Article Text

Abstract
Objective Sessile serrated lesions (SSLs) are common across the age spectrum, but the BRAF mutant cancers arising occur predominantly in the elderly. Aberrant DNA methylation is uncommon in SSL from young patients. Here, we interrogate the role of ageing and DNA methylation in SSL initiation and progression.
Design We used an inducible model of Braf mutation to direct recombination of the oncogenic Braf V637E allele to the murine intestine. BRAF mutation was activated after periods of ageing, and tissue was assessed for histological, DNA methylation and gene expression changes thereafter. We also investigated DNA methylation alterations in human SSLs.
Results Inducing Braf mutation in aged mice was associated with a 10-fold relative risk of serrated lesions compared with young mice. There were extensive differences in age-associated DNA methylation between animals induced at 9 months versus wean, with relatively little differential Braf-specific methylation. DNA methylation at WNT pathway genes scales with age and Braf mutation accelerated age-associated DNA methylation. In human SSLs, increased epigenetic age was associated with high-risk serrated colorectal neoplasia.
Conclusions SSLs arising in the aged intestine are at a significantly higher risk of spontaneous neoplastic progression. These findings provide support for a new conceptual model for serrated colorectal carcinogenesis, whereby risk of Braf-induced neoplastic transformation is dependent on age and may be related to age-associated molecular alterations that accumulate in the ageing intestine, including DNA methylation. This may have implications for surveillance and chemopreventive strategies targeting the epigenome.
- adenocarcinoma
- oncogenes
- colonic polyps
- colorectal neoplasia
- ageing
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Sessile serrated lesions (SSLs) have distinct malignant potential.
They are a common colonoscopic finding in patients of all ages, but the cancers arising occur exclusively in the elderly.
Extensive aberrant DNA methylation is common in advanced SSLs and cancers.
It is not clear whether SSLs lay dormant in the colon for decades, acquiring these alterations or whether spontaneously occurring SSLs rapidly acquire these alterations and are at risk of rapid neoplastic progression.
Evidence from the study of organoids cultured for protracted periods (Tao et al, 2019) suggests that age may influence the potential for Braf mutation to induce neoplastic transformation.
What are the new findings?
Here, we show that Braf mutation can induce rapid neoplastic transformation in the aged and extensively aberrantly methylated intestinal epithelium.
This challenges the conventional notion of serrated neoplastic evolution, where risk is thought to correlate with the length of time since SSL initiation, to one depending on the age of the patient at onset.
Age-associated DNA methylation occurs in the intestinal epithelium, and this is potentiated by Braf mutation, and target pathways that are pertinent to colorectal neoplasia.
How might it impact on clinical practice in the foreseeable future?
Our preclinical study suggests that the malignant potential of SSLs of similar size and histology is greater in older patients.
This could have implications for surveillance as SSLs are common in all age groups but older patients may benefit more from close surveillance to ensure SSLs are removed in a timely manner.
Clinical studies are necessary to validate these findings in patients.
Introduction
It is well established that the important subgroup of BRAF mutant colorectal cancers arise from sessile serrated lesions (SSLs), previously called sessile serrated adenomas. With improvements in colonoscopic detection and diagnostic methods, SSLs are now recognised to be common lesions.1–4 Unlike conventional adenomas that are much more common in older individuals, SSLs are equally represented across the age spectrum and are often found in individuals in their 30s and 40s.5 However, the BRAF mutant cancers arising from SSL occur at an older mean age than BRAF wild-type cancers arising from conventional adenomas and they rarely occur before 50 years of age outside the setting of serrated polyposis syndrome.5 6 This paradox suggests that the risk of malignant transformation of an SSL may depend on the age of the individual in which it is present as well as the characteristics of the lesion itself. Surveillance intervals for patients presenting with SSL are currently guided by criteria established for conventional adenomas,7 8 which have a very different natural history, morphology and molecular profile compared with SSL.9–11 Better understanding the drivers of progression of SSLs and particularly the role of age may help develop guidelines to inform surveillance intervals for patients following diagnosis of an SSL.
