Article Text

Abstract
Objective Primary sclerosing cholangitis (PSC) is in 70% of cases associated with inflammatory bowel disease. The hypermorphic T108M variant of the orphan G protein-coupled receptor GPR35 increases risk for PSC and ulcerative colitis (UC), conditions strongly predisposing for inflammation-associated liver and colon cancer. Lack of GPR35 reduces tumour numbers in mouse models of spontaneous and colitis associated cancer. The tumour microenvironment substantially determines tumour growth, and tumour-associated macrophages are crucial for neovascularisation. We aim to understand the role of the GPR35 pathway in the tumour microenvironment of spontaneous and colitis-associated colon cancers.
Design Mice lacking GPR35 on their macrophages underwent models of spontaneous colon cancer or colitis-associated cancer. The role of tumour-associated macrophages was then assessed in biochemical and functional assays.
Results Here, we show that GPR35 on macrophages is a potent amplifier of tumour growth by stimulating neoangiogenesis and tumour tissue remodelling. Deletion of Gpr35 in macrophages profoundly reduces tumour growth in inflammation-associated and spontaneous tumour models caused by mutant tumour suppressor adenomatous polyposis coli. Neoangiogenesis and matrix metalloproteinase activity is promoted by GPR35 via Na/K-ATPase-dependent ion pumping and Src activation, and is selectively inhibited by a GPR35-specific pepducin. Supernatants from human inducible-pluripotent-stem-cell derived macrophages carrying the UC and PSC risk variant stimulate tube formation by enhancing the release of angiogenic factors.
Conclusions Activation of the GPR35 pathway promotes tumour growth via two separate routes, by directly augmenting proliferation in epithelial cells that express the receptor, and by coordinating macrophages’ ability to create a tumour-permissive environment.
- colorectal cancer
- primary sclerosing cholangitis
- angiogenesis
- ulcerative colitis
- receptor characterisation
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. All data relevant to the study are included in the article or uploaded as online supplemental information. n/a.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
- colorectal cancer
- primary sclerosing cholangitis
- angiogenesis
- ulcerative colitis
- receptor characterisation
Significance of this study
What is already known on this subject?
A coding variant (T108M) in the G protein-coupled receptor GPR35 increases the risk for primary sclerosing cholangitis (PSC) and ulcerative colitis (UC). PSC and UC are chronic inflammatory conditions that significantly increase the risk for the development of cancer, and GPR35 is highly expressed in colorectal cancer.
GPR35 interacts with the sodium-potassium pump (Na/K-ATPase). Na/K-ATPase is the quintessential controller of a cell’s electrochemical gradient and controls Src-kinase signalling. The UC/PSC-associated GPR35T108M is hypermorphic and promotes the pump’s ion exchange and signal transduction function resulting in increased proliferation of intestinal epithelial cells. Mice lacking Gpr35 globally or conditionally in their Vln+ cells (intestinal epithelial cells) develop reduced numbers of tumours in sporadic cancer and colitis-associated cancer.
Significance of this study
What are the new findings?
Tumours lacking GPR35 are smaller compared with wild-type tumours. Deletion of Gpr35 in LysM+ cells reduces tumour growth substantially. GPR35 on macrophages increases the secretion of the angiogenic factors CXCL-1 (the murine orthologue of interleukin 8 (IL-8)) and VEGF. GPR35 on LysM+ cells promotes vessel sprouting from ex vivo cultured aortic rings which is dependent on its interaction with the Na/K-ATPase. Loss of GPR35 on macrophages reduces matrix metalloproteinase activity in tumour tissue, a prerequisite for tissue remodelling and angiogenesis. Macrophages carrying the human risk variant GPR35T108M secrete substantially more VEGF, IL-8 and matrix-metallo-proteinases (MMP2). Risk variant supernatants induce tube formation in human microvascular endothelial cells. Pharmacological pepducin-based GPR35 blockade markedly reduces tumour size and tissue levels of CXCL-1, VEGF, MMP2 and MMP9. GPR35 in macrophages is an upstream central coordinator of tissue remodelling and tumour-promotion, creating a tumour-permissive environment.
How might it impact on clinical practice in the foreseeable future?
Anti-VEGF antibodies are approved in cancer therapy to reduce tumour growth and angiogenesis, anti-IL-8 antibodies are in phase 2 clinical trials and have huge promise to slow down tumour angiogenesis.
Blocking GPR35 could be very promising in cancer therapy as this reduces the release of both, VEGF and IL-8 and reduces tumour cell proliferation by inhibiting neoangiogenesis.
Introduction
Colorectal cancer (CRC) is the leading cause of death from gastrointestinal malignancy, and the second most common cause of cancer-related death in the western world.1 Mutations in the adenomatous polyposis coli (APC) gene are a key event in both spontaneous and hereditary CRC.2 A subset of CRC is due to intestinal inflammation, referred to as colitis-associated cancer (CAC).3 CAC typically arises from long-standing ulcerative colitis (UC), and is particularly frequent in a variant of UC (often referred to as PSC-inflammatory bowel disease (IBD)) that is often present in patients with primary sclerosing cholangitis (PSC). PSC and UC are chronic inflammatory conditions of the bile ducts and the large intestine, respectively.4 5 Both diseases have an extreme cumulative risk for the development of cancer.6 7
A coding variant in GPR35 (rs3749171, leading to a T108M substitution) is associated with risk for PSC and UC.8 9 GPR35 is an orphan G protein-coupled receptor that interacts with the sodium-potassium pump (Na/K-ATPase).10 Na/K-ATPase is the quintessential controller of a cell’s electrochemical gradient, for which ~30% of cellular energy in the form of ATP is expended. The GPR35 interaction promotes Na/K-ATPase’s pump function, secondarily controlling Ca2+ homoeostasis. The UC/PSC-associated GPR35T108M variant is hypermorphic in its stimulatory activity on the pump.10 Na/K-ATPase acts as a scaffold for the non-receptor tyrosine kinase Src, and modulates its phosphorylation.11 GPR35, via its Na/K-ATPase interaction, regulates the proliferation of intestinal epithelial cells by phosphorylation and activation of Src.10 Indeed mice lacking Gpr35 develop fewer tumours in a sporadic murine cancer model (in mice carrying an Apcmin allele) and in a model of CAC induced by the mutagen azoxymethane followed by dextran sodium sulphate (AOM-DSS).10 GRP35 is known to be highly expressed in human CRC12 13 and deletion of Gpr35 selectively in the intestinal epithelium was sufficient to reduce tumour numbers.10 Kaya et al report that LPA activation of GPR35 in CX3CR1+macrophages maintains tumour necrosis factor-mediated intestinal homoeostasis and thereby reduces acute inflammation.14
