Article Text

Abstract
Objective Molecular taxonomy of tumours is the foundation of personalised medicine and is becoming of paramount importance for therapeutic purposes. Four transcriptomics-based classification systems of pancreatic ductal adenocarcinoma (PDAC) exist, which consistently identified a subtype of highly aggressive PDACs with basal-like features, including ΔNp63 expression and loss of the epithelial master regulator GATA6. We investigated the precise molecular events driving PDAC progression and the emergence of the basal programme.
Design We combined the analysis of patient-derived transcriptomics datasets and tissue samples with mechanistic experiments using a novel dual-recombinase mouse model for Gata6 deletion at late stages of KRasG12D-driven pancreatic tumorigenesis (Gata6LateKO).
Results This comprehensive human-to-mouse approach showed that GATA6 loss is necessary, but not sufficient, for the expression of ΔNp63 and the basal programme in patients and in mice. The concomitant loss of HNF1A and HNF4A, likely through epigenetic silencing, is required for the full phenotype switch. Moreover, Gata6 deletion in mice dramatically increased the metastatic rate, with a propensity for lung metastases. Through RNA-Seq analysis of primary cells isolated from mouse tumours, we show that Gata6 inhibits tumour cell plasticity and immune evasion, consistent with patient-derived data, suggesting that GATA6 works as a barrier for acquiring the fully developed basal and metastatic phenotype.
Conclusions Our work provides both a mechanistic molecular link between the basal phenotype and metastasis and a valuable preclinical tool to investigate the most aggressive subtype of PDAC. These data, therefore, are important for understanding the pathobiological features underlying the heterogeneity of pancreatic cancer in both mice and human.
- pancreatic cancer
- molecular mechanisms
- epithelial differentiation
Data availability statement
RNA-Seq accession number: PRJEB37700.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Multiple transcriptomics-based studies have identified a basal-like subtype of pancreatic ductal adenocarcinoma (PDAC) with especially poor prognosis.
Loss of GATA6 in PDAC cells is associated with altered differentiation, including ectopic expression of basal markers such as KRT14.
Aberrant expression of the ΔNp63 transcription factor can drive the expression of the basal transcriptional programme.
What are the new findings?
Loss of GATA6 expression is necessary but not sufficient for the expression of ΔNp63 and the basal phenotype.
Concomitant silencing of HNF4A and HNF1A, possibly through epigenetic mechanisms, is required for the full-blown phenotype.
Gata6 deletion in established murine tumours favours the basal and metastatic phenotype, with a lung tropism, in a next-generation model of KRasG12D-driven PDAC.
Loss of GATA6 expression is associated with features of immune escape in mouse and human PDAC cells.
How might it impact on clinical practice in the foreseeable future?
The combined analysis of GATA6, HNFs and TP63 expression in patient-derived samples will provide a more precise classification of PDAC.
Restoration of the classical PDAC phenotype may not only reduce metastatic potential but also increase immune recognition of tumour cells.
Introduction
Molecular taxonomy of tumours harbours great potential for the development of personalised medicine. In the case of pancreatic ductal adenocarcinoma (PDAC), molecular classification has revealed the existence of multiple subtypes with distinctive biological and clinical behaviour and likely specific vulnerabilities. Four PDAC taxonomies were proposed until now, differing in the number of subgroups and nomenclature.1–4 All classifications identified a PDAC subtype with loss of cell identity features, associated with significantly worse patient survival. This subtype, called ‘quasi-mesenchymal’,2 ‘basal-like’,3 ‘squamous’,1 ‘pure basal’4 or ‘basal A/B’5 showed rather homogeneous gene expression profiles across classifications.6 We will refer to this PDAC subtype as ‘basal’. A better understanding of the molecular drivers of this aggressive PDAC subtype would improve patients’ management in the context of an almost invariably lethal malignancy.
The transcription factor GATA6, a crucial regulator of acinar cell differentiation7 and suppressor of KRasG12V-driven tumorigenesis in mice,8 is highly expressed in the classical subtype2 and silenced through promoter methylation in squamous tumours.1 We confirmed that GATA6 was lost in a subset of PDACs, in association with a basal-like differentiation, and shed light on the underlying mechanism.9 GATA6 silencing resulted in epithelial-to-epithelial transition (whereby a simple epithelium expresses markers of a stratified epithelium) and epithelial-to-mesenchymal transition (EMT) while its overexpression induced mesenchymal-to-epithelial transition (MET), supporting its central role in determining the phenotype of PDACs.8 9 Consistently, loss of GATA6—as a single biomarker—identified basal tumours as efficiently as the corresponding gene signature in a cohort of patients with metastatic PDAC.10
Recent publications indicated that the basal phenotype is driven by broad epigenomic reprogramming, especially at superenhancers, controlled by ΔNp63,11–13 the shorter isoform of the TP63 transcription factor marking the basal layer of stratified epithelia. Interestingly, GATA6 itself was identified as being controlled by a superenhancer lost in basal patient-derived cells14 and EZH2-driven epigenetic silencing of GATA6 is involved in PDAC de-differentiation,15 suggesting that loss of GATA6 might be embedded in the basal programme rather than driving it.
Here, we aimed at elucidating whether loss of GATA6 is the cause or the consequence of the basal phenotype in PDAC. By combining the analysis of patient-derived samples and transcriptomics datasets, in vitro experiments with PDAC cells, and a next-generation KRasG12D -driven mouse PDAC model where Gata6 was deleted at late stages of tumorigenesis, we show that GATA6 loss is necessary, but not sufficient, for the appearance of a basal programme in PDAC. Concomitant downregulation of HNF1A and HNF4A is required for the full phenotypic switch. Additionally, Gata6 loss in preneoplastic lesions (PanINs) favoured the development of metastases in mice, possibly by promoting plasticity and immune escape of tumour cells. We demonstrate that an epithelial/progenitor transcriptional network acts as a barrier against tumour progression, and provide a molecular link between the basal gene programme in vivo and the metastatic spread in PDAC.