SSLs are uniquely hallmarked by BRAF mutation and widespread DNA methylation changes termed the CpG island methylator phenotype (CIMP).12 13 These extensive methylation changes facilitate the silencing of key tumour suppressor genes necessary for malignant transformation. This includes genes in the WNT signalling pathway, such as the SFRP family of genes, and DNA repair genes such as MLH1. Approximately half of all SSLs are CIMP-positive and the vast majority of advanced SSLs with dysplasia are CIMP-positive, demonstrating the necessity of CIMP for progression to malignancy.11 We have previously reported a striking association between CIMP and increased patient age among histologically indistinguishable SSL,14 suggesting a biological explanation for why age may be a major risk factor for progression of SSL. It remains unclear if the highly methylated SSLs that are identified in older patients have dwelled for a significant period of time, perhaps decades, or whether methylation alterations occur rapidly in the spontaneously forming SSLs in the aged intestine. This remains a key distinction preventing the translation of this knowledge into the clinic.
A causative versus synergistic role for mutant BRAF and CIMP has been debated.6 15–18 In a mouse model, we have recently demonstrated that prolonged exposure to intestinal Braf mutation in vivo induces a CIMP-like methylator phenotype, preceding the development of murine serrated adenomas and ultimately invasive cancer.19 In the wild-type intestine, low levels of DNA methylation accumulate with age for many of the same loci found to be altered with Braf mutation.19 20 This suggests that the aged intestine may provide the necessary epigenetic milieu for more rapid accumulation of DNA methylation following oncogenic mutation of Braf in older individuals. CIMP-specific loci have also been shown to accumulate with increasing passage in vitro in both cell line15 and organoid systems.18 Recently, Tao et al18 demonstrated that organoids cultured for ~12 months acquire DNA methylation alterations, accompanied by concomitant gene repression, which is reminiscent of those that occur in the CIMP. Tao et al18 showed that Braf mutation could induce immediate spontaneous neoplastic transformation in two of these organoids, by contrast Braf mutation induces neoplastic transformation over a period of 5 months when the mutation is initiated without prior culturing. This finding indicated that ageing-like alterations may shift the threshold for neoplastic transformation. However, this study was limited to two organoids, and it is not clear whether culturing accurately reflects the ageing process that occurs in vivo.
This accumulation of DNA methylation with age has been shown to be due to ‘epigenetic drift’21 22 which occurs as a result of stochastic errors in DNA maintenance methylation.23 24 Epigenetic drift is not randomly distributed and appears to have an affinity for specific CpGs.25 This has allowed for the development of epigenetic clocks that can reliably predict an organism’s age based on the DNA methylation state of a subset of CpGs throughout their genome.26 27 The epigenetic clock has provided a model for biological ageing and increases in a person’s ‘epigenetic age’ compared with their chronological age have been associated with an increased risk of disease28 29 and an overall increase in all-cause mortality.30 31
Here, we have used the conditionally active BrafV637E murine model to evaluate the effects of somatic oncogenic signalling on epigenetic drift and tumour formation across the age spectrum. We have exposed young and aged mice to mutant Braf for the same time period to provide insights into how the risk of serrated neoplasia is influenced by age.
