Article Text

Abstract
Objective Infection by HBV is the main risk factor for hepatocellular carcinoma (HCC) worldwide. HBV directly drives carcinogenesis through integrations in the human genome. This study aimed to precisely characterise HBV integrations, in relation with viral and host genomics and clinical features.
Design A novel pipeline was set up to perform viral capture on tumours and non-tumour liver tissues from a French cohort of 177 patients mainly of European and African origins. Clonality of each integration event was determined with the localisation, orientation and content of the integrated sequence. In three selected tumours, complex integrations were reconstructed using long-read sequencing or Bionano whole genome mapping.
Results Replicating HBV DNA was more frequently detected in non-tumour tissues and associated with a higher number of non-clonal integrations. In HCC, clonal selection of HBV integrations was related to two different mechanisms involved in carcinogenesis. First, integration of viral enhancer nearby a cancer-driver gene may lead to a strong overexpression of oncogenes. Second, we identified frequent chromosome rearrangements at HBV integration sites leading to cancer-driver genes (TERT, TP53, MYC) alterations at distance. Moreover, HBV integrations have direct clinical implications as HCC with a high number of insertions develop in young patients and have a poor prognosis.
Conclusion Deep characterisation of HBV integrations in liver tissues highlights new HBV-associated driver mechanisms involved in hepatocarcinogenesis. HBV integrations have multiple direct oncogenic consequences that remain an important challenge for the follow-up of HBV-infected patients.
- hepatocellular carcinoma
- hepatitis B
- cancer genetics
Data availability statement
Data are available in a public, open access repository. The sequencing data of the LICA-FR cohort reported in this paper have been deposited to the European Genome-phenome Archive (EGA) database (RNA-seq fastq files accessions (EGAS00001001284), (EGAS00001002879), (EGAS00001003310), (EGAS00001003837) and (EGAS00001004629); WES bam files accessions (EGAS00001000217), (EGAS00001001002), (EGAS00001003063), (EGAS00001003130), (EGAS00001003837) and (EGAS00001004629); WGS bam files accessions (EGAS00001000706), (EGAS00001002408), (EGAS00001002888), (EGAS00001003063), (EGAS00001003837) and (EGAS00001004629), through the International Cancer Genome Consortium (ICGC) data access committee. Data are available for reuse and can be consulted at the following address: https://ega-archive.org/studies/EGASxxx with access permission from ICGC Data Access Compliance Office.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
HBV infection is the main risk factor for hepatocellular carcinoma (HCC) worldwide.
HBV is a small DNA virus which can integrate in the human genome and thus play a role in promoting carcinogenesis.
Studies using next-generation sequencing on liver tumours from Asian patients pointed out that HBV promotes HCC development through insertional mutagenesis.
What are the new findings?
Replicating HBV DNA was more frequently detected in non-tumour tissues, and associated with a higher number of integrations.
HCC development through HBV insertional mutagenesis is linked to the presence of a viral enhancer in the integrated sequence in the proximity of a cancer-driver gene.
HBV-induced carcinogenesis can also be driven by frequent copy number alterations of cancer-driver genes associated with distant viral integrations.
The number of HBV integrations is an independent prognostic factor in HBV-related HCC.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
A high number of HBV integrations is associated with viral replication in non-tumour liver tissues and with poor prognosis in tumours, showing the importance of providing efficient antiviral therapy early during life to limit the number of HBV integrations and fight against direct HBV-related HCC development.
This study underlines that HBV-related HCC are promoted by distinct mechanisms, highlighting the heterogeneity of these tumours and the importance of molecular characterisation to identify new specific therapeutic opportunities.
Introduction
Despite the implementation of vaccination programmes and the strong decrease of new infections among children, the percentage of people living with chronic HBV infection worldwide remained as high as 3.5% of the global population in 2015.1 Moreover, only 9% of infected people were diagnosed and only 8% of them were under treatment, which makes hepatitis B a major public health threat now prioritised by WHO.1 2 The burden of HBV infection is closely related to the development of hepatocellular carcinoma (HCC), the most frequent primary liver cancer and the fourth cause of cancer death worldwide.3 Persistent HBV infection is actually responsible for >50% of all HCC cases worldwide and up to 85% in some areas where the infection is endemic.4 Even patients receiving antiviral treatments who have maintained viral suppression remain at risk of developing an HCC.5–8
HBV-related HCC occurs in the setting of cirrhosis and in normal liver,9 underlying that the virus has its own oncogenic properties besides the induction of chronic inflammation in the liver. HBV is a small 3.2 kb DNA virus that can integrate in human DNA and promote cell transformation by insertional mutagenesis or through expression of viral oncoproteins such as the protein HBx.10–12 As HBV integrations occur early during infection,13 comparing the insertions occurring in normal hepatocytes and in tumour cells may enable to identify new genomic defects associated with tumour development.
The development of next-generation sequencing led to a more precise characterisation of the role of HBV insertions in HCC initiation and development.14–18 Cancer-related genes such as TERT, CCNE1 and KMT2B have been identified as recurrently targeted by HBV insertions in HCC, with specific functional consequences and clinical outcomes.16 19–21 Thus, the presence of integrated HBV DNA in hepatocytes has been pointed out as a driver of hepatocarcinogenesis through the alteration of the expression or function of these genes.12 But up to now, such identifications of HBV insertions in HCC have been mainly performed in Asian populations where HBV genotypes B and C are the most prevalent HBV strains.22 Furthermore, as a significant proportion of HBV-related HCC does not contain any integration in a known cancer-related gene, two other independent mechanisms involving integrated HBV DNA have been proposed: induction of chromosomal instability and persistent expression of altered HBV genes.23 However, the interactions between the three mechanisms mentioned and the precise role they might play individually or synergistically in HCC initiation and development remain to be fully elucidated.
Therefore, in this work, we developed a reliable pipeline of analysis based on viral capture, to characterise HBV integrations in a cohort of 177 patients, together with genome rearrangements, clinical features and viral characteristics in tumours and their corresponding non-tumour liver tissues. We also investigated the oncogenic consequences of recurrent HBV integrations on gene expression and chromosomal instability, involved in hepatocarcinogenesis.
Materials and methods
(Extended in online supplemental materials and methods section).
Supplemental material
A series of tissue samples containing 1128 frozen HCC and 1063 non-tumour counterparts from 1128 patients was assembled (online supplemental figure 1). Clinical and serological data were collected from each centre (online supplemental table 1). Viral DNA screening was performed according to protocols previously described24 with specific HBV probes (online supplemental table 2).