Mutations in oncogenes and tumour-suppressor genes lead to unorganised proliferation and reduced cell death which initiates the development of malignancies. Tumour growth is not only determined by signal transduction pathways leading to proliferation,15 but also by neoangiogenesis.16 The tumour microenvironment (TME) consists of several cell types including leukocytes,17 18 as well as the extracellular matrix, which are all important for growth and progression of tumours.19 Tumour cells themselves have the capability to recruit cells of the TME by releasing chemokines, cytokines and proteinases.20 This cooperation between cells of the TME and malignant cells leads to tissue remodelling and angiogenesis within the tumour, resulting in enhanced proliferation and metastatic capability.21
Atypical angiogenesis does not only play an important role in tumour neo-vascularisation,22 but is also a hallmark of chronic inflammation including IBD.23 Release of CXCL-1 (KC, GROα) and/or CXCL-8 (interleukin 8, IL-8) together with VEGF initiate tumour angiogenesis and lead to tumour growth by guaranteeing sufficient supply of nutrients.24 These cytokines are also increased in IBD and augment neovascularisation in areas of tissue regeneration.23 Tumour-associated macrophages (TAMs) as part of the TME can resemble either proinflammatory M1 macrophages or M2-like macrophages with immunosuppressive and tumour-promoting properties.18 25 Increased numbers of TAMs, in particular M2-like cells, directly correlate with poor prognosis of cancer by stimulating angiogenesis, modulating tumour cell invasion, cell motility as well as persistent growth.26–28 TAMs also express proteinases including matrix metalloproteinases (MMPs), which play a key role in angiogenesis and progression of tumours.29 Neutrophils have also been demonstrated to contribute to tumour angiogenesis employing similar mechanisms.30
Here, we report that macrophage GPR35 is important in vessel growth in healthy and malignant tissue. GPR35 critically controls the size of intestinal tumours in murine spontaneous (APC min ) and CAC (AOM/DSS) models. We demonstrate that deletion of Gpr35 in macrophages recapitulates the profoundly reduced tumour size observed in Gpr35 –/– germline mutant mice, identifying macrophage GPR35 as a key driver of tumour growth. Aortic tissue from mice lacking GPR35 is strikingly less capable of developing vascular sprouts. Vessel sprouting is mainly dependent on GPR35’s interaction with the Na/K-ATPase-dependent activation of Src kinase, and does not require ligand-induced GPR35 activation.
Materials and methods
For detailed resources, please see online supplemental materials and methods and online supplemental table 1.
Supplemental material
Results
Macrophage GPR35 promotes tumour growth
We had previously reported a reduced incidence of intestinal tumours in Gpr35 –/– mice in spontaneous CRC (Apc min) and CAC (AOM/DSS) models.10 Selective deletion of Gpr35 in the intestinal epithelium (ie, in Gpr35 fl/fl;Vil-Cre or ‘Gpr35 ΔIEC’ mice) was sufficient to reduce tumour incidence in both models.10 In both CRC and CAC, individual tumours in Gpr35 –/– mice were strikingly smaller compared with Gpr35 +/+ tumours (figure 1A). Levels of CXCL-1, VEGF and IL-1β, proangiogenic mediators mainly produced by TAM,28 31 32 were substantially lower in Gpr35 –/– compared with Gpr35 +/+ tumours (figure 1B). This prompted us to ask whether GPR35 on LysM+ cells, which encompasses macrophages and neutrophils,33 might mediate a TME pathway that promotes tumour growth.17 18 We, therefore, generated Apc min allele-carrying mice with LysM-specific Gpr35 deletion (Gpr35 fl/fl;LysM-Cre;Apc min/+, ‘Gpr35 ΔMΦ;Apc min’) and compared them to their Gpr35-sufficient controls (Gpr35 fl/fl;Apc min/+, ‘Gpr35 fl/fl;Apc min’) (figure 1E). Gpr35 ΔMΦ;Apc min mice developed tumours more than three times smaller than Gpr35 fl/fl;Apc min mice (figure 1F), although the total number of tumours was not significantly different (figure 1C). Subjecting Gpr35 ΔMΦ (ie, without an Apc min allele) to AOM/DSS colitis revealed a similarly stark reduction in tumour size compared with their Gpr35 fl/fl controls (figure 1F). In this model of CAC, even the total tumour count in Gpr35 ΔMΦ mice was half of that in Gpr35 fl/fl mice (figure 1D), and associated with a higher survival rate (online supplemental figure 1D). The reduced incidence and size of tumours in Gpr35 ΔMΦ mice was not linked to protection from DSS-induced colitis, as weight loss during short-term (ie, 7 days) DSS, nor during the three cycles of AOM/DSS was indifferent between Gpr35 +/+ and Gpr35 –/–, and Gpr35 fl/fl and Gpr35 ΔMΦ mice (online supplemental figure 1A–C). Levels of VEGF, CXCL-1 and IL-1β were also markedly lower in inflammatory AOM/DSS tumours of Gpr35 ΔMΦ compared with Gpr35 fl/fl mice (figure 1G–I). Inflammatory infiltrate was reduced in Gpr35 ΔMΦ tumours compared with Gpr35 fl/fl tumours (online supplemental figure 1E). Macrophages and neutrophils express Gpr35, which is efficiently deleted in both cell types in Gpr35 ΔMΦ mice (online supplemental figure 2B). Of note, GPR35 expression extends to human peripheral blood-derived neutrophils and monocyte-derived macrophages (online supplemental file 2C). Neutrophil activation, measured by tissue MPO levels, were similar in Gpr35 ΔMΦ and Gpr35 fl/fl mice after 3 cycles of AOM/DSS, suggesting neutrophil GPR35 did not affect neutrophil activation per se in this context (online supplemental figure 1F). Altogether, this demonstrated that GPR35 in LysM+ myeloid cells potently promotes tumour growth in both CRC and CAC, and hinted toward an important role of tumour angiogenesis in this microenvironmental pathway.
