All assays included two positive contros samples and a no-template control (contained all reaction components except the genomic DNA). Oligonucleotide sequences used to confirm agrin or Rab27A recombination were
Agrin Exon 6 forward: CGGACACACATATGCTAGTGA.
Agrin Exon 34 reverse: CAAAGTGGTTGCTCTGCAGCG.
Rab27A forward: CCAGGGCACTTCCTAAAGATAAG.
Rab27A reverse 1: CACGCTGCTTCAATGAATAAGGTG.
Rab27A reverse 2: CATCCACAAAGCTCCAGATTCCAC.
The amplification protocol included an initial denaturation and enzyme activation at 95°C for 5 min followed by 30 cycles of denaturation at 95°C for 30 seconds, annealing at 56°C for 90 s, and extension at 72°C for 30 seconds and a final extension at 60°C for 10 min.
Quantitative reverse transcription-PCR
MIA PaCa2 NSCC (CD24−CD44+ and 4N) plated into 24 wells were transfected with siScramble of siLPR4 (Thermo Fisher Cat# 202503, 15 pmol) using Lipofectamine 3000 (Invitrogen) according to manual instructions. On the third day post transfection, NSCC (siScramble/siLRP-4) were treated with CSC EVs (10 μg/well) and resuspended in TRIzol (Invitrogen) 24 hours later. Oligonucleotide sequences used were:
CXCL5 forward: TCTGCAAGTGTTCGCCATAG
CXCL5 reverse: TGTCTTCCCTGGGTTCAGAG
STAT3 forward: GGCATTCGGGAAGTATTGTCG
STAT3 reverse: GGTAGGCGCCTCAGTCGTATC
CYR61 forward: GAGTGGGTCTGTGACGAGGAT
CYR61 reverse: GGTTGTATAGGATGCGAGGCT
ALX forward: GTTTGGAGCTGTGATGGAAGGC
ALX reverse: CGCTTCACTCAGGAAATCCTCC
VIM forward: AGGCAAAGCAGGAGTCCACTGA
VIM reverse: ATCTGGCGTTCCAGGGACTCAT
AREG forward: GCACCTGGAAGCAGTAACATGC
AREG reverse: GGCAGCTATGGCTGCTAATGCA
ACTB forward: GAGCACAGAGCCTCGCCTTT
ACTB reverse: ACATGCCGGAGCCGTTGTC
RNA was isolated from TRIzol (Invitrogen cell pellets), as described in the manual. cDNA synthesis from 1 μg of RNA was performed with high-capacity cDNA reverse transcription kit (Life Technologies, Cat# 4374966) according to the manual. Power SYBR green PCR master mix (Applied Biosystems) was used for the quantification PCR. The following primers were used to quantify expression levels.
Western blot
Cell protein extraction was performed using RIPA lysis buffer (VWR), both supplemented with cComplete (Roche) and phenylmethylsulphonyl fluoride (Sigma) for 30 min on ice followed by a centrifugation at 17 000 g for 30 min to remove DNA. Protein (30 µg) was used for western blot analysis after quantification using DC Protein Assay (Bio-Rad). In the experiment to evaluate levels of LRP-4 in CSC and NSCC, the same numbers of CSC and NSCC from MIA PaCa-2 were sorted directly to protein extraction buffer. All volume (total protein derived from the same number of cells) was used to perform western blot.