Methods and material
All relevant methods and materials can be found in online supplemental file.
Supplemental material
Results
GATA6 loss is necessary for the expression of the basal programme
We analysed five PDAC transcriptomic datasets with molecular classification, which revealed that GATA6 expression was consistently lower in the poorly differentiated subtypes (quasi-mesenchymal2 p=0.008, basal-like3 p=5.54e-10, squamous1 p=1.57e-11, pure-basal4 p<2e-16, basal A/B5 p<0.0001) (figure 1A). Since ΔNp63 was suggested to drive the basal transcriptional programme in PDAC,11 12 we explored its relationship with GATA6 in 4/5 of the datasets (the Collisson was excluded due to low sample size) plus the TCGA PAAD dataset. TP63 expression was negatively correlated with GATA6 expression in 4/5 datasets (figure 1B, online supplemental figure 1A). Additionally, ΔNp63-target genes16 were significantly enriched among those upregulated in GATA6low tumours (bottom quartile) in 3/5 datasets and showed a tendency in the remaining 2 (figure 1C, online supplemental figure 1B), supporting that the ΔNp63-dependent programme is induced when GATA6 is lost. We showed previously that GATA6 loss in PDAC associates with ectopic expression of the basal marker KRT14 in a small collection of patient-derived samples.9 We measured GATA6, TP63 and KRT14 expression with immunohistochemistry (IHC) in an independent larger set of 60 formalin-fixed paraffin-embedded tissues from PDAC resections (figure 1D). GATA6 expression was lost in >10% of tumour cells in 23/60 patients (38.3%). In addition, KRT14 was exclusively expressed in GATA6low tumours (16/23, 69.6% p=1.64e-09) and TP63 expression was detected in 14/23 (60.1%) GATA6low and 10/37 (27%) GATA6high tumours (p=0.014) (online supplemental figure 2A). Of note, the GATA6high/TP63pos tumours only had small foci of TP63-positive cells, which, on more detailed analysis, were found to be located in metaplastic lesions containing GATA6-negative cells in 9/10 cases (online supplemental figure 2B, black arrowhead). Moreover, we compared the frequency of basal phenotypes between the top and bottom GATA6 expression quartiles (GATA6high, GATA6low) in the five PDAC datasets with classification and observed only 2/207 GATA6high/Basal cases (online supplemental figure 1C). These data strongly indicate that GATA6 loss is necessary for the expression of the basal phenotype.
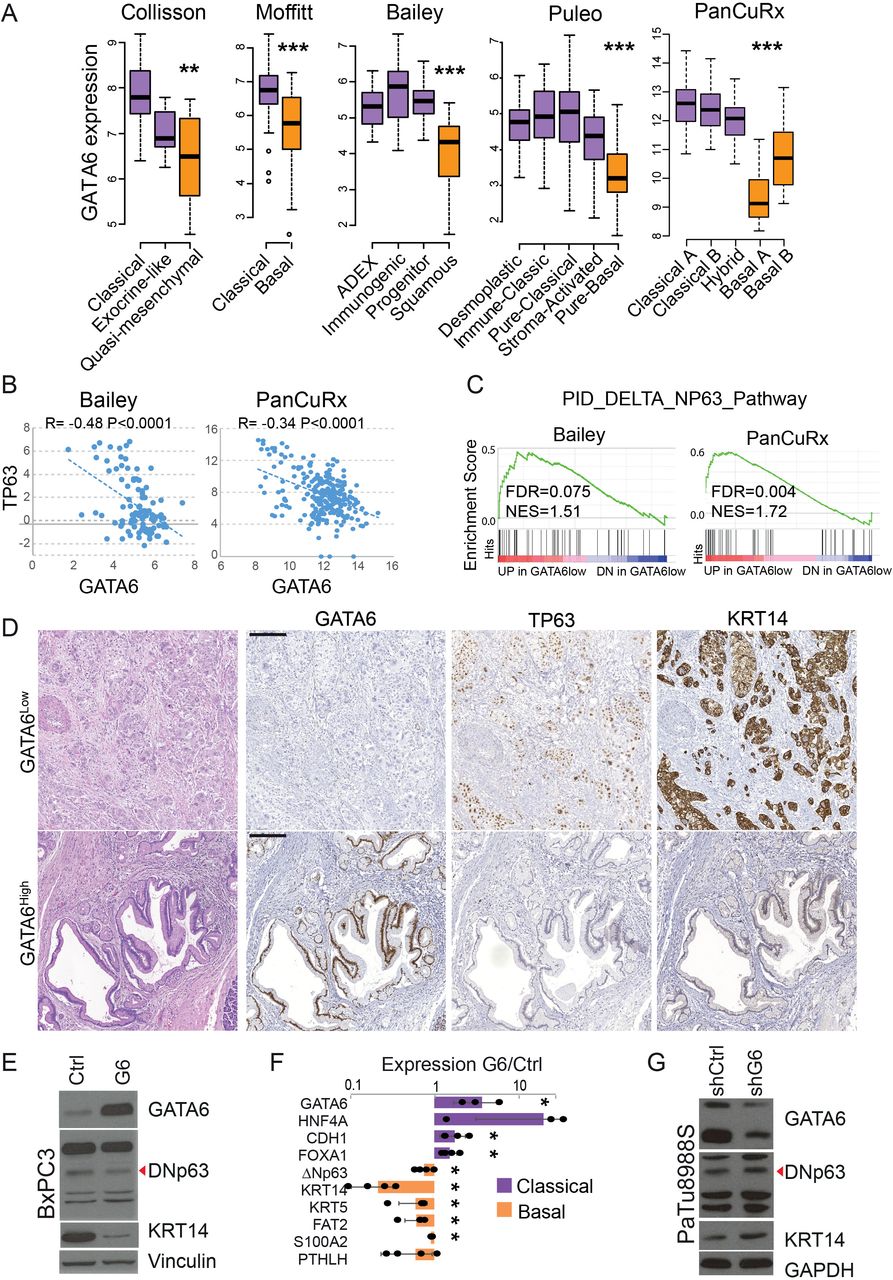
GATA6 loss is necessary for the expression of the basal-like programme. (A) Analysis of GATA6 mRNA expression in pancreatic ductal adenocarcinoma (PDAC) datasets with transcriptomics-based molecular classification. (B) Correlation between TP63 and GATA6 mRNA expression in the indicated PDAC datasets. (C) Enrichment of the gene set ‘ΔNp63 target genes’ among the genes upregulated in GATA6low versus GATA6high tumours in the indicated datasets. (D) Representative images of GATA6high and GATA6low PDAC. H&E and immunohistochemical stainings for TP63, GATA6 and KRT14. Scale bar=200 µm. (E) Expression of GATA6, ΔNp63 (red arrowhead) and KRT14 in control (Ctrl) and GATA6-overexpressing (G6) BxPC3 cells, analysed by western blotting of whole protein lysates. Vinculin was used as loading control. (F) Expression of a set of classical and basal genes in GATA6-overexpressing BxPC3 cells compared with Ctrl cells, measured by RT-qPCR. Results are shown as mean±SD of at least n=3 biological replicates. *p>0.05. (G) Expression of GATA6, ΔNp63 (red arrowhead) and KRT14 in control (shCtrl) and GATA6-silenced (shG6) PaTu8988S cells, analysed by western blotting of whole protein lysates. GAPDH was used as loading control. FRD, false discovery rate. NES, normalised enrichment score.