Supplemental material

GPR35 increases tumour size and release of proangiogenic cytokines. (A) The area of intestinal adenomas from Apc min mice either wild-type (Gpr35+/+ ) and globally deficient for GPR35 (Gpr35-/- ) (left panel) or from Gpr35+/+ and Gpr35-/- mice exposed to AOM and three cycles of DSS (right panel) were compared. N=7 tumours per genotype. (B) CXCL-1, VEGF and IL-1β levels measured in AOM/DSS tumour tissue of Gpr35+/+ and Gpr35-/- mice. N=6 tumours per genotype. (C) Mice conditionally deficient for macrophage GPR35 (Gpr35 ΔMΦ) were crossed with APCmin mice and tumours were counted when the mice reached an age of 16 weeks. N=14 mice per genotype. (D) Gpr35 ΔMΦ mice were exposed to AOM and DSS. Colon adenomas were counted in Gpr35 ΔMΦ mice and in their control Gpr35fl/fl . N=11 to 13 mice per genotype. (E) Representative images showing H&E stain of tumours from Gpr35fl/fl and Gpr35 ΔMΦ mice. (F) The area of intestinal adenomas from either Gpr35fl/fl ; APC min and Gpr35 ΔMΦ; Apc min mice (left panel) or from Gpr35fl/fl and Gpr35 ΔMΦ mice exposed to AOM/DSS (right panel) were compared. N=9 tumours per genotype. (G–I) CXCL-1 (G), VEGF (H) and IL-1β (I) levels measured in AOM/DSS tumour tissue of Gpr35fl/fl and Gpr35 ΔMΦ mice. N=6 tumours per genotype. All data represented as mean±SEM. Statistical significance was calculated using Mann-Whitney U after Kruskal-Wallis testing. * p<0.05, ** p<0.01. AOM, azoxymethane; APC, Adenomatosis Polyposis Coli; DSS, dextran sodium sulphate; IL-1β, interleukin 1β; n.s., not significant.
GPR35 deletion in LysM+ myeloid cells reduces angiogenesis
The architecture of blood vessels in AOM/DSS-induced tumours of Gpr35 fl/fl mice appeared to be different from that in tumours of Gpr35 ΔMΦ mice. We observed fewer endothelial cells, identified by CD31, which stretched along the adenomatous crypts in Gpr35 ΔMΦ mice, whereas in adenomas from Gpr35 fl/fl mice, the CD31+ neo-vasculature resembled a reticular structure (figure 2A). When these tumours were analysed for total numbers of CD31+ cells by FACS, fewer CD31+ cells were detected in Gpr35 ΔMΦ compared with Gpr35 fl/fl tumours (figure 2B). Neither primary CD31+ murine endothelial cells, nor human umbilical vein endothelial cells, human microvascular endothelial cells (HMVECs), or the murine endothelial cell lines SVEC40 and H2-11 expressed Gpr35 mRNA (online supplemental figure 2A). We also noticed that adenomas in Gpr35 ΔMΦ mice exhibited only sparse staining for CD206, the mannose receptor and a marker of ‘M2’ macrophages, compared with dense staining in Gpr35 fl/fl adenomas (figure 2A). FACS analysis of Gpr35 ΔMΦ tumours (online supplemental figure 2D) showed significantly less CD68+ cells compared with Gpr35 fl/fl tumours (figure 2B). The number of CD163+ and CD206+ cells also trended lower in Gpr35 ΔMΦ compared with Gpr35 fl/fl tumours (figure 2B). Ly6-G+ cell counts were lower in Gpr35 ΔMΦ compared with Gpr35 fl/fl tumours, consistent with lower immune cell infiltration (online supplemental figure 1E.) Bone-marrow derived Gpr35 +/+ macrophages stimulated to differentiate into paradigmatic ‘M0’, ‘M1’ and ‘M2’ phenotypes secreted substantially more of the angiogenic mediators VEGF and CXCL-1 compared with their Gpr35 –/– counterparts (figure 2C,D). Human M0, M1 and M2 macrophages, differentiated from induced pluripotent stem cells (iPSC), secreted substantially more VEGF and CXCL-8 when they were differentiated from iPSCs genetically engineered to express the hypermorphic GPR35 T108M risk variant,10 compared with those differentiated from their syngeneic non-risk parent strain (figure 2E,F). This demonstrated that GPR35-dependent angiogenic factor release is conserved across species, and the human risk variant profoundly enhances their secretion. Macrophages involved in repair and (tumour)angiogenesis are thought to resemble mainly an M2 phenotype. We, therefore, pursued endothelial tube formation assays with SVECs or H2-11 murine endothelial cells stimulated with supernatants from Gpr35 +/+ and Gpr35 –/– M2 macrophages. Conditioned media from Gpr35 +/+ M2 macrophages induced SVECs to form significantly longer branches, more junctions within the tubular network, and a total area of formed tubes significantly larger compared with conditioned media from Gpr35 –/– macrophages (figure 3A). This suggested that GPR35 controls the release of angiogenic factors in macrophages that promote endothelial tube formation, a prerequisite of neovascularisation. Tube formation only occurs in the presence of growth factors such as VEGF. Control experiments show that supernatants of BMDM contain sufficient amounts of VEGF and potential other soluble factors CXCL-1/CXCL-8. Control RPMI media was not able to induce tube formation in 2 H-11 cells, which recurred on addition of recombinant VEGF (figure 3B). Blocking VEGF with an anti-VEGF antibody reduced branch length, number of junctions and the total mesh area, indicating the importance of growth factors released by macrophages (figure 3C). Increased branching and mesh formation of 2 H-11 endothelial cells (online supplemental figure 3) when exposed to Gpr35 +/+ compared to Gpr35 –/– macrophage supernatants was associated with increased phosphorylation at Tyr1175 of the VEGF receptor (figure 3E and online supplemental figure 5A). HMVEC exposed to supernatants from human T108M M2 macrophages differentiated from iPSCs also exhibited increased branching and mesh formation compared with those from their non-risk counterparts (figure 3D). These data were consistent with GPR35-dependent secretion of factors, including VEGF, from macrophages that act on endothelial cells to promote vessel formation.

GPR35 deletion in myeloid cells reduces angiogenic potential. (A) Adenomas from Gpr35fl/fl and Gpr35 ΔMΦ mice exposed to AOM/DSS stained for CD31+ endothelial cells and CD206+ M2 macrophages. N=5 each genotype, representative confocal microscopy. Scale bars 100 µm. (B) FACS analyses of CD31+, CD68+, CD163+, CD206+ and Ly-G6+ cells in Gpr35fl/fl and Gpr35 ΔMΦ tumour tissue. N=8–12 Gpr35fl/fl tumours from 8 to 12 mice and N=7 Gpr35 ΔMΦ tumours from mice. (C) VEGF levels in M0, M1 and M2 BMDM from Gpr35+/+ and Gpr35-/- mice. N=6 per genotype. (D) CXCL-1 levels in M0, M1 and M2 BMDM from Gpr35+/+ and Gpr35-/- mice. N=6 per genotype. (E) VEGF levels in supernatants of M0, M1 and M2 human iPS cell-derived macrophages. N=6 each genotype. (F) CXCL-8 levels in supernatants of M0, M1 and M2 human iPS cell-derived macrophages. N=6 each genotype. * p<0.05, ** p<0.01. AOM/DSS, azoxymethane followed by dextran sodium sulphate; iPS, inducible-pluripotent-stem; n.s., not significant; WT, wild type.

