Article Text

Abstract
Objective Cigarette smoking is a major risk factor for colorectal cancer (CRC). We aimed to investigate whether cigarette smoke promotes CRC by altering the gut microbiota and related metabolites.
Design Azoxymethane-treated C57BL/6 mice were exposed to cigarette smoke or clean air 2 hours per day for 28 weeks. Shotgun metagenomic sequencing and liquid chromatography mass spectrometry were parallelly performed on mice stools to investigate alterations in microbiota and metabolites. Germ-free mice were transplanted with stools from smoke-exposed and smoke-free control mice.
Results Mice exposed to cigarette smoke had significantly increased tumour incidence and cellular proliferation compared with smoke-free control mice. Gut microbial dysbiosis was observed in smoke-exposed mice with significant differential abundance of bacterial species including the enrichment of Eggerthella lenta and depletion of Parabacteroides distasonis and Lactobacillus spp. Metabolomic analysis showed increased bile acid metabolites, especially taurodeoxycholic acid (TDCA) in the colon of smoke-exposed mice. We found that E. lenta had the most positive correlation with TDCA in smoke-exposed mice. Moreover, smoke-exposed mice manifested enhanced oncogenic MAPK/ERK (mitogen-activated protein kinase/extracellular signal‑regulated protein kinase 1/2) signalling (a downstream target of TDCA) and impaired gut barrier function. Furthermore, germ-free mice transplanted with stools from smoke-exposed mice (GF-AOMS) had increased colonocyte proliferation. Similarly, GF-AOMS showed increased abundances of gut E. lenta and TDCA, activated MAPK/ERK pathway and impaired gut barrier in colonic epithelium.
Conclusion The gut microbiota dysbiosis induced by cigarette smoke plays a protumourigenic role in CRC. The smoke-induced gut microbiota dysbiosis altered gut metabolites and impaired gut barrier function, which could activate oncogenic MAPK/ERK signalling in colonic epithelium.
- COLORECTAL CANCER
- BACTERIAL PATHOGENESIS
- BILE ACID METABOLISM
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets generated in the current study are available in the Genome Sequence Archive (GSA) at the National Genomics Data Center, Beijing Institute of Gemonics, Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA006099) and are publicly accessible at https://bigd.big.ac.cn/gsa/.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Gut microbiota and their metabolites have critical roles in colorectal tumourigenesis.
Cigarette smoking is a well-known risk factor of colorectal cancer (CRC), but the mechanism of how it promotes CRC development remains unclear.
What are the new findings?
Cigarette smoking increases colorectal tumourigenesis and alters microbiota composition in azoxymethane-treated mice with significant enrichment of bacterial species including Eggerthella lenta.
Metabolomic profile is markedly changed by cigarette smoking with increased biosynthesis of the procarcinogenic taurodeoxycholic acid (TDCA), which also has the most positive correlation with E. lenta.
Smoke-exposed mice display impaired colonic epithelium and enhanced oncogenic MAPK/ERK (mitogen-activated protein kinase/extracellular signal‑regulated protein kinase 1/2) and proinflammatory (interleukin 17 and tumour necrosis factor) signalling pathways.
Transplanting stools from smoke-exposed mice into germ-free mice leads to enrichments in E. lenta and TDCA, impaired colonic epithelium and increased activation of MAPK/ERK and proinflammatory pathways in recipient mice.
How might it impact on clinical practice in the foreseeable future?
Our findings indicate that cigarette smoking causes compositional alterations in microbiota and their metabolites, resulting in gut barrier dysfunction and activation of oncogenic and proinflammatory signalling pathways, contributing to colorectal tumourigenesis.
Manipulation of the gut microbiota might represent a promising prophylactic strategy against CRC in smoking population.
Disclaimer: this video summarises a scientific article published by BMJ Publishing Group Limited (BMJ). The content of this video has not been peer-reviewed and does not constitute medical advice. Any opinions expressed are solely those of the contributors. Viewers should be aware that professionals in the field may have different opinions. BMJ does not endorse any opinions expressed or recommendations discussed. Viewers should not use the content of the video as the basis for any medical treatment. BMJ disclaims all liability and responsibility arising from any reliance placed on the content.
Introduction
Colorectal cancer (CRC) is one of the most common cancers globally. Although there are many strategies for early CRC screening and prevention, its burden is expected to increase further.1 There are evidences supporting the association of lifestyles such as diet, cigarette smoking, obesity and exercise with CRC.2
Cigarette smoking increases the risk of lung cancer with about 80% of all primary lung cancers attributable to cigarette smoking.3 Smoking can also increase the risk of cancer in other organs that are not exposed to cigarette smoke directly, such as the colon, rectum, pancreas and kidney.4 Studies showed that cigarette smoking was significantly associated with CRC incidence and mortality in humans and was also observed to increase the risk of CRC development in animal models.5 6 However, the mechanism through which cigarette smoking promotes CRC initiation and progression is unknown. Increased bacterial diversity was observed after smoking cessation in humans.7 8 Reports have also shown that the microbiome and mucin structure alteration is associated with cigarette smoking.9 Moreover, the association of altered gut microbiota with CRC is well established by us10–12 and others.13 Gut microbes from patients with CRC can promote colon tumourigenesis in recipient mice.11 Nevertheless, whether the alteration of gut microbiota represents a link between cigarette smoking and CRC remains elusive.
In this study, we aimed to determine the role of cigarette smoking in CRC development using conventional and germ-free mouse models. We demonstrated that cigarette smoking could promote CRC by inducing gut microbiota dysbiosis, which influences metabolites especially taurodeoxycholic acid (TDCA). Increased gut TDCA could activate oncogenic MAPK/ERK (mitogen-activated protein kinase/extracellular signal‑regulated protein kinase 1/2) pathway in colon epithelium and then promote colonocyte proliferation. Moreover, cigarette smoking could impair gut barrier function which further facilitates TDCA to activate colonic oncogenic MAPK/ERK signalling.