To understand the hierarchical relationship between GATA6 and ΔNp63 in the regulation of the basal phenotype, we overexpressed GATA6 in BxPC3, a PDAC cell line with high levels of ΔNp63.11 12 GATA6 overexpression led to a 40% reduction of ΔNp63 protein (p=4.15e-04) and 30% reduction of the mRNA (p=0.03) (figure 1E, online supplemental figure 2D). Consistently, using reverse transcription followed by qPCR (RT-qPCR) we observed the upregulation of classical (HNF4A, CDH1, FOXA1) and downregulation of basal markers (ΔNp63, KRT14, KRT5, FAT2, S100A2, PTHLH); KRT14 was strongly reduced both at mRNA (p=6.3e-06) and protein level (p=1.42e-06) (figure 1E,F, online supplemental figure 2D). Intriguingly, we did not observe clear changes in proliferation, migration or Matrigel invasion in vitro (not shown). Conversely, ΔNp63 and KRT14 proteins were slightly induced in PaTu8988S cells (classical) after GATA6 knock-down (online supplemental figure 2E). GATA6 expression in PaTu8988S shG6 cells was similar to the basal level in BxPC3 cells, while GATA6 overexpression in the latter was close to the endogenous expression in PaTu8988S cells (online supplemental figure 2C) indicating that the range of GATA6 expression on experimental manipulation remains within endogenous physiological levels.
A reanalysis of published RNA-Seq data12 showed that TP63 knock-out in BxPC3 cells significantly induced GATA6 expression (adj. p=0.02) while ΔNp63 overexpression in PaTu8988S cells only resulted in a small, not significant, decrease in GATA6 mRNA (online supplemental figure 2F). ChIP-Seq from the same publication showed a TP63 peak downstream of GATA6 transcription start site (not shown), possibly indicating a direct repression. The lack of a GATA6 peak in the vicinity (<10 kb) of the ΔNp63 TSS in PaTu8988S cells,9 suggests indirect regulation. The intersection between TP63 ChIP-Seq peaks in BxPC312 and GATA6 ChIP-Seq peaks in PaTu8988S9 showed limited overlap (0.8% of GATA6 peaks, 10.7% of TP63 peaks), suggesting that the two transcription factors control separate programmes in basal versus classical cells. Interestingly, only 1 out of 3802 GATA6 distal peaks (>50 Mb from a TSS) was located on regions identified as ‘Squamous elements’ by Somerville et al 12 (online supplemental figure 2G). These data indicate that, while important, neither GATA6 loss nor ΔNp63 expression is sufficient to drive a full phenotypic switch in PDAC cells and support the involvement of a cooperative model of transcriptional regulation.
GATA6 cooperates with HNF1A and HNF4A to sustain the classical phenotype
To identify the crucial molecular events downstream of GATA6 loss, we analysed the PanCuRx dataset, including the largest series of all-stages PDAC samples and thus better representing the PDAC patient population than datasets only including resectable tumours. We compared GATA6low/Basal (n=41) versus GATA6low/Classical (n=21) tumours. Gene set enrichment analysis (GSEA) revealed that HNF1A and HNF4A putative target genes were enriched among the upregulated transcripts in GATA6low/Classical tumours (figure 2A). Accordingly, HNF1A and HNF4A mRNAs were significantly higher in GATA6low/Classical tumours, compared with the basal ones (figure 2B and online supplemental figure 3). Similarly, KRT14pos regions of patients’ tumours showed reduced HNF4A protein levels, compared with KRT14neg regions (figure 2C). Of note, loss of GATA6 expression was broader and more pronounced in all samples with KRT14pos regions, compared with HNF4A reduction.