GPR35 deletion in macrophages and neutrophils reduces tube formation. (A) Tube formation assay with murine SVEC endothelial cells incubated with Gpr35+/+ and Gpr35-/- M2 macrophage supernatants. Branch length, number of junctions and total mesh area were determined using ImageJ’s angiogenesis tool. N=18. (B) 2 H-11 control tube formation assays with control RPMI/10%FBS and RPMI/10%FBS containing 30 ng/mL VEGF. No macrophage supernatants present. Branch length, number of junctions and total mesh area were determined using the Image J’s angiogenesis tool. N=14 C. 2 H-11 tube formation assays with supernatants of wild-type (WT) and knock-out macrophages in the presence of blocking anti-VEGF antibody. Branch length, number of junctions and total mesh area were determined using Image J’s angiogenesis tool. N=14. (D) HMVEC tube formation assays with human microvascular endothelial cells. Endothelial cells were incubated with supernatants of iPS cell-derived macrophage supernatants (WT and T108M risk variant). Branch length, number of junctions and total mesh area were determined using Image J’s angiogenesis tool. N=10. (E) VEGF receptor 2 phosphorylation of 2 H-11 cells treated with WT or Gpr35–/– M2 supernatants. All data represented as mean±SEM. Statistical significance was calculated using Mann-Whitney U after Kruskal-Wallis testing. HMVEC, human microvascular endothelial cell; iPS, inducible-pluripotent-stem; n.s., not significant.
GPR35 promotes ex vivo endothelial sprouting
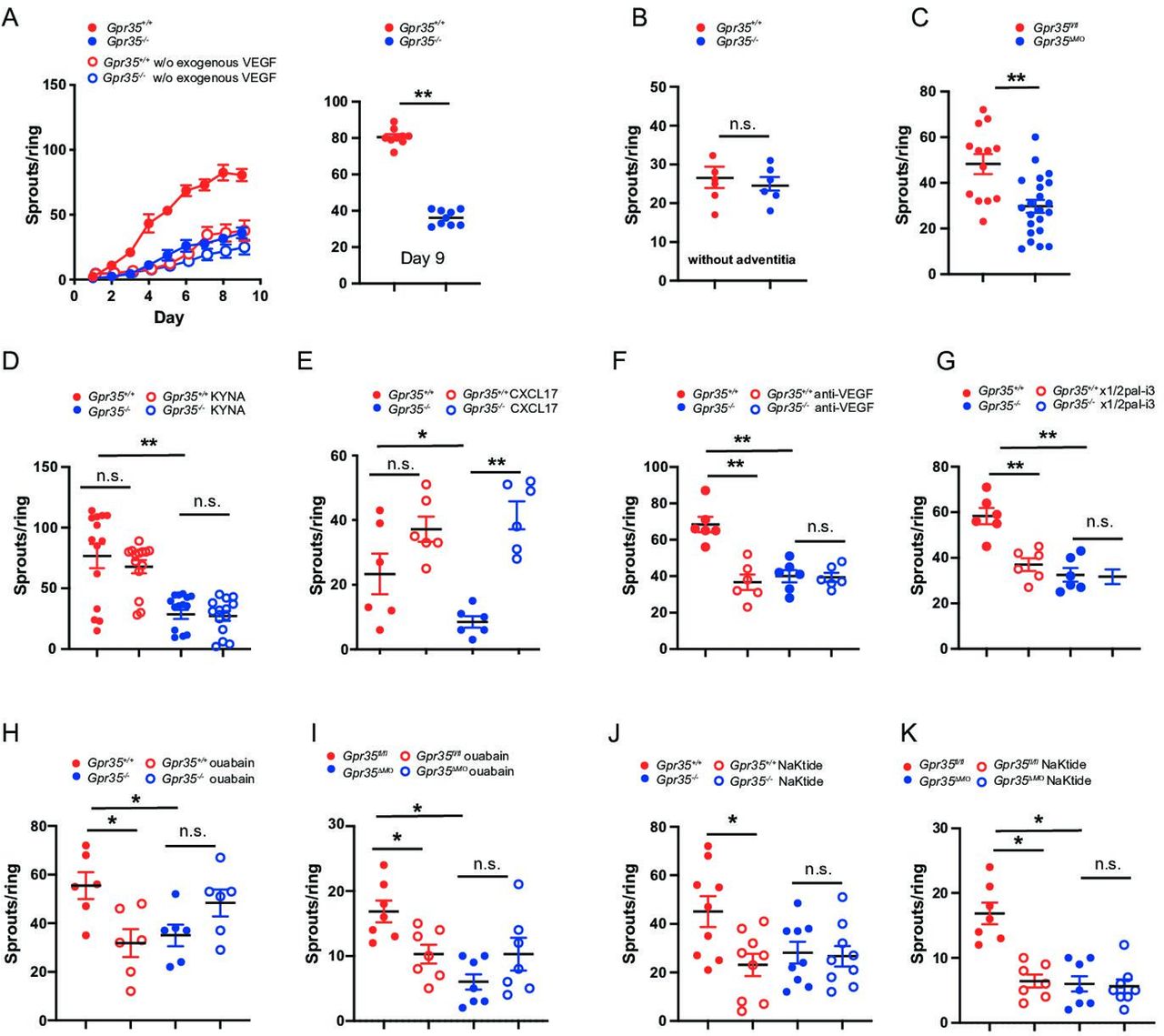
On in vitro culture, fibroblasts and macrophages are the first cells to migrate out of the aortic ring, setting the stage for the formation of neovessels. Endothelial cells typically start migrating and proliferating on day 2–3, and pericytes eventually surround the elongating sprouts.34 35 Here, we tested whether GPR35 is involved in vascular sprouting in vascular tissue and hence subjected aortic rings from wild-type and knock-out mice to an aortic sprout assay. Gpr35 –/– aortic rings developed markedly fewer vessel sprouts compared with Gpr35 +/+ rings (figure 4A). Addition of exogenous VEGF (30 ng/mL) was required (added on day 1), as only very little sprouting was triggered in wild-type and knock-out aortic rings in its absence (figure 4A). Removal of large parts of the aortic adventitia, which contains macrophages, abrogated the difference between Gpr35 +/+ and Gpr35 –/– aortic rings and let overall far fewer sprouts develop compared with when the aortic wall was left intact (figure 4B). Vessel sprouting from Gpr35 ΔMΦ compared with Gpr35 fl/fl aortic rings was equally profoundly suppressed (figure 4C) as in Gpr35–/– compared with Gpr35 +/+ aortic rings. This demonstrated that GPR35 on LysM+ myeloid cells orchestrated the outgrowth of neovessels.