Samples were run in a SDS-PAGE gel and then transferred to a 0.2 µm nitrocellulose membrane (GE HealthCare) using Tris–glycine transfer buffer. Next, nitrocellulose membranes were blocked using a 5% milk/PBS-Tween 0.1% solution for 1 hour at room temperature. Incubation with primary antibody was performed overnight at 4°C (antiagrin 1:200 Abcam Cat# ab85174, RRID:AB_1860988, anti-Rab27A 1:500 Abnova Cat# H00005873-M02, RRID:AB_519010, anti-LRP-4 1:1000, Thermo Fisher Scientific Cat# MA5-27675, RRID:AB_2735261). On the next day, membranes were washed in PBS-Tween 0.1% solution and incubated with the respective HRP-conjugated secondary antibody at 1:5000 dilution for 1 hour at room temperature (antirabbit IgG HRP-linked antibody Cell Signalling Technology Cat# 7074, RRID:AB_2099233, Anti-mouse IgG HRP-linked antibody Advansta Cat# R-05 071–500, RRID:AB_10718209). Then, membranes were washed in PBS-Tween 0.1% solution and Clarity Western ECL Substrate (Bio-Rad) was used to detect antibody-specific signal. β-actin (Sigma-Aldrich Cat# A3854, RRID:AB_262011) and vimentin (Sigma-Aldrich Cat# V5255, RRID:AB_47762) were used as loading control for Rab27A and agrin analysis, respectively.
Human cytokine array
MIA PaCa-2 Tet-On shRab27a cells were cultured for 24 hours in the presence or absence of doxycycline. Afterwards, cells were washed and cultured for an additional 48 hours in the presence or absence of doxycycline. Conditioned medium was collected and used to analyse cells’ secretome using the human cytokine array C3 (RayBiotech Cat# AAH-CYT-3–2, RRID:AB_2753199). Protocol was performed according to the manufacturer.
Signal intensity quantification was performed using FIJI (Fiji, RRID:SCR_002285), Protein Array Analyser.87 88
TCGA analysis
As a validation cohort, we downloaded the dataset of pancreatic carcinomas from TCGA (https://cancergenome.nih.gov/), composed of 176 patients with gene expression data, quantified as fragments per kilobase million. Clinical information provided by TCGA was also downloaded, allowing us namely to verify that eight cases presented a neuroendocrine type of cancer; these eight cases were removed from most of the analyses, and we only verified that we obtained the same results for overall survival as the ones displayed in The Human Protein Atlas website (that includes the entire cohort, https://www.proteinatlas.org/). By mining the protein expression data provided by the TCGA consortium, we verified that for the genes under analyses in our study, YAP protein was tested in 113 cases, and evaluated through two antibodies (from rabbit): Phospho-YAP (Ser127) Antibody #4911 (Cell Signalling Technology, Danvers, Massachusetts, USA) herein designed as YAP_pS127; YAP Antibody (H-125) sc-15407 (Santa Cruz Biotechnology, Dallas, Texas, EUA). Survival information was extracted from clinical files and information on recurrence was downloaded from cBioPortal from Cancer Genomics (https://www.cbioportal.org/).
The Kaplan-Meier method was used to generate the disease-free survival (time to the first recurrence event; deceased individuals without information on recurrence were removed from the analysis) and the overall survival (time to death from any cause). The plots were obtained in R using ‘survival’ and ‘survminer’ packages. The best separation cut-off method, also employed by the protein atlas, was used to cluster the individuals in the ‘high’ and ‘low’ gene/protein expression groups. This cut-off refers to the value, within the second quartile, that yields the lowest log-rank p value in the comparison between the groups. For the pairwise combinations of genes or gene–protein, the individually estimated cut-off values were used.
Pearson correlations between gene expressions or gene–protein expressions were performed using the ‘cor.test’ package in R. Expression outliers were removed following the Tukey’s method to which 1.5 times the IQR are added (third quartile) or subtracted (first quartile) to establish the upper and lower outlier cut-offs, respectively. Plots were generated in ‘ggplot2’ package in R.
Statistical analysis
For all analyses, significance was determined at p<0.05*, p<0.01**, p<0.001*** and p<0.0001****, representing significance between conditions. All analyses, unless specified elsewhere, were performed in GraphPad Prism (GraphPad Prism, RRID:SCR_002798) or SPSS Statistics V27 Release 27.0.1.0.(IBM, RRID:SCR_019096).