Concomitant loss of HNF1A and HNF4A is required for the expression of the basal phenotype after loss of GATA6. (A) Enrichment plot of the gene sets containing putative HNF1A and HNF4A target genes when comparing basal (low-BAS) and non-basal (low-CLA) GATA6low tumours of the PanCuRx cohort. (B) Expression of HNF1A, HNF4A and GATA6 in the different groups of patients in the PanCuRx dataset. *p<0.05, **p<0.001, ***p<0.0001. (C) Representative images of KRT14 and HNF4A staining in a pancreatic ductal adenocarcinoma (PDAC) sample. Bottom images show KRT14neg/HNF4Ahigh (brown box) and KRT14pos/HNF4low (orange box) regions. Scale bar: 500 µM. (D) Expression of HNF4A and TP63 in AsPC1 and SUIT-2 on HNF4A knock-down. Vinculin was used as loading control. (E) H3K27me3 distribution along the HNF4A locus in patient-derived xenografts-derived cell lines of the three categories. (F) The proposed model: GATA6 is the primary gatekeeper of the classical phenotype; HNF1A and HNF4A can block the full basal programme but, once lost, the ΔNp63-driven basal programme is fully expressed and drives PDAC progression toward metastasis. A negative feedback regulation driven by ΔNp63 might contribute to stabilise the basal phenotype. FDR, false discovery rate. MET, mesenchymal-to-epithelial transition; NES, normalised enrichment score.
The classical/progenitor AsPC1 and SUIT2 cells12 have lower GATA6 expression than PaTu8988S, while HNF1A and especially HNF4A are high, thus they represent a model for GATAlow/Classical tumours (online supplemental figure 2B). HNF4A knock-down was enough to induce expression of ΔNp63 in AsPC1 and in two clones of SUIT2 cells, SUIT2-007 and SUIT2-028, supporting that HNF4A represents a barrier to the basal phenotype downstream of GATA6 (figure 2D).
We previously reported the most comprehensive epigenomics data available for a collection of basal and classical patient-derived xenografts (PDX)-derived cell lines.14 We reprocessed raw data to compare the epigenetic marks over GATA6, HNF1A and HNF4A loci in GATA6low/Basal (n=4), GATA6low/Classical (n=5) and GATA6high/Classical (n=5) cell lines. Notably, while we found a marked accumulation of the heterochromatin marker H3K27me3 on the HNF4A locus in Gata6low/Basal cells, the locus was not epigenetically silenced in Gata6low/Classical ones (figure 2E, online supplemental figure 4A). GATA6 showed a similar pattern but H3K27me3 was predominantly enriched upstream of the TSS (online supplemental figure 4A). HNF1A was not highly marked with H3K27me3 in GATA6low/Basal cells (online supplemental figure 4A).
H3K27ac and H3K4me3 patterns around the TSS of all the three genes were consistent with higher transcription in GATA6low/Classical and Gata6high/Classical cells, that is, enrichment of these two markers of active chromatin was low or absent in GATA6low/Basal, with the exception of 1.037 cells, while it was high in all other cells (online supplemental figure 4B,C). These data suggest that GATA6 and HNFs are epigenetically silenced in basal cells, while classical cells retain HNFs expression even when GATA6 is low. Importantly, although a subset of GATA6high/HNF1Alow and GATA6high/HNF4Alow tumours was present in all patient-derived datasets, none of those tumours was basal, further supporting that GATA6 is sufficient to maintain classical features and that the concomitant loss of GATA6 and HNFs is required for the basal phenotype to emerge.
Development of a next-generation mouse model to delete Gata6 in PanINs
We showed previously that Gata6 deletion at tumour initiation accelerates KRasG12V-driven pancreatic tumorigenesis.8 However, KRasG12V ; Gata6P−/− mice developed tumours that were generally well differentiated and Krt14-negative (not shown). To discriminate the effects related to tumour initiation from those related to tumour progression, we turned to a next-generation mouse model. For this purpose, we bred Gata6lox/lox mice17 with the dual recombinase mice harbouring the Pdx1-Flp, FSF-KRasG12D , FSF-R26CreERT2 18 and R26Dual 19 alleles, to generate KFC mice (KRas, Flp, Cre). This new model allows to uncouple the activation of KRasG12D expression and Gata6 deletion (figure 3A).

A next-generation mouse model for conditional Gata6 deletion. (A) Schematic representation of the alleles used to generate the Gata6LateKO mouse model. (B) Representative H&E image of the pancreas of a 20-week Pdx1-Flp and KRasFSF-G12D mouse. Scale bar: 2 mm. (C) Images showing H&E and expression of GFP and Gata6, in a magnified region of the pancreas shown in B (dotted square). Black arrowhead: cells with lower Gata6. Scale bar: 100 µM. (D) Representative images of a Gata6LateKO pancreas after tamoxifen administration, showing H&E, GFP and Gata6 expression. Scale bar: 100 µM. (E) Representative images of the pancreas of two Gata6Ctrl and two Gata6LateKO mice with variable tumour size. (F) Quantification of the tumour area in Gata6Ctrl (n=37) and Gata6LateKO (n=42) mice. (G) Quantification of the Ki67-positive cells per high-magnification field in Gata6Ctrl (n=22) and Gata6LateKO (n=36) mice. (H) Age at necropsy for Gata6Ctrl (n=38) and Gata6LateKO (n=44) mice. Statistical significance for F–H was checked with Mann-Whitney U test.