Macrophage GPR35 controls vascular sprouting by interacting with the Na/K-ATPase. (A) Aortic rings from Gpr35 +/+ or Gpr35 –/– mice embedded in collagen matrix. Number of sprouts counted daily until day 9 of the experiment. Aortic rings were either embedded in media containing 30 ng/mL of VEGF (full circles) or left in OptiMEM media without growth factors (open circles) N=9 mice per genotype. (B) Vascular adventitia was removed from aortic rings from Gpr35 +/+ or Gpr35 –/–. Number of sprouts on day 9. N=6 mice per genotype. (C) Aortic rings from wildtype animals (Gpr35fl/fl ) or mice lacking GPR35 conditionally on their LysM+ cellsGpr35 ΔMΦ. N=13 for Gpr35fl/fl and 23 for Gpr35ΔMΦ mice. (D) KYNA (100 µM) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice and vascular sprouts counted on day 9. N=5 mice for each genotype. (E) CXCL-17 (20 ng/mL) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice and vascular sprouts counted on day 9. N=6 mice for each genotype. (F) Anti-VEGF antibody (100 ng/mL) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice and vascular sprouts counted on day 9. n=6 for each genotype. (G) Blocking CXCR2 pepducin x1/2pal-i3 (3 µM) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice and vascular sprouts counted on day 9. n=6 for each genotype. (H, I.) ouabain (100 µM) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice (H) and to the aortic rings of Gpr35fl/fl and Gpr35 ΔMΦ mice (I) and vascular sprouts counted on day 9. N=6 for each genotype. (J, K) pNaKtide (1 µM) was added to the aortic rings of Gpr35 +/+ or Gpr35 –/– mice (J) and to the aortic rings of Gpr35fl/fl and Gpr35 ΔMΦ mice (K) and vascular sprouts counted on day 9. N=9 for each genotype. All data represented as mean±SEM. Statistical significance was calculated using Mann-Whitney U after Kruskal-Wallis testing.* p<0.05, ** p<0.01. n.s., not significant.
KYNA, a product of tryptophan catabolism, and the chemokine CXCL-17 have been suggested as ligands of GPR35.36 37 Recombinant CXCL-17, added to aortic ring cultures, increased vessel sprouting in both Gpr35 +/+ and Gpr35 –/– aortic rings, indicating that CXCL-17 signals in a GPR35-independent manner (figure 4E). KYNA did not alter vessel sprouting, suggesting it is not involved in GPR35-mediated angiogenesis (figure 4D). Addition of a neutralising anti-VEGF monoclonal antibody on day 2 of the experiment, that is, after initial exogenous triggering, reduced vessel sprouting in Gpr35 +/+ aortic rings to levels observed in Gpr35 –/– aortic rings, in which sprouting was not affected (figure 4F). Moreover, blocking CXCL-1 signalling by blocking its receptor CXCR2 with a selective pepducin38 also reduced vessel sprouting from Gpr35 +/+, but not from Gpr35 –/– aortic rings (figure 3G). This demonstrated that GPR35 promotes vessel sprouting via VEGF and CXCR2, which tallied with higher VEGF and CXCL-1 secretion in tissue and macrophages from Gpr35 +/+ compared to Gpr35 –/– mice.
GPR35 promotes angiogenesis via the sodium-potassium-pump in macrophages
Reduced Na/K-ATPase ion pumping activity in the absence GPR35 elevates intracellular Ca2+ and lowers K+ levels.10 The endogenous cardiotonic steroid ouabain achieves a similar effect via binding to Na/K-ATPase and retaining it in an inactive (E2P) state.39 Ouabain indeed reduced vessel sprouting in Gpr35 +/+ aortas to levels observed in Gpr35 –/– aortas, whereas sprouting was not significantly changed but slightly increased in Gpr35 –/– aortas (figure 4H). This was precisely phenocopied in aortas from Gpr35 fl/fl when compared with Gpr35 ΔMΦ mice (figure 4I), demonstrating vessel sprouting was coordinated by GPR35 on myeloid cells in a Na/K-ATPase-dependent manner. Among its non-pump functions, Na/K-ATPase acts as a scaffold for Src kinase and is thereby important in signal transduction.11 40 41 The α1 type Na/K-ATPase forms a complex with the Src SH2 and kinase domain and keeps the kinase domain in an inactive state.11 A 20-amino acid sequence (pNaKtide) specifically inhibits Na/K-ATPase-associated Src activation.42 43 Addition of pNaKtide peptide decreased sprouting in Gpr35 +/+ and Gpr35 fl/fl aortas to levels observed in their respective Gpr35 –/– and Gpr35 ΔMΦ counterparts, in which pNaKtide had no further effect (figure 4J,K). When exposing Gpr35 +/+ macrophages to ouabain, no increase in Src phosphorylation was observed. This was unexpected given previously published work in cell lines that do not endogenously express GPR35.43 Src phosphorylation was indeed increased by ouabain in Gpr35 –/– macrophages (online supplemental figure 4), pointing towards an important role of GPR35 determining the Src signalling response on pharmacological inhibition of Na/K-ATPase. Altogether this demonstrated that GPR35 expressed on myeloid cells within the aortic wall promotes vessel sprouting in a manner dependent on Na/K-ATPase-triggered Src activation.