Results
Temporal CpG island hypermethylation in the intestinal epithelium of wild-type mice
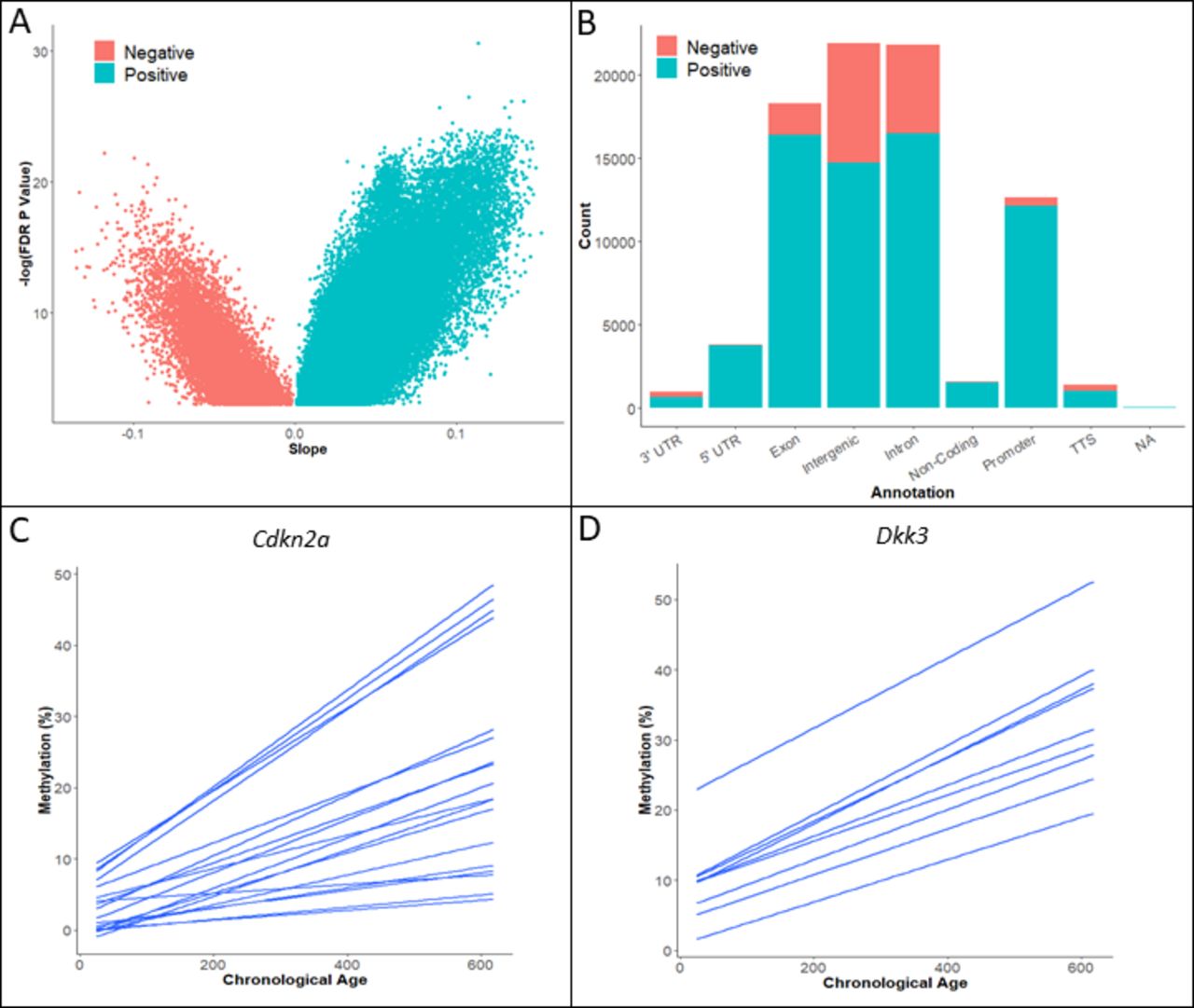
To establish the effects of ageing on the intestinal methylome in our murine model, we examined the DNA methylation status of the proximal small intestine of wild-type mice (n=25) aged between 24 days and 20 months post wean. Tissue samples were derived from the small intestine, as this is where the phenotype induced by the BrafV637 murine model, which is examined extensively in the forthcoming paragraphs, predominantly occurs (see the Extended methods section). Using reduced representation bisulfite sequencing, we captured 1 027 330 CpG sites, each covered by >10 reads in all samples (including those samples analysed in the forthcoming paragraphs). We identified 82 468 CpG sites (8.03%) where DNA methylation was significantly associated with biological ageing (type-A sites, false discovery rate (FDR)-corrected p<0.05, R2 range 0.98–0.31, figure 1A and online supplemental figure 1). Type-A (age-associated) CpGs were predominantly confined to exonic, intergenic, intronic and promoter regions (figure 1B). Age-associated hypomethylation was frequent in exonic, intergenic and intronic regions, but uncommon in promoter regions (figure 1B).