Materials and methods
Animal experiments
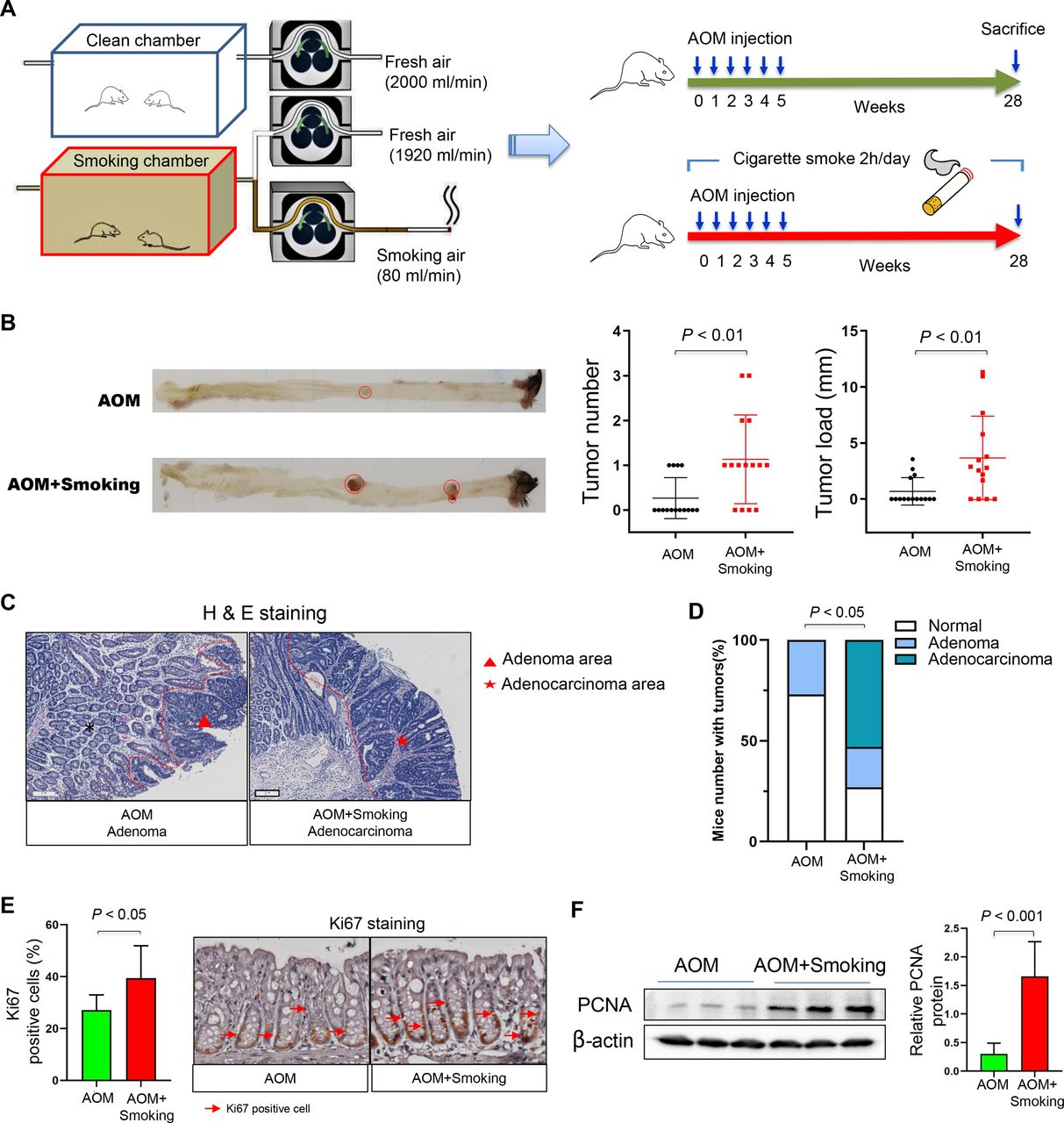
Male 10-week-old C57BL/6 mice were purchased from Animal Centre, the Chinese University of Hong Kong (Hong Kong).14 All mice were given food and water ad libitum and fed in specific pathogen-free environments. They were intraperitoneally injected with carcinogen azoxymethane (AOM) (10 mg/kg; Merck, Darmstadt, Germany) once per week for 6 consecutive weeks to induce CRC. At the beginning of AOM injection, mice were exposed to cigarette smoke (4% of 2000 mL/min airflow) or clean air 2 hours per day (15 mice per group, consisting of four cages with 3–4 mice per cage) for 28 weeks. Cigarette smoke was produced by the peristaltic tubing pump (Masterflex, Illinois, USA). Cigarette (Marlboro; Philip Morris International, Virginia, USA) with nicotine (1.0 mg/cigarette) was lighted, continuously inhaled by the pump, mixed with fresh air, then pumped into the smoking chamber. Only fresh air was pumped into the clean chamber (figure 1A). Both cigarette smoke-exposed and smoke-free mice sacrificed at the end of week 28.

Cigarette smoking increases colorectal tumourigenicity in mice. (A) Smoking or clean chamber designed for cigarette smoke exposure and schematic overview of the AOM-induced cancer model. The mixed fresh air and smoke air was pumped into the smoking chamber and fresh air was pumped into the clean chamber. Mice were placed into the chamber 2 hours daily for 28 weeks. AOM (10 mg/kg) was injected intraperitoneally once per week for 6 consecutive weeks from day 0. Mice were sacrificed at the end of week 28 (AOM group, n=15; AOM+Smoking group, n=15). (B) Representative images of colon at sacrifice. Tumour number and tumour size in the mice of AOM and AOM+Smoking group. (C) Representative images of H&E staining of adenoma in the AOM group and adenocarcinoma in the AOM+Smoking group. (D) Incidence of adenoma and adenocarcinoma in the colon of AOM-treated mice. Statistical significance was determined by Fisher’s exact test. (E) Representative images of immunohistochemistry staining of Ki67 positive cells and proportion of Ki67 positive cells in the colon. (F) Protein expression of PCNA in the colon of AOM-treated mice by western blot. Data are expressed as mean±SD. Statistical significance was determined by unpaired Student’s t-test. AOM, azoxymethane; PCNA, proliferating cell nuclear antigen.
Male 10-week-old Kunming mice were purchased from the Department of Laboratory Animal Science, the Third Military Medical University, Chongqing, China.14 All mice were given food and water ad libitum and fed in germ-free environments. To examine the direct effect of cigarette smoking-modulated-gut microbiota on colonic mucosa, 16 mice were transplanted with 200 μL of stools (0.7 g stool per 1 mL of phosphate-buffered saline) from conventional C57BL/6 mice exposed to smoke or clean air (8 germ-free mice per group, consisting of three cages with 2–3 mice per cage). Oral gavage of stool samples was performed once at week 0 and stool samples of germ-free mice were collected once a week.11 Mice were sacrificed at week 20 following the gavage. Next, to determine the direct contribution of smoking-modulated gut microbiota on tumourigenesis, 30 mice were randomly transplanted with stools from conventional C57BL/6 mice exposed to smoke or clean air (15 germ-free mice per group, consisting of three cages with 5 mice per cage). The mice also received AOM to induce colorectal neoplasia. The regimen of AOM was the same as described for conventional mice. Mice were sacrificed at week 28 after gavage.