Supplemental material
HBV viral capture was performed for 177 frozen HCC and 170 matched non-tumour liver tissues using single-stranded biotinylated probes (SeqCap EZ Designs, Roche NimbleGen, Madison, Wisconsin, USA) to target 40 references distributed on the eight main HBV genotypes and four human regions (TERT promoter, CCNE1, CCNA2 and KMT2B; online supplemental table 3). DNA libraries of 1 kb fragments were prepared and multiplexed before following the double capture steps with SeqCap-EZ-HyperCap Workflow (Roche). Sequencing was performed with Illumina MiSeq instrument to produce paired-end reads of 2×250 nucleotides.
Analysis of integration events was performed following a pipeline described in a flowchart (online supplemental figure 2). Briefly, sequencing data were aligned on HBV and human references (online supplemental table 3). HBV copy number per cell was evaluated from the mean read coverage of the HBV genome, including integrated and episomal HBV (online supplemental figure 3A). Paired and chimeric reads were extracted using htslib package to identify HBV/human junction breakpoints and assess clonality of integration events. The 8809 integration events were classified in three categories using the k-means method: (1) ‘clonal integrations’ (>48% of cells), (2) ‘unique integrations’ (<3% of cells) and (3) ‘subclonal integrations’ (3%–48% of cells) (online supplemental figure 3B). For 20 pairs of tumours and non-tumour tissues, integration events identified with viral capture and whole genome sequencing (WGS) were comparable (online supplemental figure 3C,D). Integration breakpoints were annotated for replication timing in HepG2 (ENCODE),25 top 20% highly expressed genes in HBV-positive livers, common and rare fragile sites,26 repetitive sequences and CpG islands and chromatin structure in adult liver (ROADMAP).27 Insertions were named ‘classic’ if the two breakpoints clustered within 25 kb in opposite direction, ‘local inversion’ if two breakpoints clustered within 25 kb in the same direction, ‘large rearrangements’ if only one breakpoint was identified and ‘cluster of insertions’ if more than two breakpoints were identified.
Genomic DNA sequencing of driver genes were performed in 265 tumours and their adjacent liver tissues with high coverage WGS (n=62) or whole exome sequencing (WES, n=203) and re-sequenced at TERT promoter with Sanger or MiSeq.
Detection of HBV episomal form was performed with specific DNAse/TaqMan-based assay24 in 162 tumours and 155 non-tumour liver tissues (online supplemental table 2).
Viral mRNA and specific genes mRNA screening was performed by quantitative reverse transcriptase (qRT)-PCR using 10 probe sets covering 8 HBV regions from the main HBV genotypes, and probe sets to detect HCV, HDV or TERT expression (online supplemental table 2).
HBV replicative forms were considered as present in one tissue if both HBV episomal DNA and HBV pregenomic RNA (pgRNA) forms were identified. When episomal forms were detected in the absence of pgRNA, the tissue was considered as containing ‘episomal not replicative HBV DNA’.
RNA-sequencing (RNA-seq) and transcriptomic analyses were performed in 265 tumours (130 HBV-positive and 135 HBV-negative) and 24 HBV-positive non-tumour tissues using Illumina TruSeq or Illumina TruSeq Stranded mRNA kit on HiSeq2000 by IntegraGen, Evry, France.19 We used the Bioconductor limma package28 to test for differential expression in all expressed genes with an in-house adaptation of the gene set enrichment analysis (GSEA) method.29 Tumours were classified in G1-G6 transcriptomic groups as previously described by qRT-PCR of 190 genes using BioMark PCR system.30 31
HBV variants analysis from viral capture was performed according to nomenclature previously described.22 32
Cell culture, transfection and dual luciferase assay were performed in HuH7 cells purchased from the American Type Culture Collection as previously described.33
Long-read sequencing were performed in three HBV-positive tumours (#1151T, #1597T, #1994T) using the Single Molecule Real Time technology, PacBio-SMRTcell system (ICGex platform, Curie Institute, France) and the Consensus Circular Sequencing algorithm (Pacific Biosciences). Whole genome mapping was performed to analyse the same samples by Bionano Genomics, La Jolla, California, USA.
Statistical analyses were performed with R V.3.6.0 (http://www.R-project.org), various statistical tests were applied with respect to the type of variable. Survival analysis was performed in patients treated for a primary HCC tumour by R0 liver resection as previously described.34
Results
HBV integrations occur in open chromatin regions and relate to viral replication in the liver
We performed HBV viral capture on 177 HCC and 170 non-tumour liver tissues from 177 HBV-positive patients to analyse HBV integrations in the human genome with a new bioinformatics pipeline and identify precisely the structure of insertion events and their clonality (see ‘Materials and methods’ section and online supplemental table 1). In 84% of non-tumour liver tissues (143 out of 170), we identified 6610 HBV integration breakpoints at HBV/human junctions corresponding to unique (82%), subclonal (17%) or clonal (1%) events, involving similarly all HBV genotypes (figure 1A; online supplemental figure 4; online supplemental table 4). In the human genome, HBV integration breakpoints were enriched in active and open chromatin regions, and they were close to early replicated and highly expressed genes. Integrations were also more frequent in simple repeats, in particular in telomeric and subtelomeric regions, but not in Alu motifs, fragile sites or long/short interspersed nuclear elements (LINE/SINE; online supplemental figure 5A). Reconstruction of a selection of 394 events of subclonal or clonal integrations in non-tumour tissues showed that they all contained a large part of the whole HBV sequence (median size=2912 nucleotides) and 42% of them were bordered by 1-to-8 nucleotide homology between the targeted human sequence and the integrated HBV sequence, suggesting the involvement of microhomology-mediated end joining (online supplemental figure 5B,C).