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE84,85 partner repository with the dataset identifier PXD023529 and 10.6019/PXD023529. All the codes to reproduce the data alignment and statistical analysis are publicly available at GitHub repository (GitHub, RRID:SCR_002630, https://github.com/fjcamlab/exonet_MS).
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Serum samples from pancreatic ductal adenocarcinoma (PDAC) patients were obtained from Hospital Beatriz Ângelo, Lisboa, Portugal, as well as from PDAC patients from Centro Hospitalar de São João (CHSJ), Porto, Portugal. Informed consent was obtained from all patients included in this study in both Hospitals. Serum collection was approved by the ethical committees of CHSJ, Porto, Portugal (ID number CES 327-15) and Hospital Beatriz Ângelo, Lisboa, Portugal (ID number 1372/2015_CMOEB). Participants gave informed consent to participate in the study before taking part. All mice were housed under standard housing conditions at the i3S animal facility, and all animal procedures were reviewed and approved by the i3S Animal Welfare and Ethics Body. Animal protocol was approved by Direção Geral de Alimentação e Veterinária (ID 015225). Consent from the CHSJ ethical committee was obtained to establish PDX models of PDAC. Human studies were conducted in accordance with the Declaration of Helsinki ethical guidelines.
Acknowledgments
We thank Sofia Quintas, Patricia F Vieira and Joana Martins for technical support, and Celso Reis for the anti-human MUC1 antibody.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
- Data supplement 14
- Data supplement 15
- Data supplement 16
- Data supplement 17
- Data supplement 18
Footnotes
CFR and NB contributed equally.
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors Conceptualisation: SAM and CFR; methodology: CFR, NB and SAM; formal analysis: CFR, NB, CD, FC-L, HO, BC and LP; investigation: CFR, NB, IB, BA, AC-P, SS and SC; resources: CAM, PM-R, BM, DG, TK, GM, RM, FC, MC, RK, JLC and SAM; data curation: CFR, NB, IB, CD, CAM, FC-L, PM-R, BM, SS, BC and LP; writing, original draft: CFR, NB and SAM; writing, review and editing: SAC, CAM, JCM and SAM; visualisation: BA; supervision: SAM; funding acquisition: SAM; Guarantor:SAM.
Funding The work was supported by NORTE-01–0145-FEDER-000029, Norte Portugal Regional Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund and national funds through FCT—Foundation for Science and Technology POCI-01–0145-FEDER-32189. Programa Operacional Regional do Norte and co-financed by European Regional Development Fund under the project "The Porto Comprehensive Cancer Center" with the reference NORTE-01-0145-FEDER-072678 - Consórcio PORTO.CCC – Porto.Comprehensive Cancer Center. CFR is supported by FCT (SFRH/BD/131461/2017), NB by (SFRH/BD/130801/2017), IB by FCT (SFRH/BD/144854/2019), and BA by FCT (PD/BD/135546/2018). DG’s contribution was supported by the NCI (R21 CA179907). We acknowledge the support of the i3S Scientific Platforms: Translational Cytometry, Animal Facility, Bioimaging and Histology and Electron Microscopy are members of the national infrastructure PPBI - Portuguese Platform of Bioimaging (PPBI-POCI-01–0145-FEDER-022122). Proteomics was performed at the Proteomics Facility of The Spanish National Center for Biotechnology (CNB-CSIC), ProteoRed, PRB3-ISCIII, supported by grant PT17/0019.
Competing interests SAM holds patents in the area of exosome biology. TK is a founder of Abcam plc and Storm Therapeutics and is on the scientific advisory board of Foghorn Therapeutics. RK holds patents in the area of exosomes biology and are licensed to Codiak Biosciences, Inc. RK is stockholder in Codiak Biosciences, Inc. RK is a consultant and scientific adviser for Codiak Biosciences, Inc. The other authors declare no potential conflict of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.