At sacrifice, solid neoplastic lesions were carefully counted for tumour number and measured for tumour load (sum of mean diameters of all tumours in each mouse; mean diameter [major diameter+minor diameter]/2).14 Animal experiments were carried out in compliance with the regulations of the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and The Third Military Medical University, China.
Metagenomic sequencing and analysis
Genomic DNAs of stool samples were extracted using Quick-DNA Fecal/Soil Microbe Miniprep Kit (Zymo Research, Irvine, California, USA). DNA library preparation and shotgun metagenomic sequencing were performed by Novogene,15 Tianjin, China, which generated 34 457 567±3 248 626 (mean±SD; min: 24 136 603, max: 43 985 236) paired end reads. Taxonomic profile of the microbiota was obtained using Kraken2 (v2.0.8-beta) algorithm after host DNA removal and reads quality filtering by KneadData (v0.7.2). The standard Kraken 2 database comprising complete NCBI RefSeq genomes of bacterial, archaeal and viral domains, and a collection of known vectors was used as reference. The differential abundance analysis of bacterial species was performed by multivariate statistical model MaAsLin2.16 Bacterial species with (1) abundance >0.05% in at least one sample; (2) adjusted p value <0.05 (false discovery rate (FDR) corrected) and (3) fold change (FC) >1.5 in abundance between smoke-exposed and smoke-free were considered statistically significant. After rarefying to minimum library size, 9 664 368 reads, we calculated the alpha and beta diversity using phyloseq R package. Alpha diversity was measured by Chao1, Fisher and Shannon statistics. Beta diversity was accessed by Arrhenius z distance, and the principal coordinates analysis (PCoA) was used for ordination analysis. Community dissimilarities were tested by permutational multivariate analyses of variance (PERMANOVA) with 1000 iterations using the Arrhenius z distance. To measure the association of Eggerthella lenta with CRC, we analysed metagenomic sequencing data of human stool samples including 185 patients with CRC and 204 normal controls, from our previous publication.17
Additional methods are provided in online supplemental information.
Supplemental material
Results
Cigarette smoke promotes colorectal tumourigenesis in mice
To study the effect of cigarette smoking on colorectal tumourigenesis, we exposed AOM-treated C57BL/6 mice to clean air or cigarette smoke (figure 1A). Colorectal tumour number and tumour size were both significantly larger in cigarette smoke-exposed mice than smoke-free control mice (both p<0.01; figure 1B). The presence of colon adenoma and adenocarcinoma was confirmed by a pathologist with microscopic histological examination (figure 1C). Exposure to cigarette smoke significantly enhanced the incidence of colonic tumours (p<0.05) (figure 1D). We observed increased proliferation of epithelial cells in the colon of cigarette smoke-exposed mice compared with smoke-free control mice, indicated by significantly higher number of Ki-67-positive cells (figure 1E) and higher expression level of cell-proliferating protein marker proliferating cell nuclear antigen (PCNA) (figure 1F).
Cigarette smoke alters the gut microbiota composition and microbial interplay in mice
As the mice lived together for a long time, we first evaluated whether there was cage effect on microbial communities using 16S sequencing. We found that the cage effect did not impact on the microbiome communities at different time points (p>0.05, PERMANOVA) (online supplemental figure 1). However, the significant effect of smoking on the microbiome community occurred at the end of the experiment (end time point) (online supplemental figure 1), indicating that long-term smoking is a major factor influencing microbiota. Then, shotgun metagenomic sequencing analyses of faecal samples were performed to determine potential alteration of gut microbiota induced by cigarette smoke exposure. At baseline (initial time point), there was no significant differences of microbiome between smoke-exposed and smoke-free control mice in terms of alpha and beta diversity (online supplemental figure 2A,B). After 28 weeks, we observed that cigarette smoke-exposed mice had significantly lower alpha diversity compared with smoke-free control mice (figure 2A and online supplemental figure 2C). PCoA analysis (beta diversity) showed significantly different clustering of gut microbiota in smoke-exposed mice compared with smoke-free control mice (p<0.01, PERMANOVA; figure 2B). Twenty bacteria (p<0.05, FC>1.5) were altered significantly in cigarette smoke-exposed mice (figure 2C). Among these, E. lenta and Staphylococcus capitis were enriched while gut-beneficial bacteria including Lactobacillus reuteri,18 Parabacteroides distasonis19 and Bacteroides dorei20 were depleted with cigarette smoke exposure. We confirmed higher abundance of E. lenta in the cigarette smoke-exposed mice by quantitative PCR (p<0.01; figure 2D). We further evaluated the association of E. lenta with human CRC using our published dataset.17 E. lenta was significantly more abundant in patients with CRC (n=185) compared with normal subjects (n=204). Among these patients with CRC, smokers (n=30) had higher abundance of E. lenta compared with non-smokers (n=87) (p<0.05) (online supplemental figure 3A,B).
Supplemental material
Supplemental material

Cigarette smoke modulates the gut microbiota of mice. (A) Fisher statistic (alpha diversity) and (B) PCoA analysis (beta diversity) in AOM+Smoking and AOM group. Significance of alpha and beta diversity were accessed by two-tailed Mann-Whitney U test and PERMANOVA, respectively. (C) Differentially bacteria between AOM+Smoking and AOM group. Differences in abundance were detected by using a multivariate statistical model (p<0.05 (FDR corrected), FC >1.5, MaAsLin2). (D) The abundance of species Eggerthella lenta between AOM+Smoking and AOM group was validated by quantitative PCR. (E) Ecological network among differentially bacteria in AOM+Smoking group and in AOM group. Correlations were measured by SparCC method. Correlations with difference in correlation strengths between AOM+Smoking and AOM group >0.6 were selected for visualisation. AOM, azoxymethane; FC, fold change; PCoA, principal coordinates analysis; PERMANOVA, permutational multivariate analyses of variance.
The interaction among microbes may contribute towards disease progression. We investigated ecological networks of interaction among bacteria with differential abundance between smoke-exposed mice and smoke-free control mice (figure 2E). We observed that the co-occurrence and co-excluding interactions among bacteria were significantly different between smoke-exposed mice and smoke-free control mice. Co-exclusive correlations were observed between E. lenta and two probiotic bacteria: Lactobacillus jensenii and Lactobacillus crispatus (figure 2D), suggesting antagonistic associations of enriched E. lenta with depleted protective bacteria in cigarette smoke-exposed mice.