GPR35-deficient tumours produce less MMPs
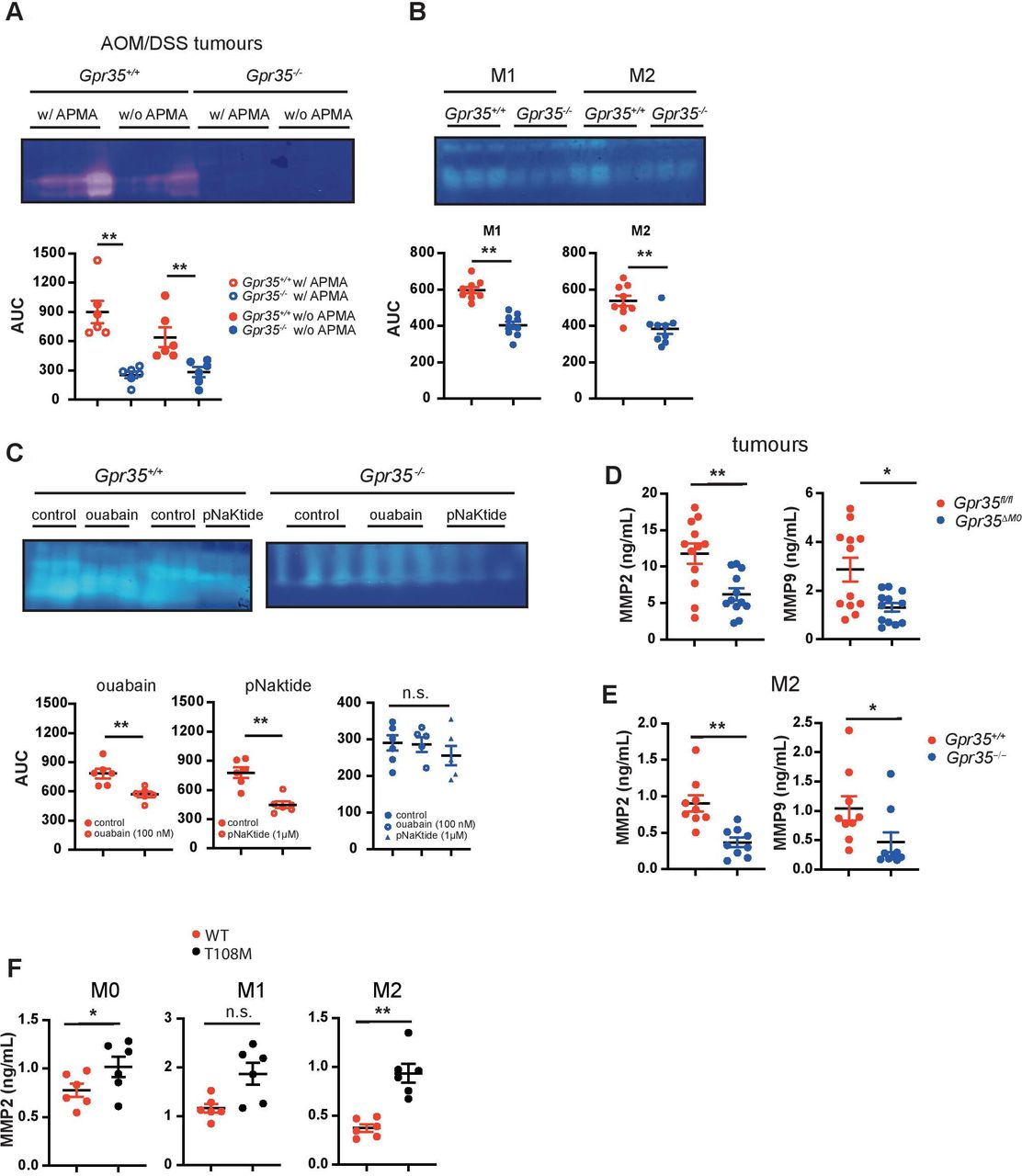
A link between MMP-9-positive TAMs and endothelial cells responding to angiogenic induction has been established.44 To assess MMP activity, we performed gelatine zymography of extracts from tumours arising in the AOM/DSS model. Gpr35 –/– tumours exhibited much lower gelatinolytic activity compared with Gpr35 +/+ tumours (figure 5A). Addition of 4-aminophenylmercuric acetate (APMA), which activates pro-MMPs to active zymogens, to tumour extracts did not further increase their gelatinolytic activity (figure 5A). This indicated that essentially all available MMP activity in tumours is already in the active form. Macrophages can release MMPs and tissue inhibitors of MMPs. While native supernatants from Gpr35 –/– or Gpr35 +/+ M1 and M2 BMDMs did not exhibit MMP activity (data not shown), addition of APMA to supernatants revealed substantial activity in Gpr35 +/+ and much lower activity in Gpr35 –/– macrophage supernatants (figure 5B). Ouabain and pNaKtide treatment of Gpr35 +/+ M2 macrophages reduced their supernatants’ MMP activity (figure 5C), whereas it did not affect MMP activity in Gpr35 –/– supernatants (figure 5C). Differences in zymography were detected in gelatine- but not casein gels. We, therefore, measured the gelatinases MMP2 and MMP9 in tumour tissue and supernatants from BMDM. Gpr35 ΔMΦ tumours contained substantially less MMP2 and MMP9 (figure 5D) compared with AOM/DSS-induced tumours from Gpr35fl/fl mice. M2 macrophages from Gpr35 –/– mice correspondingly contained less MMP2 and MMP9 compared with their Gpr35 +/+ counterparts (figure 5E). Moreover, supernatants from human iPSC-derived M0 and M2 macrophages carrying the T108M risk variant contained higher amounts of MMP2 compared with those from non-risk variant cells, with MMP2 levels in T108M variant M1 macrophages levels also trending higher (figure 5F). Altogether, this implied that GPR35 controlled via Na/K-ATPase-dependent Src activation the level of MMP activity in tumours, with the T108M variant yielding highest levels.

Lack of GPR35 results in reduced MMP levels. (A) MMP levels in Gpr35 +/+ and Gpr35 –/– adenomas. Tissue extract MMPs were activated with APMA. Area under the curve was analysed using image J. N=6 tumours per genotype. (B) MMP levels in M1 and M2 BMDM supernatants from Gpr35 +/+ and Gpr35 –/– mice. Supernatants were APMA-activated. area under the curve was analysed using image J N=9 mice per genotype. (C) MMP levels in M2 macrophages pretreated with ouabain (100 µM) or pNaKtide (1 µM). Area under the curve was analysed using image J N=6 mice per genotype. (D) Total (inactive zymogen and active enzyme) MMP2 and MMP9 levels in tumour tissue from Gpr35fl/fl and Gpr35 ΔMΦ mice. (E) MMP2 and MMP9 levels from M2 murine BMDM supernatants. (F) Total (inactive zymogen and active enzyme) MMP2 levels from supernatants of human iPS cell-derived macrophages. Macrophages were polarised to M0, M1 and M2 macrophages. All data represented as mean±SEM. Statistical significance was calculated using Mann-Whitney U after Kruskal-Wallis testing. * p<0.05, ** p<0.01/ APMA, aminophenylmercuric acetate; MMP, matrix-metallo-proteinase; n.s., not significant; WT, wild type.