Flp-dependent recombination efficiency varied widely, ranging from <5% to >90% of the pancreas, and no malignant lesions were observed in pancreata having <30% of recombination, measured by IHC for the GFP reporter (not shown). We included in our analyses only mice where Flp-mediated recombination reached at least 30% of pancreatic epithelial cells. By 20 weeks of age, KFC mice developed throughout the pancreas multiple low-grade and high-grade PanIN lesions that stained positive for GFP (figure 3B,C). In Gata6wt mice, Gata6 was detected in most epithelial cells. Occasionally, Gata6 expression was reduced in PanINs (figure 3C, black arrowhead) suggesting that spontaneous Gata6 loss might occur at this time point. We therefore administered tamoxifen (TMX) around 20 weeks of age to induce the deletion of Gata6 in KRasG12D-expressing cells and generate Gata6LateKO KFC mice. Successful TMX-induced recombination was verified by Gata6 IHC in all Gata6LateKO mice (figure 3D). GFP expression was retained in Gata6neg cells, suggesting that GFP is highly stable in pancreatic cells. Importantly, no recombination was detected in Gata6loxP/loxP mice not receiving TMX, as assessed by Gata6 IHC (not shown), thus excluding leakiness. Mice were sacrificed at 65 weeks or when moribund. From a cohort of 82 mice, 43 were Gata6LateKO and 39 were controls (Gata6Ctrl); the latter included 28 Gata6wt/wt and 3 Gata6wt/loxP mice receiving TMX and 8 Gata6loxP/loxP mice not receiving TMX. Gata6Ctrl and Gata6LateKO mice developed highly heterogeneous tumours of widely varying sizes, and no significant difference in tumour size or density of Ki67 positive cells was observed (figure 3E–G). The experimental design did not allow for Kaplan-Maier survival analysis and we did not observe that Gata6LateKO mice became moribund significantly earlier than controls (figure 3H).
Gata6 loss in tumours favours the basal phenotype, metastases and lung tropism
We used Krt5 and Krt14 expression as a proxy for the basal phenotype in mouse PDAC, since the Tp63 staining did not give reliable results (not shown). Expression of both markers was highly concordant (p=4.64e-08, online supplemental figure 5A). The proportion of Krt5/14pos tumours was higher in Gata6LateKO mice than in Gata6Ctrl mice (28/44 (63.6%) versus 16/38 (42.1%)) (figure 4A). Importantly, among Gata6Ctrl mice, 15/16 Krt5/14pos tumours were Gata6neg (Gata6Loss). When comparing tumours based on Gata6 expression, 43/63 (68.2%) Gata6neg tumours (Gata6LateKO+Gata6Loss) and only 1/19 Gata6pos (5.2%) were basal (Krt5/14pos) (p=3.4e-06, figure 4A). There was no significant difference in tumour grade between Gata6Ctrl and Gata6LateKO mice, while Gata6neg and basal tumours were significantly more often of grade 2 and 3 (online supplemental figure 5B, p=0.0132 and 0.034, respectively). This ultimately confirmed that GATA6 loss is necessary but not sufficient for the expression of the basal phenotype.

Gata6 loss in tumours leads to a basal-like phenotype and increased metastatic potential with lung-specific tropism. (A) Expression of Gata6 and Krt14 in representative Gata6Ctrl and Gata6LateKO pancreatic ductal adenocarcinomas, detected by IHC (left) and quantification of Krt5 and Krt14 expression in tumours classified either by genotype (Gata6Ctrl n=38 and Gata6LateKO n=44) or by Gata6 expression (Gata6pos n=19 and Gata6neg n=63). *p<0.05, ***p<0.001. Scale bar: 200 µm. (B) Representative H&E images of liver and lung metastases in a Gata6Ctrl and a Gata6LateKO mouse and quantification of metastasis occurrence in Gata6Ctrl (n=38) and Gata6LateKO (n=44) mice. **p<0.01. Scale bar: 5 mm left/centre, 100 µm right. (C) Distribution of metastases to the liver (LiMet) or to the lung (LuMet) in Gata6LateKO mice. (D) Quantification of E-cadherin IHC in Gata6LateKO liver (n=19) and lung (n=30) metastases, **p<0.01.
Patients with basal PDACs have worse outcome.1–4 9 Congruently, we observed that significantly more Gata6LateKO mice had clear signs of disease progression as reflected by significantly more metastases (30/43, 69.8%) than the Gata6Ctrl controls (8/39, 20.5%) (p=8.5e-06, figure 4B). Importantly, all 8 Gata6Ctrl mice with metastases had Gata6neg tumours (primary and metastatic). Gata6neg tumours were also more proliferative, as shown by Ki67 staining (p=2.93e-04, online supplemental figure 5C). These data indicate that Gata6 is an efficient suppressor of metastasis in murine KRasG12D-driven PDAC.
Among the Gata6LateKO mice, 15/30 (50%) had only lung metastases, 4/30 (13.3%) had only liver metastases and 11/30 (36.7%) had both (figure 4C). This result differs from findings in patients, where the liver is the most common site of metastases.20 There is evidence that tumour cells are heterogeneous and must, in addition, be highly plastic to form metastases.21 22 This degree of plasticity influences the organotropism of PDAC metastatic cells, whereby cells that cannot fully revert the epithelial phenotype colonise preferentially the lungs.23 Among Gata6LateKO mice, liver metastases were significantly more often E-cadherinpos than lung metastases as detected by IHC (16/19, 84.2% E-cadherinpos LiMet; 2/30, 6.7% E-cadherinpos LuMet, p=3.69e-08) (figure 4D, online supplemental figure 5D). This observation is consistent with data showing undetectable E-cadherin in primary cells derived from a lung metastasis24 and might indicate that Gata6LateKO cells are more limited in their ability to efficiently reactivate the epithelial programme.