Cigarette smoke alters gut microbiota-related metabolites in stool
We determined alterations in faecal metabolites after smoke exposure by liquid chromatography with mass spectrometry (MS)/MS analysis of mice stool. Orthogonal partial least squares discriminant analysis showed that faecal metabolic profile in smoke-exposed mice was significantly different from smoke-free mice (figure 3A). Forty-one metabolites (adjusted p<0.05) were altered in stool of cigarette smoke-exposed mice compared with smoke-free control mice (figure 3B). The altered metabolites were enriched or depleted in different metabolomic signalling pathways (figure 3C). Bile acid biosynthesis was the top enriched pathway in cigarette smoke-exposed mice compared with smoke-free mice. In the gut, bacteria convert primary bile acids to secondary bile acids which are in part absorbed by terminal ileum and colon. Among these secondary bile acids, TDCA is known to be procarcinogenic.21 22 The abundance of TDCA was increased significantly in smoke-exposed mice (figure 3B). The elevated stool TDCA in cigarette smoke mice was further confirmed by targeted MS (p=0.0094) (figure 3D).
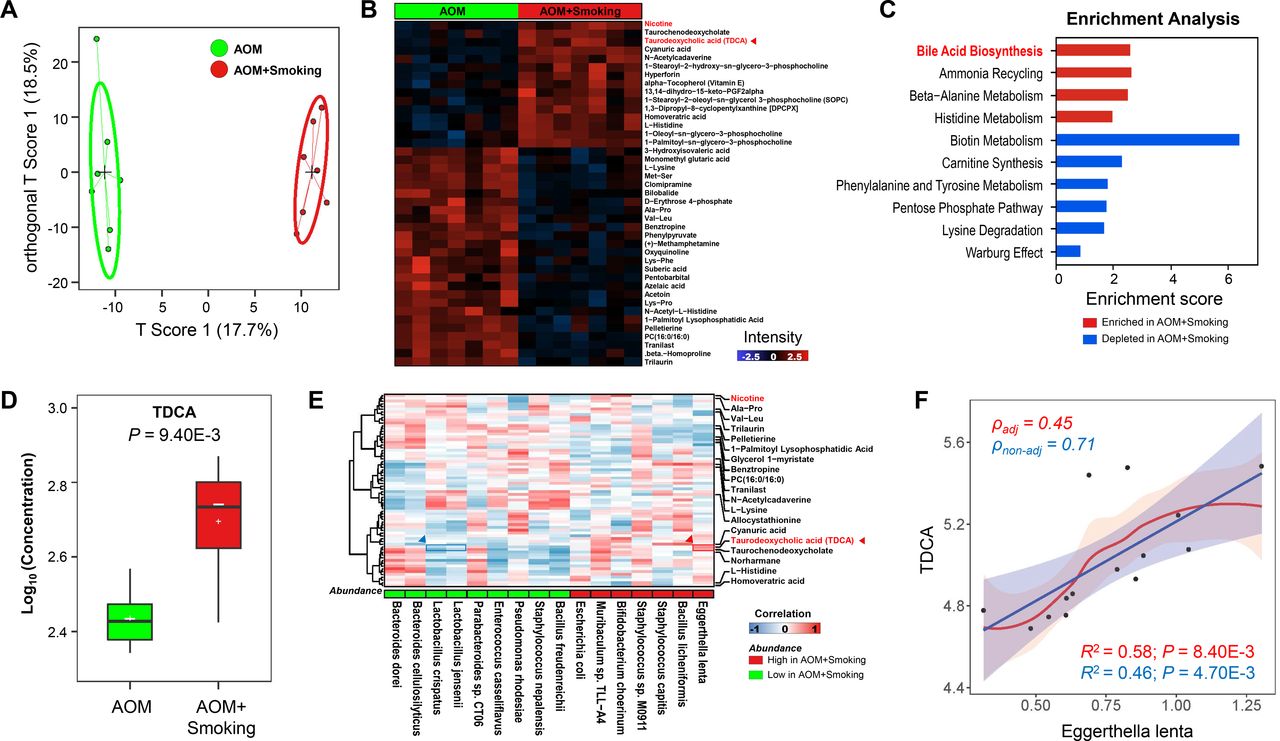
Cigarette smoke alters gut microbiota-related metabolites in stool. (A) Stool metabolic profile was significantly different between AOM+Smoking and AOM group by OPLS-DA method which is a supervised multiple regression analysis for identifying discernible patterns among different groups. The x-axis captures the variation between the groups, while the y-axis captures the variation within the groups. (B) Differentially metabolites between AOM+Smoking and AOM groups, p<0.05, two-tailed Mann-Whitney U test. (C) Enrichment analysis of differentially metabolites between AOM+Smoking and AOM group. Enrichment score >1 were included. (D) The concentration of TDCA in the stool in AOM+Smoking and AOM groups was measured by targeted mass spectrometry assay, p<0.05, two-tailed Student’s t-test. (E) Association analysis of bacteria with differentially metabolites by partial’s Spearman correlation. (F) Linear association between TDCA and Eggerthella lenta by linear model with and without correction (smoke exposed/smoke free). AOM, azoxymethane; OPLS-DA, orthogonal partial least squares discriminant analysis; TDCA, taurodeoxycholic acid.
To determine potential association of microbiota with metabolites, we performed correlation analysis between bacteria and metabolites by partial Spearman correlation. We observed that E. lenta had the most positive correlation with TDCA, while two probiotic bacteria; L. jensenii and L. crispatus which were depleted in smoke-exposed mice, were negatively correlated with TDCA (figure 3E). E. lenta possesses the ability to deconjugate primary bile acids and expresses bile acid epimerising enzymes 3β-hydroxysteroid dehydrogenase (3β-HSDH); hence, it participates in secondary bile acids synthesis and influences the level of TDCA in stool.23–25 We therefore measured the abundance of genes encoding 3β-HSDH enzymes and observed significantly higher abundance of the genes in smoke-exposed compared with smoke-free mice (online supplemental figure 3C). In keeping with this, we further confirmed that the abundance of E. lenta was positively correlated with the concentration of TDCA in stool (figure 3F). Thus, gut microbial dysbiosis and altered metabolites may work together to contribute to colon tumourigenesis.
Supplemental material
Cigarette smoke impairs gut barrier function
To investigate the impact of cigarette smoking on gut barrier function, we analysed the expression levels of colon tight junction proteins, claudin-3 and Zonula occludens-1 (ZO-1), and levels of serum lipopolysaccharide (LPS). Cigarette smoke markedly decreased the levels of claudin-3 and ZO-1 as determined by western blot (figure 4A) and immunofluorescence staining (figure 4B). The impaired tight junction was further confirmed under electron microscopy (EM) examination by a pathologist (figure 4C). Meanwhile, we found that the levels of serum LPS were increased significantly in smoke-exposed compared with smoke-free mice (figure 4D). These results together indicate that cigarette smoke causes impaired gut barrier function.