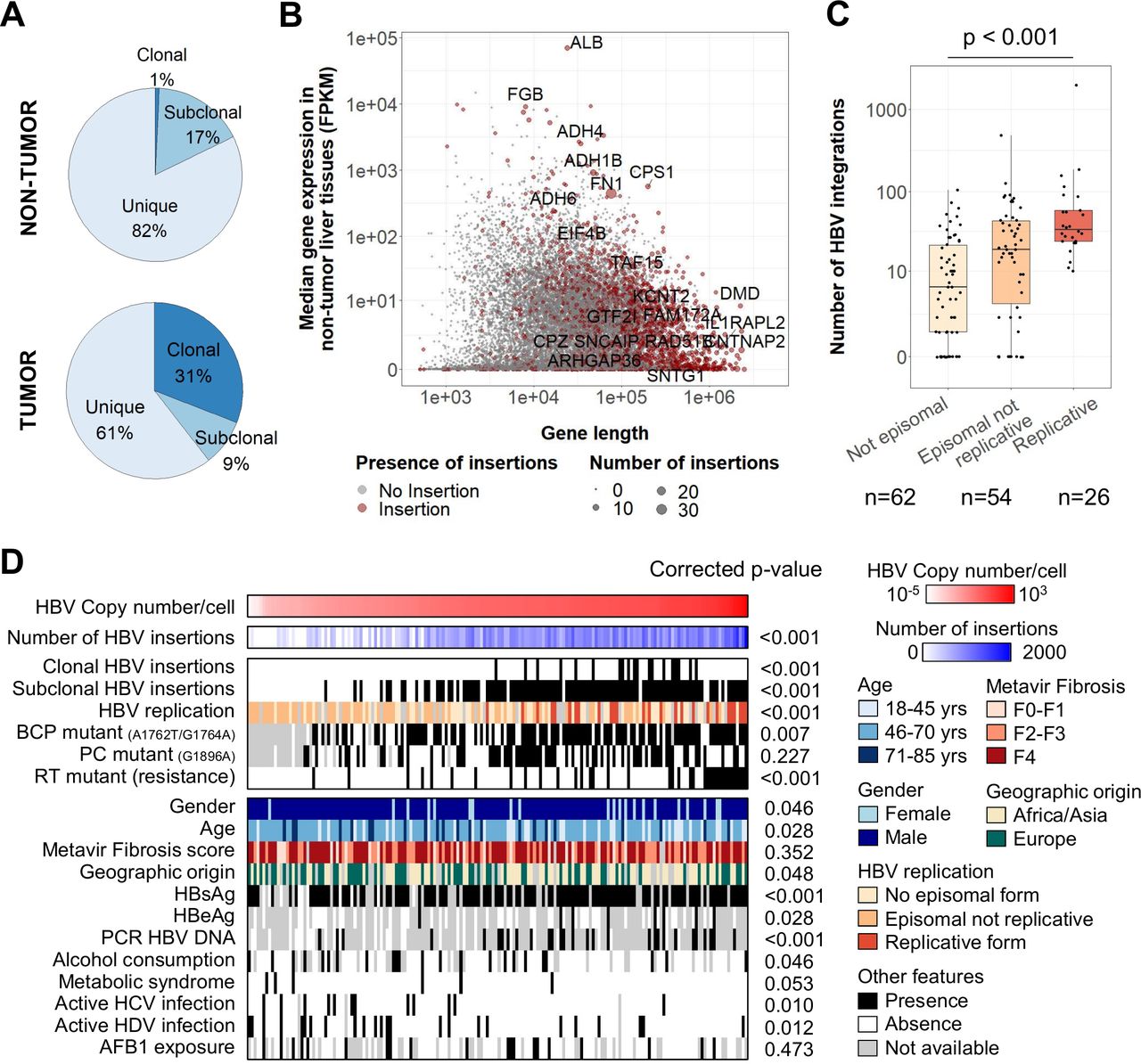
HBV integrations in non-tumour tissues are associated with viral replication and more frequent in large highly expressed genes. (A) Repartition of clonality between all HBV integration events detected in viral capture (n=8809). (B) Coding genes with or without HBV integrations according to the gene length and the median gene expression in non-tumour liver tissues. Genes with recurrent clonal or subclonal HBV integrations are annotated. (C) Number of HBV integrations identified in non-tumour samples (n=142) according to the presence of episomal HBV DNA and replicative HBV DNA (Jonckheere’s trend test). (D) Correlations between HBV copy number/cell in 170 non-tumour liver tissues assessed by viral capture and clinical or molecular features. Positivity for hepatitis B surface antigen (HBsAg), hepatitis B e antigen (HBeAg), HBV DNA (by PCR) in the patients’ serum and duration of antiviral treatments were obtained from clinical data. Wilcoxon signed-rank, Kruskal-Wallis or Pearson’s correlation statistical tests were applied with respect to the type of variable. P values were adjusted for multiple testing using the Benjamini-Hochberg method (false discovery rate). AFB1, aflatoxin-B1; BCP, basal core promoter; FPKM, fragments per kilobase of exons per million reads; PC, PreCore; RT, reverse transcriptase.
In the non-tumour liver tissues, recurrent clonal/subclonal insertions were identified in 20 genes with the following characteristics: highly expressed (7/20, fragments per kilobase of exons per million reads >100, p<0.001) or very large genes (11/20, >100 kb, p<0.001), such as FN1 (14 cases), CPS1 (4 cases), KCNT2 or ADH1B (3 cases), ADH4 or ADH6 or ALB (2 cases, figure 1B). Including all insertion events in FN1, 78 HBV breakpoints were distributed all over the gene, mainly in introns (72/78) and without specific hotspots (online supplemental figure 6A). RNA-seq analysis of 26 HBV integrations in the FN1 locus identified two major types of fusion transcripts: (1) out-of-frame HBx-FN1 transcripts in the same or in the opposite direction (9/26) and (2) in-frame HBs-FN1 transcripts generated by a cryptic splice site at position 458 in the S ORF (11/26; online supplemental figure 6A). Overall, the diversity of the fusion transcripts HBs-FN1 starting at different exons of FN1 and the global lack of overexpression of the genes targeted by HBV integrations indicated that most of the clonal/subclonal insertions in non-tumour liver tissues did not argue for a functional effect (online supplemental figure 6B). Interestingly, clonal insertions suggesting a local proliferation of non-neoplastic hepatocytes were observed in 11% of the 170 non-tumour samples, all in fibrotic livers (F2-F4), with a decreased inflammatory response and decreased nuclear factor-κB/tumour necrosis factor signalling, detected by RNA-seq of a subset of non-tumour tissues (online supplemental figure 6C).
In 170 analysed non-tumour liver tissues, high HBV copy number per cell was significantly associated with female gender, young age, African or Asian geographic origin and positivity for hepatitis B surface antigen (HBsAg), hepatitis B e antigen (HBeAg) and HBV DNA in the serum. In contrast, presence of cofactors of chronic liver disease such as active HCV or HDV infection, alcohol consumption or metabolic syndrome was associated with lower copy number of HBV and less HBV integrations. Only 18% of the samples contained replicative HBV, they were enriched in genotype A and showed a higher number of integrations, underlying that HBV replication and integration were linked processes (figure 1C,D; online supplemental figure 4). Also, samples with high HBV copy number showed frequent mutations of the basal core promoter (BCP; A1762T/G1764A) or of the RT region: mutations known to favour viral replication32 (figure 1D).