Gata6LateKO primary tumour cells are more proliferative and chemoresistant
We successfully established primary cell lines from Gata6pos (n=5), Gata6LateKO (n=15) and Gata6Loss (n=6) tumours. While Gata6pos and Gata6LateKO cell lines were homogeneously positive and negative for Gata6, respectively, Gata6Loss lines displayed a more heterogeneous expression pattern (figure 5A). ΔNp63 mRNA was significantly higher in Gata6LateKO and Gata6Loss cells (figure 5B) and a similar trend was observed for a set of basal markers (Runx3, S100a2 and Krt14) but not classical/progenitor markers (Pdx1 and Hnf4a, online supplemental figure 6A) indicating that these cells preserve some basal features in vitro. Gata6LateKO and Gata6Loss cells were significantly more proliferative than Gata6pos cells (figure 5C). The migratory capacity of KFC cells was highly variable and no statistical differences were observed, although some of the Gata6LateKO and Gata6Loss cells showed high migratory potential (online supplemental figure 6B). No difference was observed in the invasive capacity in vitro (online supplemental figure 6C).

Primary cells from Gata6LateKO tumours are more proliferative and chemo-resistant in vitro. (A) Representative immunofluorescence images of primary KFC tumour cells isolated from Gata6pos, Gata6LateKO and Gata6Loss mice. Top: expression of Gata6 (green). Bottom: merged Gata6 (green), E-cadherin (red) and DAPI (blue) stainings. (B) Expression of ΔNp63 measured by retrotranscription + qPCR (RT-qPCR). (C) Proliferation of primary KFC cells from the indicated groups, represented as fold increase in cell number 48 hours after seeding. Gata6pos n=4, Gata6LateKO n=15, Gata6Loss n=5. (D) H3K27ac enrichment at the promoter of ΔNp63, detected by ChIP-qPCR in primary KFC cells. Data are represented as % of input chromatin. Gata6pos n=4, Gata6LateKO n=7, Gata6Loss n=3. (E) Graphs representing the IC50 values measured for primary KFC cells on treatment with 5-FU and Gemcitabine in cytotoxicity assays. Gata6pos n=4, Gata6LateKO n=8, Gata6Loss n=6. (B–E) Each dot represents the average value of at least three independent experiments for each tumour cell line. *p<0.05, **p<0.01.
We then investigated the contribution of epigenetic changes to our observations, since this mechanism was shown to control the emergence of the basal phenotype in PDAC cells.11 13 17 ChIP-qPCR for H3K27ac, a marker of open chromatin, showed higher enrichment on the ΔNp63 promoter in Gata6LateKO and Gata6Loss cells (figure 5D). No significant differences were observed for the promoters of a subset of basal (Runx3, S100a2, Krt14) and classical (Pdx1 and Hnf4a) genes (online supplemental figure 6D). Based on this analysis, Gata6 deletion does not cause widespread remodelling of chromatin accessibility in KFC cells.
Finally, we evaluated the relationship between GATA6 status and the response of tumour cells to chemotherapeutic agents commonly used for the treatment of PDAC. Patients with low GATA6low or basal-like PDAC respond worse to 5-FU-based adjuvant treatments.9 10 Consistently, Gata6LateKO cell lines were significantly more resistant to 5-FU than Gata6pos cells (figure 5E). In contrast to the findings in patients, however, Gata6LateKO cells were also more resistant to gemcitabine (figure 5E). Gata6Loss cells had a mixed behaviour and no clear conclusion could be drawn. These observations indicate that the KFC cell line panel generated in this study recapitulates some features of the human disease, including the high inter-patient heterogeneity.
GATA6 loss favors cell plasticity and immune escape
To pinpoint the mechanism underlying Gata6 basal-suppressive and metastasis-suppressive function, we performed RNAseq analysis of Gata6pos, Gata6LateKO and Gata6Loss primary tumour cells. A recent multi-omics analysis of primary mouse PDAC cells identified two transcriptomics-based clusters: a mesenchymal cluster C1 (Mes-C1) and a more epithelial cluster C2 (Epi-C2), including three subclusters C2a, C2b and C2c.25 Reference cell lines from that analysis were included in our experiment.25 KFC cells could be assigned to three clusters: C1, C2a and C2b/c. All but one Gata6pos lines fell into cluster Epi-C2b/c and one was assigned to the Epi-C2a cluster. In contrast, all lines assigned to the Mes-C1 cluster were either Gata6LateKO or Gata6Loss cells, supporting the strong anti-EMT role of GATA6 (figure 6A, online supplemental figure 7A). Eight of the Gata6LateKO cell lines included in the RNAseq analysis were isolated from primary tumours that had metastasised. Interestingly, the Mes-C1 cluster included only cells from lung-tropic tumours, while the Epi-C2b/c cluster only included cells from tumours that also generated liver metastases (figure 6A). These data further supports that PDAC cells with a strong mesenchymal phenotype colonise preferentially the lungs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GATA6 loss favours cell plasticity and immune escape. (A) Hierarchical clustering of KFC cells and reference cell lines according to RNAseq analysis. Gata6pos, Gata6LateKO and Gata6Loss primary cells were assigned to the described clusters Mes-C1 (mesenchymal), Epi-C2a (epithelial) and Epi-C2b/c (epithelial). (B) Gene set enrichment analysis of genes differentially regulated between Gata6LateKO and Gata6pos cells. (C) Enrichment of the gene set ‘MHC I mediated antigen processing and presentation’ in KFC cells. (D) Quantification of CD8α-positive T cells in GATA6High (n=37) and GATA6Low (n=23) patient-derived tumours, detected by IHC. Representative images of CD8 IHC in a CD8low and a CD8high sample are shown. *p<0.05. Scale bar: 500 µM. (E) Correlation between GATA6 and CD274 (coding for PDL-1) mRNA levels in the indicated datasets. FDR, false discovery rate. NES, normalised enrichment score.