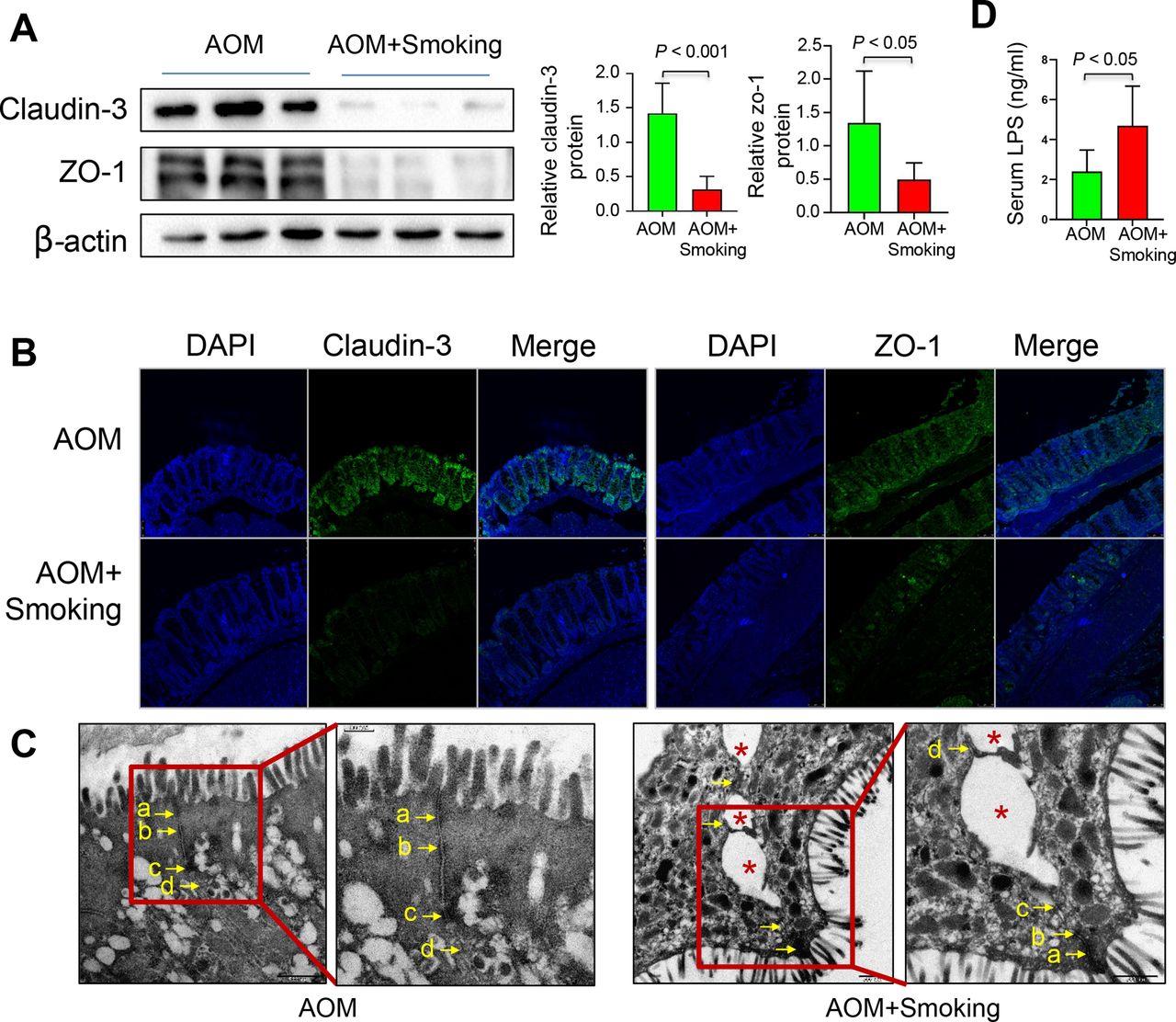
Cigarette smoke impairs the gut barrier function. (A) Protein expression of claudin-3 and ZO-1 in the colon of the AOM-treated mice by western blot. (B) Protein expression of claudin-3 and ZO-1 in the colon of the AOM-treated model by immunofluorescence staining. (C) The representative images of the structure of the colorectal gut barrier of AOM-treated mice. Arrows point to cell-cell junction under electron microscope. (a) Tight junction; (b) adherens junction; (c) desmosome; (d) gap junction. An asterisk indicates a disrupted cell junction. (D) LPS concentration in the serum of mice in the AOM and AOM+Smoking group. Data are expressed as mean±SD. Statistical significance was determined by unpaired Student’s t-test. AOM, azoxymethane; DAPI, 4′,6-diamidino-2-phenylindole; LPS, lipopolysaccharide.
Cigarette smoke enhances oncogenic MAPK/ERK signalling in colonic epithelium
To gain molecular insights into the protumourigenic effect of cigarette smoking, we profiled the expressions of cancer-associated genes in colonic epithelium using Mouse Cancer Pathway Finder PCR Array. We observed 19 upregulated genes and 7 downregulated genes in smoke-exposed mice compared with smoke-free control mice (figure 5A, online supplemental table 2). Enrichment analysis indicated that mitogen-activated protein kinases (MAPK) signalling pathway was the top activated pathway by cigarette smoke (figure 5B). Previous studies reported that TDCA could activate the ERK subfamily of MAPK pathway,26 27 thus we evaluated MAPK/ERK activation in colonic epithelium of mice exposed to cigarette smoke compared with control mice. Activation of MAPK/ERK signalling by smoking was confirmed by the elevated level of phospho-ERK1/2, a key mediator protein in MAPK/ERK pathway (figure 5C). Moreover, we observed positive correlation between ERK phosphorylation level and TDCA level (online supplemental figure 4A). These findings suggest that cigarette smoke induces ERK1/2 phosphorylation and activates MAPK/ERK signalling pathway to promote colon tumourigenesis.