Supplemental material
Supplemental material

(A) DNA methylation profiling of wild-type intestinal mouse DNA. We identified significant age-associated DNA methylation was identified in ~8% of CpG sites by linear regression analysis. Positive slope values indicate progressive DNA hypermethylation with age. (B) Age-associated DNA hypermethylation is most common in exons, introns, intergenic regions and gene promoters. Hypomethylation is common in exons, introns and intergenic regions, however, it is extremely rare in gene promoters. (C, D) Exemplar loci of age-associated DNA methylation of colon cancer tumour suppressors Cdkn2a (C), Dkk3 (D). Each line represents a single CpG site in the promoter region. Regression results for (C) and (D) are found in online supplemental table 5. NA, not applicable; TTS, transcription start site; UTR, untranslated region.
Supplemental material
Tumour suppressor genes and regulators of intestinal differentiation are putative targets of intestinal age-associated DNA methylation
We cross-referenced the loci containing age-associated promoter hypermethylation with known tumour suppressor genes from the TSGene 2.0 database and report type-A promoter methylation of 105 tumour suppressor genes, including previously reported colonic tumour suppressors such as Cdkn2a (figure 1C), Dkk3 (figure 1D), Sfrp2 and Dcc.
Type-A methylation in gene promoters was enriched for genes involved biological processes associated with differentiation (p=1.19×10-40, observed/expected (O/E) ratio: 1.92) and development (p=2.05×10-48, O/E ratio: 1.75). By contrast, immune-related molecules were under-represented (p=1.77×10-9, O/E ratio: 0.39). Next, we performed enrichment analysis at pathway level using pathways curated by Protein analysis through evolutionary relationships (PANTHER).32 Pathways level enrichment analysis identified a significant enrichment type-A methylation at genes in eight pathways, including the WNT signalling cascade (p=9.11×10-4, O/E ratio: 2.09, table 1) and Slit/Robo signalling (p=5.37×10-4, O/E ratio: 6.42, table 1).
Type-A DNA methylation at gene promoters targets specific signalling pathways
Tao et al18 evaluated methylation differences in intestinal organoids cultured for 2 and 14 months. We compared our type-A CpGs to those identified in the Tao et al18 analysis and report that 44.12% of type-A CpGs were also identified in their in vitro ageing system. Of all hypermethylated regions reported in their study, we report only 28% were hypermethylated in our in vivo system. The limited overlap in our differentially methylated CpGs may be due to the culture environment altering the methylation profile of the organoids, result from gender differences in animals, as the Tao study relied on organoids from male animals, or from the differing origin of the tissue samples (organoids from colon vs tissue from the small intestine).
Next, we performed gene expression analysis via RNA-Seq on wild-type animals aged 10 days (n=3) and 14 months (n=4) to evaluate the transcriptional consequences of type-A methylation changes. In 10-day wild-type animals, type-A genes were more likely to be expressed when compared with the wider transcriptome (online supplemental figure 2A,B), and the average expression of type-A genes was higher than non-type-A genes (online supplemental figure 2C). Of the 1208 type-A genes, 16.8% were not expressed (mean fragments per kilobase per million reads (FPKM)=0), and an additional 56.8% were lowly expressed (FPKM <1). This is keeping with observations of Tao et al in an organoid system of prolonged culturing that mimicked ageing.18 We performed differential expression analysis on type-A genes between 10-day and 14-month-old wild types to identify transcriptional changes that may have been caused by temporal methylation changes. We identified differential gene expression in 14.5% of type-A genes (figure 2A and online supplemental table 1), including key WNT signalling pathway genes such as Sfrp2 (LogFC: −6.81, p=1.65×10-6) and Wnt7B (LogFC: −3.7, p=0.034). We also analysed genes whose expression was identified as associated with prolonged organoid culture.18 Of the 16 genes assessed by Tao et al,18 6 displayed significant differential expression in 14-month wild-type animals compared with their 10-day-old counterparts (online supplemental table 2, p<0.05); however, all displayed upregulation (Log2FC: 0.45–1.3), which is in contrast with Tao et al, which observed downregulation. As these are predominantly differentiation and lineage-associated markers, this may indicate that the differentiation state of normal intestinal epithelia is maintained more readily in vivo when compared with organoid culture.