Complex HBV integrations and viral genome rearrangements are frequent in HCC
In 88% of 177 analysed HBV-related HCC, we identified HBV integrations in human genome totalising 2199 breakpoints at HBV/human junctions, and 31% of them corresponded to clonal events (figure 1A; figure 2A). A vast majority (82%) of tumours harboured at least one clonal HBV integration, compared with only 11% in non-tumour liver tissues (p<0.001, figure 2B). Overall, the number of HBV integrations was highly correlated with the HBV copy number per cell (figure 2C). However, whereas tumours showed a higher HBV copy number per cell, they harboured a lower number of HBV breakpoints per sample than in their corresponding non-tumour tissues (mean 12 vs 39, p<0.001) (figure 2D; online supplemental figure 7). This may reflect the high diversity of unique HBV integrations in a large number of hepatocytes during the infection, and the decrease in diversity induced by clonal expansion of transformed cells. In both types of tissues, the most frequent hotspot of HBV integration was observed in the C-terminal region of the HBx gene around the DR1 sequence (1817–1836) corresponding to the ends of the double-strand linear DNA form of HBV (figure 2E). In tumours, only a part of the HBV genome was detected, corresponding to integrated HBV DNA with frequent truncation of the HBx gene (online supplemental figure 8A). In addition, HBV genomes in tumours contained a higher number of structural variants within the viral sequence (deletions, duplications, inversions; figure 2F) and only 3% of these structural variants in tumours were observed in the adjacent liver tissues (17/514). Finally, the localisation and orientation of HBV/human junctions in the human genome showed frequent chromosome rearrangements in tumours, suggesting a more complex integration process or postintegration structural modifications than in non-tumour liver tissues (online supplemental figure 8B).
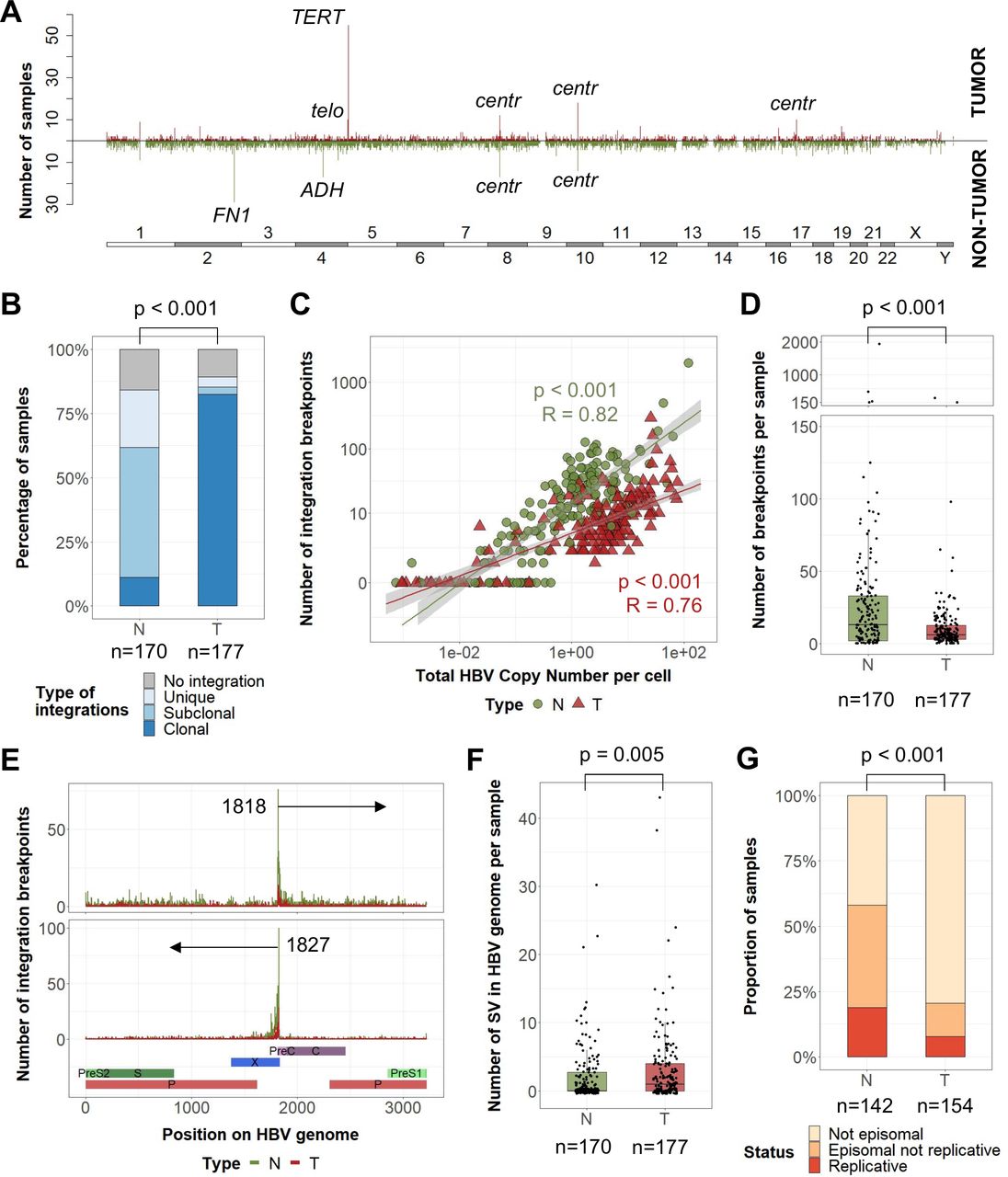
Tumours and non-tumour tissues have different HBV integration profiles and structures of HBV sequences. (A) Pan-genomic view of genomic locations of all HBV integration breakpoints in tumours (up) or non-tumour tissues (down) in 347 HBV-positive samples (non-tumour liver, n=170, and tumour samples, n=177). A line corresponds to a 1M-bin region. (B) Proportion of non-tumour tissues and tumours harbouring only unique integrations, at least one subclonal integration or at least one clonal integration (χ2 test). (C) Correlation between the HBV copy number per cell and the number of HBV integration breakpoints (Pearson’s correlation). (D) Number of HBV integration breakpoints per sample (Wilcoxon signed-rank test). (E) Localisation of HBV integration breakpoints along the HBV genome in tumours and non-tumour samples according to the orientation of the integrated sequence. (F) Number of structural variants in HBV genome per sample (Wilcoxon signed-rank test). (G) Proportion of non-tumour and tumour samples containing replicative HBV DNA, episomal non-replicative HBV DNA or no HBV episomal form (χ2 test). centr, centromeric; SV, structural variant; telo, telomeric.
HBV replication was less frequent in tumours (7%) than in non-tumours (18%, p<0.001) (figure 2G). In addition, analysis of HBV transcripts revealed that even in tumours containing an episomal form of the virus, truncated HBx transcripts derived from integrated HBV DNA were predominant over complete transcripts, suggesting that episomal forms in tumours account for only a small proportion of HBV DNA (online supplemental figure 8C,D). Moreover, the majority of tumours with clonal HBV integrations showed a high mRNA expression level in the S region (supplementary figure 8E). Finally, by analysing the variant allele frequency of the HBV variants in the viral genome, the same major genotype was identified in the tumours and their corresponding non-tumour liver tissues in 138 out of 140 cases. Surprisingly, we observed a negative selection of HBV variants affecting HBeAg production (A1762T and G1764A BCP variants, G1896A PC variant) and antiviral resistance-associated mutations (RT region) in the tumours, suggesting that emergence of variants in HBV sequence aimed to improve the virus fitness but did not give specific advantage for tumour development (online supplemental figure 8F). Overall, these results suggest that HBV integrations in non-tumour and tumour liver tissues may reflect different viral dynamics and selection processes not directly correlated.