Supplemental material
Supplemental material

Cigarette smoke enhances the expression of oncogenic MAPK/ERK pathway and proinflammatory pathway in colonic epithelium. (A) Differential expressed genes of colonic epithelium in the AOM+Smoking group compared with the AOM group by Mouse Cancer Pathway Finder PCR Array analysis (FC=AOM+Smoking/AOM, positive log2(FC)=higher expression in AOM+Smoking group and negative log2(FC)=higher expression in AOM group). FC between AOM+Smoking and AOM >2 was included. (B) The altered cancer signalling pathways in the AOM+Smoking group compared with AOM group by enrichment analysis. Enrichment scores >1 were included. The arrows represent the direction of enrichment, calculated by comparing the upregulated and downregulated genes in the pathway. The differentially expressed genes in MAPK signalling pathway were shown in network. (C) Protein expression of ρ-ERK1/2 in the colon of the AOM-treated mice by western blot. (D) Differential expressed genes of colonic epithelium in the AOM+Smoking mice compared with AOM mice by Mouse Inflammatory Response and Autoimmunity Array analysis. (E) The altered inflammatory signalling pathways in the AOM+Smoking group compared with AOM group by enrichment analysis. Enrichment score >1 were included. The differentially expressed genes in TNF and IL-17 signalling pathways were shown in network. (F) Gene expression of Cxcl2, Il-17a and Il-10 by quantitative RT-PCR. Data are expressed as mean±SD. **p<0.01, *p<0.05; statistical significance was determined by two-sided unpaired Student’s t-test. p values were adjusted by FDR (online supplemental tables 2,3). AOM, azoxymethane; ERK, extracellular signal‑regulated protein kinase; FC, fold change; Il, interleukin; MAPK, mitogen-activated protein kinase; RT-PCR, reverse transcription PCR; TNF, tumour necrosis factor.
Cigarette smoke enhances expressions of proinflammatory signalling genes
Gut bacteria dysbiosis is closely associated with inflammation which links oncogenic factors and tumourigenesis. We therefore profiled expressions of proinflammatory genes using the Mouse Inflammatory Response and the Autoimmunity PCR Array. We observed 27 upregulated genes and 5 downregulated genes in cigarette smoke-exposed mice compared with smoke-free control mice (figure 5D, online supplemental table 3). Altered pathways including proinflammatory interleukin 17 (IL-17) signalling and tumour necrosis factor (TNF) signalling pathways were induced by cigarette smoke (figure 5E). Quantitative reverse transcription PCR (RT-PCR) confirmed increased expressions of proinflammatory Il-17a, Cxcl2 and decreased expression of anti-inflammatory Il-10 in smoke-exposed mice compared with smoke-free control mice (figure 5F). Moreover, the relative mRNA expression of Il-17a was positively correlated with the abundance of E. lenta in the colon (online supplemental figure 5). These findings suggest that cigarette smoke promotes inflammation in colon tumourigenesis.
Supplemental material
Faecal microbiota transplantation in germ-free mice recapitulate the alteration of gut microbiota in smoke-exposed conventional mice
To confirm the direct role of altered gut microbiota by cigarette smoke on colorectal tumourigenesis, we performed faecal microbiota transplantation in germ-free mice. Germ-free mice were gavaged with stools either from cigarette smoke-exposed or smoke-free conventional mice. Mice were harvested and examined after 20 weeks (figure 6A). We performed shotgun metagenomic sequencing analyses to explore the colonisation of cigarette smoke-altered microbiota. Similar to the conventional AOM mice model, alpha diversity was significantly decreased in germ-free mice gavaged with stools from cigarette smoke-exposed mice (GF-AOMS) compared with those gavaged with stools from cigarette smoke-free mice (GF-AOM) (figure 6B and online supplemental figure 2C). Beta diversity analysis again showed significant segregation of gut microbiota between these two groups of germ-free mice (GF-AOMS vs GF-AOM, p<0.01, PERMANOVA) (figure 6C). Further analysis revealed 34 differentially abundant bacteria (figure 6D). Consistently, the abundance of E. lenta was significantly increased and P. distasonis was significantly decreased in GF-AOMS mice (p<0.05, FC>1.5). We confirmed significantly higher abundance of E. lenta in GF-AOMS group than GF-AOM group by quantitative PCR (p<0.001, figure 6E).

Alteration of gut microbiota in germ-free mice with faecal microbiota transplantation from smoke-exposed conventional mice. (A) Schematic overview of the germ-free mice model. Germ-free mice were orally gavaged with stool from AOM+Smoking and AOM groups (n=8/group). Mice were sacrificed at the end of week 20. (B) Fisher statistic (alpha diversity) and (C) PCoA analysis (beta diversity) in GF-AOMS and GF-AOM group. Significance of alpha and beta diversity was accessed by two-tailed Mann-Whitney U test and PERMANOVA, respectively. (D) Differentially bacteria between GF-AOMS and GF-AOM groups. Differences in abundance were detected by using a multivariate statistical model (p<0.05 (FDR corrected), FC >1.5, MaAsLin2). (E) The abundance of species Eggerthella lenta between GF-AOMS and GF-AOM group was validated by quantitative PCR. (F) Consistent alteration in bacteria abundance (p<0.05, smoke-exposed mice vs smoke-free mice; GF-AOMS mice vs GF-AOM mice) in two mice model (germ-free mice and AOM mice). The FC in abundance between smoking and non-smoking was calculated. Red points represent germ-free mice model and yellow points represent conventional mice model. (G) Network modules were conserved after gavage feeding in the germ-free mice when comparing with the donor. Correlations were measured by SparCC method and network modules were extracted based on first-order neighbourhoods of bacteria. (H) The concentration of TDCA in the stool between GF-AOMS and GF-AOM groups measured by targeted mass spectrometry assay, p<0.05, two-tailed Mann-Whitney U test. AOM, azoxymethane; FC, fold change; GF-AOM, germ-free mice gavaged with stool from AOM donor mice; GF-AOMS, germ-free mice gavaged with stool from AOM+Smoking donor mice; PCoA, principal coordinates analysis; PERMANOVA, permutational multivariate analyses of variance; TDCA, taurodeoxycholic acid.
We further tested consistence of microbiota alterations between the two mice models (germ-free mice and conventional AOM mice). We found that bacteria with increased abundance in smoke-exposed AOM mice compared with smoke-free AOM mice were consistently increased in GF-AOMS mice compared with GF-AOM mice, whereas bacteria with decreased abundance in smoke-exposed AOM mice were consistently reduced in GF-AOMS mice compared with GF-AOM mice (figure 6F). Importantly, we observed that the abundance of E. lenta was increased while probiotic P. distasonis had lower abundance in both smoke-exposed AOM mice and GF-AOMS mice. Correlation analysis revealed that ecological network modules were conserved after faecal transplant in germ-free mice when compared with the donor mice (figure 6G). Moreover, stool TDCA level also increased significantly in GF-AOMS compared with GF-AOM mice (figure 6H).