We performed GSEA with all possible comparisons. We found ‘EMT’ as the most highly enriched gene set among the genes upregulated in Gata6LateKO or Gata6Loss cells compared with Gata6pos. On the other hand, an ‘apical junctions’ gene set was enriched among the genes upregulated in Gata6pos versus Gata6LateKO cells (figure 6B). GSEA showed additional similarities between the Gata6LateKO and the Gata6Loss cells, when compared with the Gata6pos cells, including the enrichment of the ‘KRAS signaling_DN’ gene set among the upregulated genes. In contrast, the ‘Hypoxia’, ‘p53-pathway’ and ‘metabolism-related’ gene sets were downregulated (figure 6B, online supplemental figure 7B). Moreover, we also observed significant differences between Gata6LateKO and Gata6Loss cells (online supplemental figure 7B,C). Finally, when comparing Gata6LateKO cells with epithelial or mesenchymal features, EMT was clearly upregulated in Mes-Gata6LateKO, while two ‘MYC targets’ gene sets, cell cycle-related gene sets and DNA-repair-related ones were upregulated in Epi-Gata6LateKO (online supplemental figure 7D), confirming that Gata6LateKO cells are diverse. Therefore, heterogeneity in pancreatic cells is a defining characteristic not only of human but also of genetically engineered mice used to model this disease.
Interestingly, the major histocompatibility complex (MHC) class I genes H2-d1 and H2-k1 and the immunoproteasome gene Psmd8 were among the most significantly downregulated genes in the Gata6LateKO cells, suggesting that Gata6 loss might induce immune escape, thereby supporting higher metastatic potential. Congruently, the gene set ‘MHC I Mediated Antigen Processing and Presentation’ was significantly enriched among genes downregulated in the Gata6LateKO cells (figure 6C). IHC analysis of GATA6low patient tumours revealed a significantly decreased infiltration of CD8α+ T cells compared with GATA6high tumours (figure 6D). To expand these observations, we explored the available patient-derived datasets for evidence of GATA6 involvement in immune escape. The Puleo cohort showed the most consistent results: MHC I-mediated antigen processing and presentation and the estimated abundance of CD8+ T cells26 were significantly lower in GATA6low tumours (online supplemental figure 8A,B). Furthermore, the T cell checkpoint activator PDL-1 (encoded by the CD274 gene) was negatively correlated with GATA6 expression in 3/5 datasets (figure 6E and online supplemental figure 8C). Interestingly, CD274 and several genes related with antigen processing and presentation (PSMD8, PSMD9, B2M) had GATA6 peaks on the promoter in the ChIP-Seq we performed in PDAC cells,9 suggesting that GATA6 might directly regulate a subset of them. No significant decrease in tumour infiltration of Cd8+ cells was observed in Gata6LateKO mice compared with Gata6Ctrl, but a trend was observed when comparing metastatic versus non-metastatic tumours (online supplemental figure 8D). Taken together, our data suggest that GATA6 loss in PDAC can facilitate immune escape, favouring metastasis.
Discussion
Transcriptomic-based tumour taxonomy has revealed important differences and commonalities among PDACs. A detailed understanding of the molecular events driving the different phenotypes, particularly the highly aggressive basal one, will increase the chance of a successful translation of basic knowledge into clinical intervention.
Here, we show that loss of GATA6, a major regulator of epithelial identity, is necessary, but not sufficient, for the acquisition of the basal phenotype in patient-derived samples and in a next-generation mouse model in which Gata6 deletion was induced at the time of KRasG12D-driven high-grade PanIN formation (Gata6LateKO).
Multiple lines of evidence link GATA6 loss to the basal phenotype.1 2 9 10 Our data from the Gata6LateKO mice ultimately identify GATA6 as a molecular gatekeeper restricting cell plasticity to maintain lineage-specific programmes in PDAC. GATA6 loss is necessary to allow the expression of the basal programme, but additional downstream or parallel events are required. Analysis of patient-derived samples revealed that HNF1A and HNF4A might act as further molecular barriers to maintain the classical gene programme when GATA6 expression is lost, possibly through the regulation of a shared subset of genes. GATA6, HNF1A and HNF4A are all involved in cell fate determination of the pancreatic lineage during development and are highly expressed in the classical/progenitor PDACs.1 27–30 HNF4A, in particular, was recently shown to support the classical phenotype by repressing the basal-related genes SIX1 and SIX4, among others.31 However, Hnf4a deletion in mouse PDAC organoids was not enough to induce the full basal phenotype, suggesting that GATA6 and HNF4A have overlapping but not identical roles in maintaining lineage identity31 and that GATA6 has a stronger antibasal function. Consistently, GATA6 loss in patient-derived samples was always broader and more pronounced than HNF4A loss, supporting a chronological hierarchy during PDAC progression towards basality (figure 2E). Non-basal GATA6low tumours in patients displayed intermediate levels of GATA6 expression, suggesting the existence of a threshold below which HNF1A and HNF4A expression is lost and the full basal programme is established. Such a threshold does not exist in mouse tumours in our model, where both Gata6 alleles are lost at the genomic level. Therefore, loss of HNFs expression is likely the result of multiple regulatory events. Importantly, HNF4A was epigenetically silenced in GATA6low/Basal PDX-derived cells, suggesting that epigenomic remodelling is a crucial event downstream of GATA6 loss, to allow the full classical-basal switch. Importantly, the hierarchy suggested by our work, where HNFs act as a second barrier to preserve cell lineage identity, might be contingent on downregulation/loss of GATA6 loss. Increasing evidence indicates that the classical-basal phenotypes are a continuum and that multiple transcription factors contribute in a context-dependent manner. A more systematic analysis is required to precisely delineate the reciprocal relationship of these and other lineage identity factors in PDAC.
The relationship between GATA6 and ΔNp63 in controlling PDAC phenotypes is complex. Our data and the reanalysis of available datasets suggest that neither GATA6 loss nor ΔNp63 expression are sufficient for a full phenotype switch, but both events are necessary and there is evidence of a cross-regulation whereby GATA6 downregulation allows for expression of ΔNP63, which in turn contributes to keep GATA6 inhibited. Similar regulatory relationships might be true for HNF4A and HNF1A, revealing a complex interplay among these transcription factors and supporting the existence of a continuum—rather than a dichotomy—of phenotypes.