Human chromosome rearrangements were recurrently delimited by HBV integrations
Among 504 clonal integrations identified from the capture, WES or WGS sequencing of 121 HCC, 179 events (36%) precisely matched boundaries of chromosome copy number alterations (CNA) in the human genome, showing a direct relationship between viral insertion events and chromosome structural rearrangements (figure 3A). CNA-associated integrations were clonally selected, suggesting that these specific alterations are positively selected and constitute cancer driver events (figure 3A). Indeed, three major types of CNA bordered by HBV integrations were observed in >10 samples: (1) large deletions of the chromosome 17p including TP53 (15 tumours) and frequently associated with centromeric insertions (8/15), (2) focal gains of TERT at 5p (14 tumours) and (3) large gains at chromosome 8q including MYC (13 tumours) and related to centromeric insertions (4/13) (figure 3B; see examples #1733T and #1597T in figure 3C,D).

HBV integrations induce chromosomal rearrangements and distant driver oncogenic alterations. (A) Proportion of HBV integration breakpoints (n=1436) associated with a copy number alteration (CNA) in 121 HBV-positive tumours (χ2 test). (B) Pan-genomic view of the number of HBV integration breakpoints according to their association with a CNA, split by chromosome arms. The three genomic regions containing the higher number of HBV integrations associated with CNA are annotated. Fisher’s exact tests were performed to compare the number of integrations with or without CNA and p-values were adjusted for multiple testing. (C) Translocation-like event in tumour #1733T: HBV integration is associated with a focal gain on chr5p and a large deletion on chr17p. (D) Duplication-inverted-like event in tumour #1597T, reconstructed with long-read sequencing: HBV integration is associated with a focal amplification including TERT.
To investigate how integrations and chromosome alterations were associated, we analysed three selected cases with long-read sequencing (PacBio) and Bionano mapping to generate genomic assemblies and investigate rearrangements at a larger scale. In tumour #1151T, we identified two different HBV integrations on chromosome 8q that together with a whole duplication of the human genome resulted in a large eight copies amplification of 57 Mb that included MYC oncogene. We reconstructed the two distinct integration events: (1) HBV integration resulted in an inter-chromosome t(8q21;17p11) translocation inducing a large 17p deletion and (2) HBV integration located in the 8q centromeric region bordering a duplication-inverted-like event potentially reflecting an isochromosome iso(8q) (online supplemental figure 9A). In tumour #1597T (figure 3D), we identified a TERT amplification at eight copies associated with multiple structural variants including two HBV integrations located within chromosome 1q (online supplemental figure 9B).
Surprisingly, even if HCC with integration-associated rearrangements had a significantly higher number of both HBV integrations and CNA in their genome, these two features were not correlated (online supplemental figure 10A), meaning that only a subset of the viral integrations were associated with chromosome rearrangements, in particular integrations located at centromeric or telomeric regions. Interestingly, centromeric integrations were enriched in young patients with African origin (online supplemental figure 10B); telomeric HBV integrations usually occurred directly or in proximity to telomere repeats (eg, #4229T and #4268T in online supplemental figure 10C) and were enriched in tumours with HBV replication and a high number of CNA (online supplemental figure 10A). As shown in tumours #1994T using Bionano sequencing, telomeric HBV integration can support the translocation of a large chromosome region at the extremity of another chromosome (online supplemental figure 10D). Overall, 41% of clonal HBV integrations identified in tumours were involved in large rearrangements of the human genome and 48% of them were associated with CNA. Functionally, integration-associated rearrangements frequently altered a cancer driver gene, either at distance when associated with recurrent TP53 or MYC alterations, either locally with TERT alterations.
HBV genomes can integrate in cancer driver genes with cis-activating consequences