GPR35 increases endothelial transmigration of monocytes
Circulating CCR2+ inflammatory monocytes enter the TME, proliferate and differentiate into TAMs,45 a process that requires endothelial adhesion and transmigration.46 We therefore investigated transmigration by studying myeloid cells harvested from 5-day bone marrow cultures, at which point cells exhibit a monocyte phenotype.47 Mimicking monocyte transmigration, we plated endothelial cells onto Transwell membranes and used supernatants from Gpr35 +/+ or Gpr35 –/– M2 macrophages as chemoattractant. Gpr35 +/+ or Gpr35 –/– monocytes added to the upper compartment of the chamber migrated through the endothelial cell layer towards the macrophage supernatants. Compared with Gpr35 +/+ monocytes, transmigration of Gpr35 –/– monocytes was reduced (figure 6A, left panel). Fewer Gpr35 –/– than Gpr35 +/+ monocytes adhered to monolayers of H2-11 murine endothelial cells (figure 6A, right panel). Hence GPR35 deficiency in monocytes impaired their ability to adhere to, and transmigrate through, endothelial cells. In converse experiments, we exposed endothelial cells to supernatants of either Gpr35 +/+ or Gpr35 –/– M2 macrophages, and monocytes of either genotype migrated towards wild-type macrophage conditioned media. Monocytes adhered less to endothelial cells that were exposed to Gpr35 –/– M2 macrophage supernatant than to those exposed to Gpr35 +/+ supernatant (figure 6B right panel). Transmigration of monocytes through an endothelial monolayer incubated with Gpr35 –/– M2 macrophage conditioned media was correspondingly reduced when compared with migration through endothelial cells exposed Gpr35 +/+ supernatants. Fewer Gpr35 –/– than Gpr35 +/+ monocytes migrated through monolayers treated with either Gpr35 –/– or Gpr35 +/+ macrophage supernatants (figure 6B, left panel). This demonstrated that GPR35 on macrophages controls the release of factors that promoted adhesion and transendothelial migration of monocytes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lack of GPR35 decreases endothelial transmigration. (A) Right panel: Gpr35 +/+ and Gpr35 –/– monocytes transendothelial migration towards conditioned media from Gpr35 +/+ and Gpr35 –/– M2 macrophages in the lower compartments of migration chambers. Untreated endothelial cell monolayer. Left panel: adhesion of Gpr35 +/+ and Gpr35 –/– monocytes to 2 H-11 murine endothelial cells. Untreated endothelial cell monolayer. N=6 mice per genotype. (B) Left panel: transendothelial migration of monocytes through monolayers of 2 H-11 murine endothelial cells exposed to conditioned media from Gpr35 +/+ and Gpr35 –/– M2 macrophages. right panel: adhesion of Gpr35 +/+ and Gpr35 –/– monocytes to 2 H-11 murine endothelial cells treated with conditioned media from Gpr35 +/+ and Gpr35 –/– M2 macrophages. N=6 mice per genotype. (C) Upper panel: VCAM-1 expression in 2 H-11 cells treated with conditioned media from Gpr35 +/+ and Gpr35 –/– M2 macrophages pretreated with control RPMI, ouabain (100 µM) or pNaKtide (1 µM). Lower panel: VCAM-1 expression in Gpr35fl/fl and Gpr35 ΔMΦ AOM/DSS tumours. (D) ERK1/2 phosphorylation of 2 H-11 cells treated with conditioned media from Gpr35 +/+ and Gpr35 –/– M2 macrophages pretreated with control RPMI, ouabain (100 µM) or pNaKtide (1 µM). (E) Src and ERK1/2 phosphorylation in Gpr35 +/+ and Gpr35 –/– AOM/DSS tumours. N=3 tumours per genotype. (F) Numbers of macroscopic and microscopic tumours of either g35i2 or vehicle treated mice with AOM/DSS induced tumours. N=8–9 mice/group. (G) AOM/DSS tumour tissue from g35i2 or vehicle treated mice probed for Src and ERK1/2 phosphorylation. N=3 tumours per genotype. (H) CXCL-1, VEGF, MMP2, MMP9 and inflammatory infiltrate in tumour tissue from g35i2 or vehicle-treated wild-type mice. N=6 per genotype for CXCL-1, VEGF, MMP2 and MMP9 ELISA assays, and n=8 for histological assessment of inflammatory cell infiltrate. All data represented as mean±SEM. Statistical significance was calculated using Mann-Whitney U after Kruskal-Wallis testing. AOM/DSS, azoxymethane followed by dextran sodium sulphate; n.s., not significant.
VCAM-1 is a major endothelial cell adhesion molecule that facilitates monocyte transmigration from the peripheral blood into the surrounding tissue.48 VCAM-1 expression was higher in H2-11 endothelial cells exposed to Gpr35 +/+ compared with Gpr35 –/– M2 macrophage supernatants (figure 6C). This increase was abrogated when M2 macrophages were treated overnight with ouabain or pNaKtide and then washed, prior to collecting supernatants over the ensuing 24 hours (figure 6C and online supplemental figure 5B). In contrast, ouabain or pNaKtide treatment of Gpr35 –/– M2 macrophages did not affect endothelial VCAM-1 expression. This demonstrated that increased VCAM-1 induction on endothelial cells was dependent on GPR35, Na/K-ATPase and Src activity in macrophages. Consistent with such GRP35-dependent regulation, VCAM-1 expression in tissue was higher in Gpr35fl/fl compared with Gpr35 ΔMΦ mice (figure 6C). Activation of extracellular signal-regulated kinases (ERK) 1 and 2 by VCAM-1 is critical for endothelial activation, transmigration and angiogenesis.48 Conditioned media from Gpr35 –/– M2 macrophages induced less ERK1/2 phosphorylation in H2-11 endothelial cells compared with Gpr35 +/+ supernatants (figure 6D and online supplemental figure 5C). Similar to VCAM-1 expression, elevated ERK1/2 phosphorylation in H2-11 endothelial cells exposed to Gpr35 +/+ macrophages was prevented by pretreatment of macrophages with ouabain or pNaKtide (figure 6D). In contrast, endothelial ERK1/2 phosphorylation was not affected when triggered by supernatants of inhibitor-treated Gpr35 –/– macrophages (figure 6D). Src and ERK1/2 phosphorylation was correspondingly lower in Gpr35 –/– compared with Gpr35 +/+ tumours (figure 6E and online supplemental figure 5D). This hence demonstrated that GPR35-modulated activation of Na/K-ATPase, and Na/K-ATPase-dependent Src activation, in macrophages controlled Erk1/2 phosphorylation and VCAM-1 expression on endothelial cells via transmissible signals.
Gpr35 blockade with a GPR35 specific pepducin reduces tumour size in the AOM/DSS-induced CAC model
We have previously shown that treatment of wild-type mice with a GPR35-selective, inhibitory pepducin (g35i2) reduced the number of inflammatory colon tumours.10 In the AOM/DSS model, the number of macroscopically visible tumours in g35i2-treated wild-type mice decreased fivefold, whereas the number of microscopic tumours decreased only threefold (figure 6F). Src Tyr416 phosphorylation in AOM/DSS tumours was substantially lower in g35i2- compared with control-treated wild-type mice, and was accompanied by reduced Thr202/Thr204 phosphorylation of ERK1/2 (figure 6G). Furthermore, CXCL-1, VEGF, MMP2 and MMP9 levels as well as immune cell infiltration were significantly decreased when wildtype mice were treated with the g35i2 compound (figure 6H). Altogether this demonstrated that pharmacological GPR35 blockade reduced tumour size, involving the Na/K-ATPase—Src pathway delineated herein.