While the in vivo findings and the correlations observed in patient-derived samples were highly consistent, in vitro modulation of GATA6 and ΔNp63 expression in cell lines yielded variable results suggesting highly context-dependent effects including roles for the stroma and the immune system. In particular, BxPC3 are KRAS wt, thus representing a rare subset of patients with PDAC. The different behaviour we observed in BxPC3 (in this work) and L3.6pl cells (in our previous work9) after GATA6 overexpression might reflect the contribution of mutant KRAS. These differences might indicate that BxPC3 and L3.6pl cell lines do not faithfully represent the complexity of the basal-like PDACs. Indeed, only 3/6 PDX originally defined as basal-like14 shared the H3K27ac pattern of BxPC3 and L3.6pl.11 Our panel of primary mouse tumour cell lines represents a valuable tool for understanding the basal phenotype, adding to the PDX-derived cells described previously.14 Repression of basality is intrinsic in the epithelial programme governed by GATA6, HNFs, and likely other transcription factors. It remains to be determined whether the reconstitution of the lost repressive barriers would revert the phenotype, once it is established. Different levels of epigenetic regulation might determine such reversibility.
We additionally show that Gata6 loss dramatically increases the rate of metastasis, thus providing a molecular link between the basal programme and the metastatic potential of PDAC. A recently published transcriptomic dataset of PDAC encompassing all-stages confirmed that the basal phenotype is highly enriched among metastatic tumours.5 A thorough characterisation of primary cell lines isolated from mouse tumours revealed that Gata6 loss results in higher plasticity and possibly immune evasion, both characteristics of metastatic cells.
Cellular plasticity is one hallmark of metastatic cells. While EMT is required to initiate metastatic spread, the reverse process—MET—is necessary for the growth of metastases at distant sites and cells that cannot revert the EMT are not able to grow metastases in mouse models.21 22 The degree of plasticity seems to play an important role in defining the mode of dissemination32 and the organotropism of metastases, with more epithelial-like cells forming metastases preferentially the liver and more mesenchymal-like cells favouring lung metastases.23 Gata6LateKO mice preferentially developed lung metastases, mostly E-cadherin-negative, indicating that Gata6-KO cells might not be able to revert to a fully differentiated status. In patients, however, it is conceivable that GATA6 expression might be reactivated during tumour evolution or under the selective pressure of therapy.
Disseminating tumour cells must overcome multiple hurdles during their path to the metastatic site, among them the immune surveillance. Suppression of antigen processing and presentation is one immune evasion mechanism that tumour cells have hijacked from viruses.33 34 We observed that GATA6 loss in tumours from patients and mice decreased the expression of the antigen processing and presentation machinery and that infiltration of CD8-positive T cells was reduced in GATA6low tumours in patients, possibly indicating a more efficient immune evasion. Accordingly, Gata6 knock-out favoured T cell-mediated tumour cell killing in an in vivo CRISPR screening,33 suggesting that the immunogenicity gene programme is embedded within the GATA6-dependent epithelial cell identity programme. We reported similar findings for another master regulator of the epithelial cell identity, NR5A2, which actively inhibits an inflammatory gene expression programme in the normal pancreas.35 The polycomb repressive complex 2 (PRC) was recently shown to promote PDAC de-differentiation and metastasis through GATA6 repression15 andindependent results indicate that PRC2 can silence the MHC class I antigen presentation pathway.34 We observed GATA6 peaks on EZH2 and EED promoter in our published ChIP-Seq, and their transcripts were mildly downregulated in RNA-Seq on GATA6 silencing.9 These data point to an indirect role of GATA6 in modulating the antigen processing and presentation machinery, possibly through PRC2 and establish an unexpected link between identity maintenance programmes and immune recognition.
In summary, we show here that a GATA6-centred gene regulatory network functions as a gatekeeper of cell identity and blocks cell plasticity and immune evasion, thus providing a molecular link between the basal-like phenotype and metastasis. The Gata6LateKO mouse model is therefore a valuable preclinical tool to study the most aggressive subtype of PDAC.
Data availability statement
RNA-Seq accession number: PRJEB37700.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the local ethics committee of the Medical University of Vienna (‘Ethikkommission’, protocol no. 1753/2014).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors PM designed the study and secured funding, analysed the experiments and prepared the manuscript. BK performed most of the experiments and participated in writing the manuscript. NH managed the mouse colony and performed experiments. VI, SP and DH-B performed some experiments. JM performed some bioinformatics analyses. RO, SM and RR performed and analysed the RNA-Seq experiment. HPD and JS did the pathology assessments. MS and EG provided access to resection tissues. GAL and RAU performed the analysis of epigenomic data from PDX-derived cells. BS and DS provided the dual recombinase mouse strain. FXR provided the Gata6 flox mice and participated in the experimental design and manuscript preparation.
Funding Work in the lab of PM was supported by the grant P27361-B23 from the Austrian Science Grant (FWF) and by contributions from the Fellinger Krebsforschung foundation and the Ingrid Shaker-Nessmann Krebsforschungsvereinigung foundation. Patients were not involved in the design of this study. RAU and GAL were supported by The Linda T. and John A. Mellowes Endowed Innovation and Discovery Fund, the Theodore W. Batterman Family Foundation, Inc and NIH Grants: R01 DK52913 and R01 CA178627. Work in the lab of FXR was supported, in part, by grant RTI2018-101071-B-I00 (Ministerio de Ciencia, Innovación y Universidades, Madrid, Spain). CNIO is supported by Ministerio de Ciencia, Innovación y Universidades as a Centro de Excelencia Severo Ochoa SEV-2015-0510.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.