In tumours, HBV integrations followed a different distribution along the human genome when compared with non-tumour tissues and HBV copy number/cell was associated with the number of clonal events (figure 2A; figure 4A). These clonal insertions were enriched around cancer driver genes, suggesting a strong functional selection of cells containing such events (online supplemental figure 11A). However, among the 229 genes harbouring clonal integrations in their vicinity (±25 kb), only three genes were recurrently found in more than two HCC: TERT (n=48), CCNE1 (n=4), KMT2B (n=3). Four other genes had HBV integrations in two samples: AHRR, NRG1, TRIM16L and ST18 (figure 2A; online supplemental table 4).35
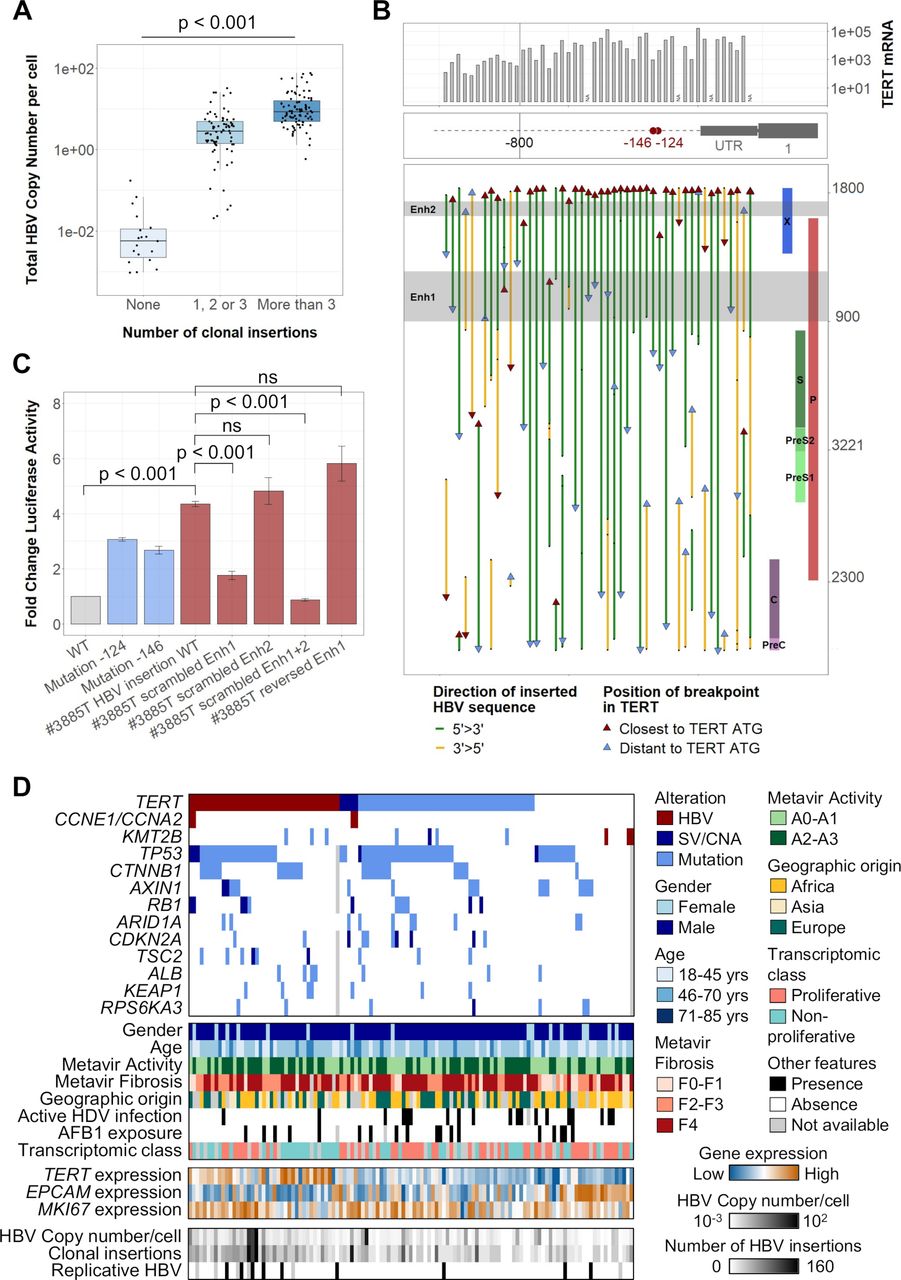
HBV integrations in the TERT promoter induce a strong activation of TERT promoting hepatocellular carcinoma (HCC) development. (A) HBV copy number/cell in 177 tumours from the capture series according to their number of clonal HBV integration breakpoints (Jonckheere’s trend test). (B) Integrated HBV sequences located in the TERT promoter in 48 tumours harbouring clonal HBV integrations at this locus. The position of the breakpoints, of the viral enhancer regions and the orientation of the integrated sequences are annotated along the HBV genome. mRNA expression (-ddCT) from qRT-PCR data are represented above. (C) The impact of HBV integration in the TERT promoter was evaluated using promoter luciferase assays in Huh7 liver cell lines. Constructs of TERT promoter containing different HBV-integrated sequences with or without scrambled enhancer regions were compared with the WT promoter and to the TERT promoter with mutations at the −124 or −146 hotspots. Error bars correspond to SD of three independent transfections for each plasmid (Student’s t-test). (D) Molecular profile of 121 HBV-positive tumours with alterations in 14 HCC-associated genes (HBV integration, SV/CNA or mutation), according to clinical and other molecular features. mRNA expression of TERT, EPCAM and MKI67 were obtained from RNA-seq. AFB1, aflatoxin B1; Enh, enhancer; CNA, copy number alteration; SV, structural variant; WT, wild type; ns, not significant.
All HBV integrations at the TERT locus were located in the promoter region or in the 5’ untranslated region (figure 4B; online supplemental figure 11B). Most of the promoter integrations were in the same 5’>3’ orientation and contained both viral enhancer regions (35 out of 48). Increased mRNA TERT expression was higher for viral integrations situated closer to the TERT ATG (<800 bp) but was independent of the orientation of the inserted sequence (figure 4B, online supplemental figure 11C). Moreover, HBV insertions induced a higher overexpression than promoter mutations at the −124 and −146 hotspots and structural variants inducing CNA without viral insertions (online supplemental figure 11D). Modelling TERT promoter integration identified in tumour #3885T in luciferase reporter vector (online supplemental figure 11E,F), we confirmed that TERT activation was caused by the two viral enhancers with enhancer 1 providing a stronger activation compared with enhancer 2 (figure 4C). HBV insertions in TERT promoter were exclusive from mutations or structural variations of the same region, and more frequent in tumours with a higher HBV copy number per cell (p<0.001) driven by more HBV clonal insertions (p<0.001) and more HBV replication (p=0.01; figure 4D).
We previously described two tumours with HBV insertions in CCNE1 or CCNA2 genes as part of a homogenous group of HCC (CCN-HCC) characterised by a signature of specific chromosome rearrangements (templated-insertion cycles) induced by stress replication.19 We identified three additional HBV integrations in the promoter of CCNE1 using viral capture and RNA-seq showed a significant overexpression of normal CCNE1 transcripts (online supplemental figure 12A,B). Three KMT2B integrations were also identified between exons 3–6, which increased the mRNA expression and altered the transcript structure through alternative splicing or intron retention (online supplemental figure 12A,C). Overall, TERT promoter integrations were enriched in non-proliferative tumours (p=0.007; figure 4D). In contrast, tumours harbouring HBV integrations in CCNE1, CCNA2 or KMT2B were more frequently identified in patients without cirrhosis (5/8) with African or Asian origin (6/8), belonging to the proliferative class of HCC, and associated with G1 (for KMT2B-integrated HCC) or G3 (for CCN-HCC) transcriptomic subgroup. Interestingly, CCN-HCC showed TERT promoter alterations whereas KMT2B-integrated HCC did not show TERT promoter or any other driver genes alterations, suggesting different processes of carcinogenesis (figure 4D).