Discussion
Here, we report that GPR35 on macrophages is a major promoter of tumour growth by facilitating tumour neovascularisation. We show that this activity is dependent on Na/K-ATPase, and Na/K-ATPase-dependent activation of the non-receptor tyrosine kinase Src within macrophages. The profound reduction in tumour growth observed in both spontaneous and inflammation-induced intestinal tumour models on macrophage-selective deletion of Gpr35 identifies a novel major mechanism, and vividly highlights the critical role of myeloid cells in tumour development.18 We had previously reported that intestinal epithelial cell-specific deletion of Gpr35 reduces intestinal tumour incidence and reduces epithelial cell proliferation.10 Our discovery here adds a major separate tumour-promoting mechanism of GPR35 that originates from TAMs and neutrophils. The UC/PSC-linked T108M polymorphism of GPR35, which is hypermorphic and increases Na/K-ATPase pump activity,10 profoundly increases the angiogenic mechanisms as we experimentally demonstrate in human macrophages differentiated from gene-edited iPSCs.8 UC is another strong risk factor for the development of CRC.49 PSC is the autoimmune disease with the highest lifetime risk of developing cancer,7 primarily cholangiocellular carcinoma but also CRC.7 GPR35 expression has also been linked to gastric cancer,50 and its increased expression in tumour tissue has been strongly associated with poor prognosis in colon cancer and non-small cell lung cancer.51 52
Neoangiogenesis is imperative for tumour growth and progression. This is therapeutically exploited by VEGF neutralisation (eg, bevacizumab in CRC53), and by IL-8/CXCR2 blockade that is currently investigated in clinical trials.54 55 The TME, and TAMs in particular, play an eminent role in tumour angiogenesis.18 TAMs appear to derive from circulating inflammatory monocytes that transmigrate into tumour tissue.56 TAMs with a predominantly ‘M2-like’ phenotype stimulate angiogenesis by releasing angiogenic cytokines such as VEGF and IL-8.18 27 GPR35 deletion does not affect differentiation into the paradigmatic ‘M1’ and ‘M2’ macrophage phenotypes in vitro.10 Tumours in mice with macrophage-specific or germline Gpr35 deletion are smaller, contain fewer CD31+ endothelial cells and CD206+ M2-like TAMs, and exhibit lower expression of VEGF and CXCL-1, altogether implying reduced angiogenesis as a major component of reduced tumour growth when GPR35 is absent. Enhanced monocyte endothelial transmigration in vitro, and aortic sprout formation ex vivo, further corroborate the critical role of macrophage GPR35 in coordinating an angiogenic response. MMPs, in particular MMP-2, MMP-9 and MMP-14,57 facilitate tissue remodelling, a vital process during neo-angiogenesis and tumour progression. MMPs are mostly produced by TAMs, released into the extracellular space as inactive zymogen, and subsequently cleaved and activated by other proteinases.29 MMP-9, for example, directly releases glycocalyx-bound VEGF from cell membranes and decreases the amount of syndecan-4 which binds and inactivates VEGF.58 The substantially reduced MMP activity in Gpr35 –/– tumours corroborates a role of this GPCR as an upstream regulator of tumour tissue remodelling.
GPR35 forms a complex with Na/K-ATPase and promotes its pumping activity, independent of pharmacological or candidate physiological ligands.10 Since Na/K-ATPase ascertains electrophysiologic homoeostasis in a cell, reduced pumping activity in GPR35’s absence increases baseline intracellular Ca2+, which is maintained via secondary-active transport, to levels otherwise only observed on receptor-mediated signalling events.10 Elevated Ca2+ in turn affects Ca2+-dependent signalling processes broadly.10 59 Intriguingly, our results infer a critical role of macrophage Na/K-ATPase pumping activity in (neo)angiogenesis. Teleologically, increasing nutrient and oxygen supply via neo-angiogenesis via this mechanism fits with the notion that maintaining the electrochemical gradient is highly energy-intensive. Increased angiogenic activity of GPR35-sufficient myeloid cells is further dependent on Na/K-ATPase-dependent Src phosphorylation. Cardiotonic steroids such as ouabain block Na/K-ATPase pump function by keeping the pump in its E2P state,60 and this pharmacological inhibition of pumping activity had been linked to increased Src phosphorylation.43 In contrast, in macrophages expressing GPR35, pNaKtide-inhibitable and hence Na/K-ATPase-dependent Src activation is linked to increased pumping activity.10 This relationship of increased Na/K-ATPase activity with increased Src activation is also evident in the aortic sprout assays we report here. This posits interesting questions on the structural basis of GPR35-dependent promotion of pumping activity and Src activation.
In summary, we identify GPR35 on myeloid cells as upstream coordinator of tumour tissue remodelling, reinforcing the critical role of TAMs and neutrophils in cancer progression. The GPR35–Na/K-ATPase–Src pathway is precisely targetable with GPR35-selective pepducins.10 Its upstream role in tumour tissue remodelling renders GPR35 an attractive target, even in cases where tumour cells themselves do not express the receptor.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. All data relevant to the study are included in the article or uploaded as online supplemental information. n/a.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @ester.pagano, @tomhemmingk
Correction notice This article has been corrected since it published Online First. The author, Dr Kaneider's, name has been updated.
Contributors Conceptualisation: NK and AK. Methology: NK, EP, GS, JEE and SS. Validation: NK, GS, JEE and EP. Formal analysis: NK and EP. Investigation: NK, GS, JEE, EP and LMH. Resources: LMH. Writing—original Draft. NK and AK, visualisation: NK and GS. Supervision: NK, AK and THK. Funding Acquisition: NK, AK and THK. External mentor: FB to EP.
Funding This paper was supported by the Wellcome Trust (Clinical Research Career Development Fellowship to NK 216630/Z/19/Z, Career Re-entry Fellowship to NK 103077/Z/13/Z, Senior Investigator Award 106260/Z/14/Z to AK, PhD training fellowship for clinicians UNS59491 to JEE), the European Research Council under the European Community’s Seventh Framework Programme Consolidator Grant no 648 889 to AK, and Scientia Fellowship (FP7-PEOPLE-2013-COFUND) grant agreement no 609 020 to GS, the Addenbrooke’s Charitable Trust (ACT 25/16A) to JEE, and an UniNA and Compagnia Di San Paolo ‘STAR programme for young researchers’ fellowship to EP. This work was further supported by the NIHR Cambridge BRC. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.