Cigarette smoke-altered microbiota increases colonocyte proliferation and tumourigenesis in germ-free mice
GF-AOMS mice had increased proliferation of colonic epithelial cells compared with GF-AOM mice, which was indicated by higher proportion of Ki-67 positive cells (figure 7A) and higher expression level of PCNA (figure 7B). In addition, we performed the AOM treatment in germ-free mice and gavaged with stools either from cigarette smoke-exposed (GFAOM-AOMS) or smoke-free mice (GFAOM-AOM) (online supplemental figure 6A). Consistently, GFAOM-AOMS mice exhibited increased colon tumour number (p<0.05) and tumour size (p<0.05) compared with GFAOM-AOM mice (online supplemental figure 6B). These results in germ-free mice are consistent with observations in conventional mice, thus suggesting that the altered microbiota and metabolome by cigarette smoke could directly promote colonocyte proliferation and tumourigenesis.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Altered microbiota by cigarette smoke increases colonocyte proliferation, impaired gut barrier function and enhances oncogenic MAPK/ERK and proinflammatory genes expression in germ-free mice. (A) Representative images of immunohistochemistry staining of Ki67 positive cells and proportion of Ki67 positive cells in the colon of germ-free mice. (B) Protein expression of PCNA in the colon of germ-free mice by western blot. (C) Protein expression of claudin-3 and ZO-1 in the colon of germ-free mice by western blot. (D) LPS concentration in the serum of mice in the GF-AOM and GF-AOMS group. (E) Electron microscope showing the structure of the colorectal gut barrier of germ-free mice. Arrows point to cell-cell junction. (F) Differential expressed genes of colonic epithelium in the GF-AOMS group compared with the GF-AOM group by Mouse Cancer Pathway Finder PCR Array analysis. (G) The altered cancer signalling pathways in the GF-AOMS group compared with the GF-AOM group by enrichment analysis. Enrichment scores >1 were included. The arrows represent the direction of enrichment, calculated by comparing the upregulated and downregulated genes in the pathway. The differentially expressed genes in MAPK signalling pathway were shown in network. (H) Protein expressions of ERK1/2 in the colon of mice in the GF-AOM and GF-AOMS group by western blot. (I) Differential expressed genes of colonic epithelium in the GF-AOMS group compared with the GF-AOM group by Mouse Inflammatory Response and Autoimmunity Array analysis. (J) The altered inflammatory signalling pathways in the GF-AOMS group compared with the GF-AOM by enrichment analysis. Enrichment scores >1 were included. The differentially expressed genes in TNF and IL-17 signalling pathways were shown in network. (K) Gene expression of Il-17a, Cxcl2 and Cxcr2 by quantitative RT-PCR. (a) Tight junction; (b) adherens junction; (c) desmosome; (d) gap junction. An asterisk indicates a disrupted cell junction. Data are expressed as mean±SD. *p<0.05; statistical significance was determined by two-tailed unpaired Student’s t-test. p values were adjusted by FDR (online supplemental tables 4,5). AOM, azoxymethane; ERK, extracellular signal‑regulated protein kinase; GF-AOM, germ-free mice gavaged with stool from AOM donor mice; GF-AOMS, germ-free mice gavaged with stool from AOM+Smoking donor mice; Il, interleukin; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; PCNA, proliferating cell nuclear antigen; RT-PCR, reverse transcription PCR; TNF, tumour necrosis factor
Altered microbiota by cigarette smoke impairs gut barrier function in germ-free mice
We next investigated whether altered microbiota by cigarette smoke could affect the gut barrier function in germ-free mice. We observed decreased expressions of claudin-3 and ZO-1 and increased levels of serum LPS in GF-AOMS mice (figure 7C,D). Impaired gut barrier function in GF-AOMS mice was also confirmed by EM which showed widening of intercellular junctions in apical junctional complex and paracellular gap in colon (figure 7E). These results in germ-free mice were in line with conventional mice, thus indicating that alteration in microbiota and metabolome by cigarette smoke could impair gut barrier function which might further contribute to tumourigenesis.
Altered microbiota by cigarette smoke enhances oncogenic MAPK/ERK pathway in the colonic epithelium of germ-free mice
To confirm the molecular mechanism of cigarette smoke-altered microbiota in promoting colonocyte proliferation, we performed Cancer Pathway Finder Array on the colonic epithelium of stool-gavaged germ-free mice. We identified nine upregulated genes and three downregulated genes in GF-AOMS mice compared with GF-AOM mice (figure 7F, online supplemental table 4). These differential expression genes were mainly enriched in MAPK/ERK signalling pathway. The activation of MAPK/ERK signalling was confirmed by the elevated protein expression of phospho-ERK1/2 in GF-AOMS mice compared with GF-AOM mice (figure 7G and H). Moreover, we observed positive correlation between ERK phosphorylation level and TDCA level (online supplemental figure 4B). Similarly, using Inflammatory Response and Autoimmunity PCR Array, we found that 16 proinflammatory genes were upregulated in GF-AOMS compared with GF-AOM mice (figure 7I, online supplemental table 5), and these genes were also mainly enriched in TNF and IL-17 signalling pathways (figure 7J). Upregulation of the key proinflammatory genes in TNF and IL-17 pathways including Il-17a and C-X-C motif chemokine ligand 2 (Cxcl2), and C-X-C motif chemokine receptor 2 (Cxcr2) were confirmed in GF-AOMS mice compared with GF-AOM mice by quantitative RT-PCR (figure 7K). The consistent findings from both conventional and germ-free mice models indicate that altered-microbiota by cigarette smoke directly induced colonic proinflammatory TNF and IL-17 signalling and oncogenic MAPK/ERK signalling. Thus, altered gut microbiota contributes to the protumourigenic role of cigarette smoke in colorectal carcinogenesis.
Discussion
Here we establish for the first time, a novel gut microbiota-mediated mechanism of cigarette smoke-induced colorectal tumourigenesis. Cigarette smoking is a risk factor for CRC. However, the mechanistic understanding of cigarette smoking-induced CRC is limited. Despite several studies showing association between gut microbiota and cigarette smoking, it remains unclear whether the altered microbiota by exposure to cigarette smoking plays an important role in the initiation and progression of CRC. To fill this critical gap, we performed experiments involving long-term exposure of mice to cigarette smoke using AOM-induced CRC mouse model and germ-free mice transplanted with cigarette smoke-altered microbiota.