Integrated analysis between clinical features, HBV and human genomic alterations
To integrate all the genomic alterations identified in the tumours, we analysed a subgroup of 130 HBV-positive HCC compared with 135 HBV-negative tumours sequenced in WGS or WES and RNA-seq19 34 (see online supplemental table 1 for description of the series). Patients with HBV HCC were younger, enriched in African or Asian origin, and they showed specific cofactors: HDV infection or carcinogen exposure signatures (figure 5A, first column). Indeed, the two sporadic mutational signatures 22 and 24, characteristics of acid aristolochic (AA) and aflatoxin B1 (AFB1) exposure respectively, were more frequent within HBV-positive patients. Transcriptomic and genomic analysis revealed an association between HBV infection and G3 transcriptomic class, more frequent TP53 mutations and less CTNNB1 mutations in tumours.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
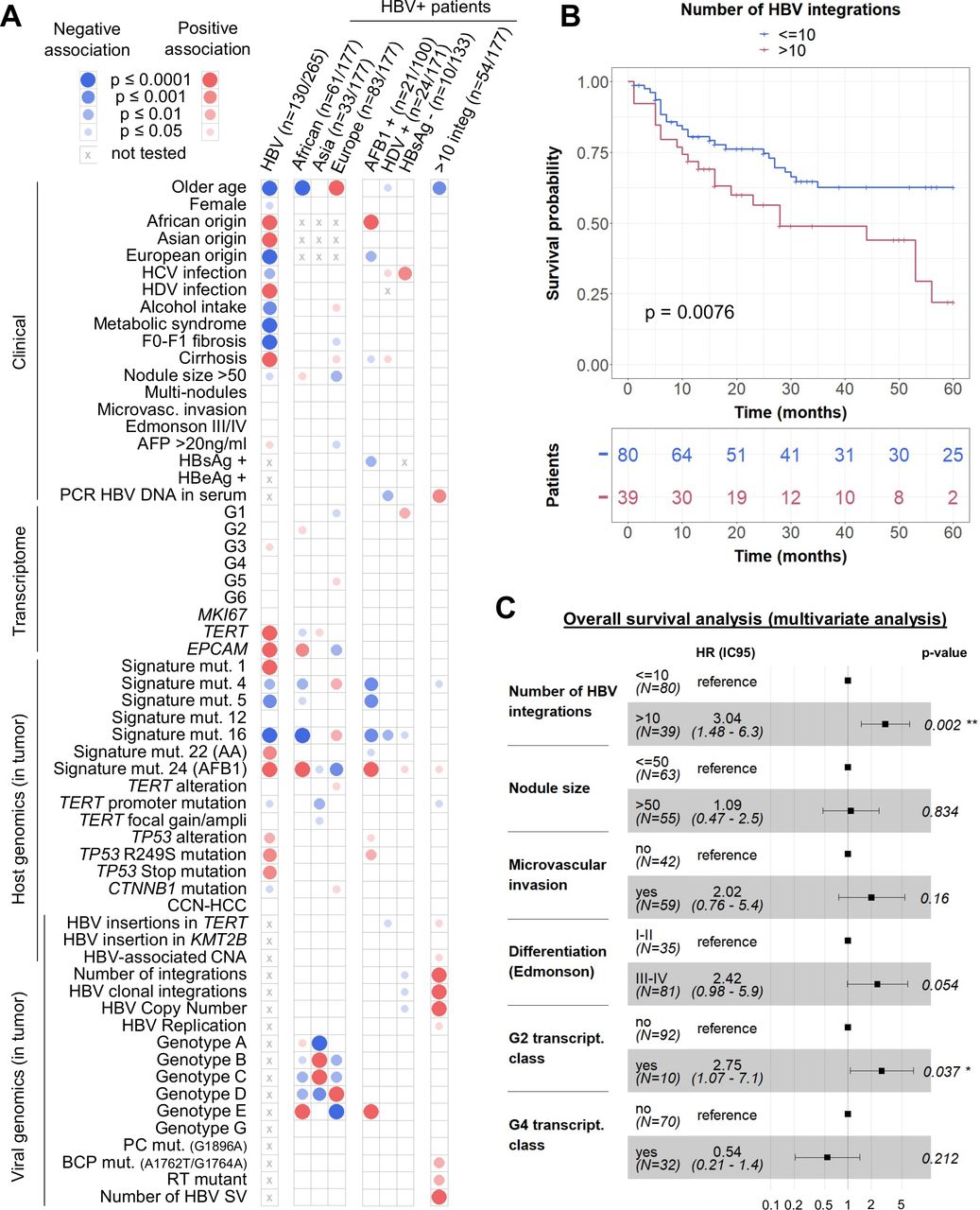
Integrative analysis reveals that a high number of HBV integrations is associated with poor survival of patients. (A) Mosaic plot: association of HBV infection with patient, tumour and viral characteristics in a series of 265 HCC (next-generation sequencing series) and association of geographic origin, aflatoxin B1 exposure, HDV infection, hepatitis B surface antigen (HBsAg) negativity or number of HBV integrations with patient, tumour and viral characteristics in a series of 177 HBV-positive HCC (capture series). Blue and red circles indicate negative and positive associations, respectively. Colour intensities represent different levels of statistical significance. Statistical analysis was performed using χ2 test, Wilcoxon signed-rank test or Pearson’s correlation with respect to the type of variable. (B) Kaplan-Meier curves for 5-year overall survival from 119 patients after curative R0 resection. (C) Multivariate Cox regression model for overall survival analysis. AA, aristolochic acid. *p<0.05; **p<0.01.
In addition, in the series of 177 HBV-positive patients analysed in capture, 60 had an African origin; they were younger with frequent AFB1-mutational signature in HCC characterised by a high expression of progenitor markers such as EPCAM (figure 5A). No specific viral feature identified in tumours were significantly associated with the geographic origin of patients, except for the HBV genotype. Among patients with cofactors, whereas AFB1 exposure was strongly associated with TP53 R249S mutation, HBV/HDV-related HCC harboured less HBV-associated alterations in TERT promoter or any other driver genes. Finally, 10 patients had a negative HBsAg serology, they were negative for HBV DNA in the serum but positive in the liver; their tumours showed a tendency for low numbers of HBV copies per cell and of HBV integrations and these HCC were enriched in G1 transcriptomic group. Of note, two of these HCCs had a clonal HBV integration in the TERT promoter (#4229T and #4265T).
Tumours with a high number of HBV integrations were associated with a poor prognosis, independently from other features such as tumour size, microvascular invasion, differentiation status and transcriptomic groups (figure 5B–5C; online supplemental table 5). This association between the high number of integrations in tumours and poor survival was independent from the other features of HBV infection. Interestingly, these patients were significantly younger and with tumours harbouring a high proportion of HBV integrations affecting cancer driver genes such as TERT (p=0.02) through an insertion in the promoter, TP53 (p<0.001) or MYC (p=0.03) through HBV-associated CNA (figure 5A).
Discussion
This study presents an integrative analysis of HBV genomes in non-tumour and tumour liver tissues of a large cohort of patients mainly coming from Europe and Africa. On one hand, HBV integrations in non-tumour tissues reflect an active viral replication and the expansion of hepatocytes in liver tissues with a lower expression of inflammation-associated genes. On the other hand, HBV-integrated sequences in tumours highlight a functional selection of cells harbouring HBV-associated structural rearrangements or insertional mutagenesis as driver alterations.
Our method of identification of HBV integrations based on viral capture coupled with WGS and WES on frozen tissues enabled to characterise integrations based on their clonality defining precisely the detected events as ‘clonal’, ‘subclonal’ or ‘unique’ with a quantitative method. In contrast with previous studies and in accordance with the clonal cell expansion during carcinogenesis with selection of functional genomic alterations, here, more clonal HBV insertions were identified in tumours compared with the non-tumour liver tissues.14 15 21 36 In the same line, we observed an important enrichment of HBV integrations in simple repeats, underlying the importance of precisely filtering insertional events detected in viral capture to remove duplicates. Therefore, our study based on a highly confident description of HBV integrations provides a comprehensive view of the integration process in liver tissues.
In non-tumour liver, we confirmed that integration events occur more frequently in regions of the human genome with open chromatin,21 37 resulting in part from microhomology-mediated end joining.15 38 39 More importantly, viral replication is related to the number of integrations reflecting either an increased number of infected cells, either a multisteps integration process within a single cell. Both hypotheses are probably occurring concurrently in the liver. A recent in vitro study13 has shown that the integration rate within the first week after infection is not altered by replication suppression, suggesting that the majority of integration events occurs at primo-infection. However, all NGS-based studies, including ours, identified the presence of several clonal integrations in some cellular clones. Therefore, several HBV integrations must accumulate at a low rate in a single cell, promoted by active replication.