In this study, we demonstrated that cigarette smoking could promote colon tumourigenesis by modulating the composition of gut microbiota and induce gut microbiota dysbiosis. In particular, E. lenta that has been reported to be associated with CRC28 29 was significantly enriched in mice exposed to cigarette smoke. On the other hand, P. distasonis that was depleted in mice exposed to cigarette smoke had been demonstrated to increase colonic expression of tight junction proteins and attenuate colorectal tumourigenesis.19 Lactobacillus (L. reuteri, L. jensenii and L. crispatus), known for its capability of suppressing infection or proliferation of pathogenic bacteria, had lower abundance in mice exposed to cigarette smoke. We further demonstrated the antagonistic relationship between enriched E. lenta and depleted Lactobacillus species in mice exposed to cigarette smoke. Taken together, gain of pathobionts and loss of protective bacteria together at least in part contribute to the CRC tumourigenesis induced by cigarette smoking.
We also identified that bile acid biosynthesis pathway was the top significantly enriched metabolic pathway in the stool of smoke-exposed mice. Primary bile acids are synthesised in liver and secreted into gut via bile duct. Gut commensal microbiota transforms primary bile acids to secondary bile acids which are then mostly absorbed by terminal ileum and colon with a few being excreted in stool.30 Due to their tumour-promoting properties, bile acids are receiving increased attention.30–32 Among the secondary bile acids derived from gut microbiota, we confirmed that TDCA was significantly increased in the stool of smoke-exposed mice. Previous studies have demonstrated that TDCA could promote colorectal and oesophageal adenocarcinoma.21 22 TDCA instillation into the lumen of colon increased the frequency of N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG)-induced colorectal neoplasm.22 We further found that changes in stool metabolites were significantly correlated with alteration in abundance of gut microbes, in which TDCA had the most positively correlation with E. lenta. This result was supported by previous studies which showed that E. lenta possesses the ability to deconjugate primary bile acids and expresses bile acid epimerising enzymes 3β-HSDH. These features allow it to participate in the synthesis of secondary bile acids, thus influencing TDCA concentration in stool.23–25
We further evaluated gut barrier function in mice and identified that it was impaired in mice exposed to cigarette smoke. Expressions of claudin-3 and ZO-1, two important tight junction proteins, were reduced in cigarette smoke-exposed mice. Impaired intestinal tight junctions could increase colon permeability, thereby allowing more gut noxious metabolites such as TDCA to get into the intercellular space of colonic epithelial cells,33 34 activating the MAPK/ERK signalling pathway.
We next investigated the molecular mechanism through which cigarette smoke could promote colon tumourigenesis and revealed significant enrichment and activation of MAPK/ERK pathway induced by cigarette smoke. This result is consistent with the function of TDCA which has been reported as a ligand to activate MAPK/ERK pathway.26 27 The activation of MAPK/ERK pathway is important for cell proliferation35 and was proved to be involved in the pathogenesis, progression and oncogenesis of human CRC.36 Moreover, we found that proinflammatory IL-17 and TNF pathways were induced by cigarette smoke. IL-17 has been shown to promote the development of colon cancer.37 Previous study showed that E. lenta was enriched in patients with IBD and it could induce IL-17a, by the cardiac glycoside reductase 2 enzyme,38 supporting our finding that E. lenta may activate the proinflammatory cytokine IL-17 pathways to induce tumourigenesis. Activation of TNF-α pathway induces colitis and colorectal carcinogenesis.39 These findings collectively indicated that activation of oncogenic MAPK signalling and proinflammatory IL-17 and TNF signalling pathways triggered by smoke-associated gut dysbiosis and altered metabolites contribute to cigarette smoke-associated colon tumourigenesis.
The direct role of the smoke-altered gut microbiota in tumourigenesis was further investigated in germ-free mice with faecal microbiota transplantation. Cigarette smoke-altered gut microbiota alone could increase proliferation of colonic epithelial cells in germ-free mice. The gut microbial composition in recipient germ-free mice was similar to the gut microbiota composition in donor smoke-exposed conventional mice. In particular, E. lenta was significantly enriched in both smoke-exposed AOM mice and in GF-AOMS mice. Cigarette smoke-altered gut microbiota also increased stool TDCA level and induced activation of MAPK/ERK, IL-17 and TNF signalling pathways in colonic epithelium of GF-AOM mice. These findings collectively suggest that cigarette smoke alter the gut microbiota, which promotes colon tumourigenesis through increasing TDCA level in colon and further activating oncogenic MAPK/ERK, IL-17 and TNF signalling pathways in colonic epithelium. Moreover, the cigarette smoke-altered gut microbiota impaired colonic epithelium as evidenced by decreased expressions of tight junction proteins claudin-3 and ZO-1. Gut barrier dysfunction may facilitate the carcinogenic TDCA to enter the intercellular space of colonic epithelium and further activate the oncogenic MAPK/ERK signalling pathway.
This study demonstrated the promoting effect on CRC by cigarette smoking though modulating the composition of gut microbiota. Smoking cessation was associated with improved CRC-specific survival compared with continued smokers.40 Thus, smoking cessation is one of the practical approaches for preventing CRC by rebuilding a healthy gut microbiome at least.
In conclusion, this study for the first time, demonstrated that cigarette smoke promotes colon tumourigenesis by inducing gut microbiota dysbiosis (online supplemental figure 7). The smoke-induced gut microbiota dysbiosis could increase TDCA level in colon, which then activates oncogenic MAPK/ERK, IL-17 and TNF signalling pathways in colonic epithelium. Moreover, gut barrier dysfunction caused by gut microbiota dysbiosis may further facilitate TDCA to activate MAPK/ERK signalling pathway.
Supplemental material
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets generated in the current study are available in the Genome Sequence Archive (GSA) at the National Genomics Data Center, Beijing Institute of Gemonics, Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA006099) and are publicly accessible at https://bigd.big.ac.cn/gsa/.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies were performed in accordance with guidelines approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and The Third Military Medical University, China.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
XB, HW and WL are joint first authors.
Twitter @Hongyan Gou
Contributors XB was involved in study design, performed experiments and drafted the manuscript; WL performed bioinformatics analysis and revised manuscript; OOC revised the manuscript; CLiu provided bioinformatic support. LZ, CLi, YZ, GW and HG performed animal experiments; WK performed histological evaluation; HW performed the germ-free animal experiment; EKN commented on the study; JY designed, supervised the study and revised the manuscript. JY was responsible for the overall content of this study.
Funding This project was supported by National Key R&D Program of China (No: 2020YFA0509200/2020YFA0509203); RGC Theme-based Res Scheme Hong Kong (T21-705/20-N), RGC Collaborative Research Fund (C4039-19GF, C7065-18GF), RGC-GRF Hong Kong (14163817), Vice-Chancellor’s Discretionary Fund of the Chinese University of Hong Kong.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.