The number of HBV integrations may increase over time within hepatocytes in normal liver and as in vitro and in vivo studies have shown that HBV integrations occur early after infection,13 40 41 a majority of events could be considered as passenger. Nevertheless, the presence of clonal integrations (ie, present in all cells of a given frozen sample) reflects the expansion of hepatocytes and this process appears to be different in non-tumour tissues and in tumours. In non-tumour tissues, it might be explained by a selective pressure induced by chronic inflammation and the emergence of cirrhotic nodules in the liver but this process seems to be independent from integrations and viral replication as it has already been suggested.42–44 Indeed, in our study, clonal integrations in these tissues do not argue for functional consequences but are located in regions recurrently targeted by integrations (large, highly expressed genes), and are not associated with viral replication. Yet, as non-tumour tissues harbouring clonal integrations showed a downregulation of genes involved in inflammatory response, it suggests that some hepatocytes with a selective advantage to escape immune antiviral response might undergo active proliferation and obtain a protective profile against malignant transformation.
Although the majority of HBV integrations are passenger events and do not have any functional consequences, some can be driver and promote HCC initiation in cirrhotic or non-cirrhotic livers. While integrations had already been associated with CNA in previous studies,14 45 46 we identified for the first time recurrent structural rearrangements associated with viral insertions in HCC. Through integrations often located around centromeric or telomeric regions, HBV is involved in large complex structural rearrangements frequently inducing gains of chr8q (including MYC locus) and chr5p (including TERT locus) and losses of chr17p (including TP53 locus). Thus, in tumours harbouring integration-associated rearrangements, HBV integrations are associated with alterations of cancer-driver genes located at distance. As HBV DNA integration occurs at double-strand breaks,23 47 the presence of HBV-integrated sequences delimitating CNA may result from opportunistic mechanism. However, our data showed no correlation between the numbers of HBV integrations and of CNA in tumours; it highlighted the role of specific alterations due to integration-associated rearrangements in promoting selection of hepatocytes and HCC development.
HBV integrations may also drive carcinogenesis by altering the closest gene from the viral-integrated sequence through insertional mutagenesis. Our study confirmed that the TERT promoter is the main HBV integration hotspot in HCC, as one-third of HBV-related HCC harboured clonal integration at this hotspot. In these tumours, strong activation of TERT was due to the presence of the viral enhancers as it had already been reported,16 48 at proximity of the ATG start codon and in an orientation-independent manner. In addition, HBV integrations in CCNA2 or CCNE1 genes have already been investigated in a previous study of our group.19 HBV-integrated sequence is either altering directly the structure of the protein (for CCNA2) or inducing a strong overexpression due to its enhancer sequences (for CCNE1). Alterations of one of these two genes induce strong proliferation, replicative stress and a signature of rearrangements directly promoting the development of a tumour belonging to a specific subgroup of HCC developed on non-cirrhotic livers (CCN-HCC). Finally, even if further investigations are needed to fully understand the functional consequences, tumours with HBV integrations in KMT2B are also mainly developing on non-cirrhotic liver with a high expression of progenitor markers, suggesting another strong direct mechanism induced by HBV-integrated sequence.
Overall, this study underlines the complexity of the interplay between HBV integrations, HBV replication and chromosomal instability in hepatocarcinogenesis, in addition to other cofactors. On one side, aflatoxin B1 exposure, mainly present in Africa, may initiate directly HBV-related HCC development through TP53 R249S mutation.49 On the other side, HDV infection limits HBV replication50 and viral integrations but accelerates chronic inflammation and fibrosis in young patients. Importantly, our series showed that the number of HBV integrations in tumours is the only marker within viral features associated with poor prognostic.
In conclusion, structural rearrangements associated with HBV integrations are a mechanism to drive carcinogenesis by altering cancer-driver genes at distance, different from cis-activating insertional mutagenesis. This underlines the heterogeneity of HBV-related HCC and the importance of molecular characterisation to identify new specific therapeutic opportunities.
Data availability statement
Data are available in a public, open access repository. The sequencing data of the LICA-FR cohort reported in this paper have been deposited to the European Genome-phenome Archive (EGA) database (RNA-seq fastq files accessions (EGAS00001001284), (EGAS00001002879), (EGAS00001003310), (EGAS00001003837) and (EGAS00001004629); WES bam files accessions (EGAS00001000217), (EGAS00001001002), (EGAS00001003063), (EGAS00001003130), (EGAS00001003837) and (EGAS00001004629); WGS bam files accessions (EGAS00001000706), (EGAS00001002408), (EGAS00001002888), (EGAS00001003063), (EGAS00001003837) and (EGAS00001004629), through the International Cancer Genome Consortium (ICGC) data access committee. Data are available for reuse and can be consulted at the following address: https://ega-archive.org/studies/EGASxxx with access permission from ICGC Data Access Compliance Office.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the institutional review board (IRB) committees.
Acknowledgments
The authors would like to thank Alain Nicolas, Sylvain Baulande and Sonia Lameiras at Institut Curie for their help in analysing PacBio results and setting up viral capture. The authors would like to thank Quentin Bayard and Karl Hong for their help in analysing Bionano sequencing results. The authors would also like to thank Gabrielle Couchy, Iadh Mami, Bénédicte Noblet, Massih Ningarhari, Jill Pilet and Jie Yang for their help in molecular biology experiments.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Zucmanrossi
Contributors JZ-R conceived and directed the research. CP and JZ-R designed the study and wrote the manuscript. CP and TLB performed the experiments. CP, SI, TZH, SC, EL and JZ-R analysed and interpreted the data. JC, VP, J-FB, J-CN and GA provided essential biological resources and collected clinical data. All authors approved the final manuscript and contributed to critical revisions to its intellectual context.
Funding This work was supported by ANRS (French national agency for research on AIDS and viral hepatitis). The group is supported by the Ligue Nationale contre le Cancer (Equipe Labellisée), Labex OncoImmunology (investissement d’avenir), grant IREB, Coup d’Elan de la Fondation Bettencourt-Shueller, the SIRIC CARPEM, FRM prix Rosen, Ligue Contre le Cancer Comité de Paris (prix René et André Duquesne) and Fondation Mérieux. CP was supported by a fellowship from ANRS (French national agency for research on AIDS and viral hepatitis). TLB was supported by an “Attractivite IDEX" fellowship from IUH, TZH by a fellowship from Cancéropole Ile de France and Fondation d'Entreprise Bristol-Myers Squibb pour la Recherche en Immuno-Oncologie, and SC by CARPEM and the Labex OncoImmunology.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.