Supplemental material
Supplemental material
Supplemental material

RNA-sequencing was performed on 10-day-old wild-type animals (n=3) and 14-month-old wild-type animals (n=3) to assess transcript expression. Differential gene expression of DNA methylation type-A genes in the small intestine of 10-day-old wild-type animals versus 14-month-old wild-type animals using DeSeq2. P values were adjusted using the false discovery rate method to limit type I errors. Negative log fold changes (FC) represent differential downregulation in 14-month-old wild types and vice versa.
Immune-related signalling is dysregulated in the aged intestine
To identify other signalling pathways that are putative targets of ageing in the wild-type intestinal mucosa, we analysed whole transcriptome in relation to age, irrespective of DNA methylation changes. We identified 810 genes that have altered expression in the aged small intestine (p<0.05, figure 2B), of which 537 display increased expression with age and 273 show a decrease in expression. We performed enrichment analyses on age-associated genes with increased expression and decreased expression independently to identify any pathway level changes that occur with age in the intestine. Downregulated genes were associated with various immune-related biological processes, including response to virus (O/E: 9.26, p=1.68×10-11), the immune effector process (O/E: 3.61, p=1.06×10-4) and defence response (O/E: 2.55, p=1.99×10-4). Likewise, enriched biological processes in overexpressed genes reflected altered immune response (O/E: 4.81, p=4.21×10-57). These data indicate that altered immune response may also contribute to age-associated changes.
Braf mutation potentiates age-related DNA methylation changes
Next, we sought to assess the effects of prolonged oncogenic MAPKinase signalling on age-associated DNA methylation. We introduced the intestinally specific oncogenic Braf V637E mutation in animals at wean and assessed temporal changes in DNA methylation in the small intestine from 10 days to 14 months post activation (n=37). There was a significant association between age and DNA methylation in 16.8% of CpG residues (172 217 CpG sites, FDR<0.05 R2=0.18–0.97), which included 81.3% of type-A CpGs identified in wild-type animals.
We hypothesised that the rate of DNA methylation changes in age-associated loci common to both wild-type and Braf mutant animals may differ according to Braf mutation status. Of the 67 056 shared age-associated loci, we identified significantly different rates of methylation accumulation over time in 46 581 CpG residues (69.5%, online supplemental figure 1). We denote these CpGs type-AB; age-associated but modified by oncogenic Braf mutation). CpGs in which we observe temporal changes in the setting of Braf mutation, but not in the wild-type small intestine, were termed type-B CpGs (temporal but specific to Braf mutation). These are discussed in the coming paragraphs. 91.2% of type-AB CpGs accumulated DNA methylation at a greater rate with Braf mutation, while 8.8% of CpGs accumulated methylation changes more slowly (figure 3A depicts the rate of change in these CpG sites).

(A) Braf mutation induces a widespread acceleration in the rate of type-A DNA methylation changes. The rate of change of DNA methylation (%/day) in CpG sites that were significantly different between BrafV637 and wild-type animals (type-AB CpGs, age associated but modified by Braf) as assessed by analysis of covariance. The red line depicts a hypothetical equal rate of change between groups. The bottom-left and top-right quadrants show concordant hypomethylation and hypermethylation, respectively, occurring at different rates according to Braf status. The top-left and bottom-right show methylation events that are discordant by Braf status. Dark blue ellipses indicate the density of CpGs at a given location on the graph. High number of overlapping data are shown by short distances between ellipses. (B) The genomic distribution of type-AB CpGs. (C) The genomic distribution of type-B CpGs. NA, not applicable; TTS, transcription start site; UTR, untranslated region.
For gene promoters associated with these 46 581 CpG sites, we observed an enrichment for genes in the cadherin signalling pathway (O/E: 4.41, p=4.32×10-7, table 2), the WNT signalling pathway (O/E: 2.65, p=2.29×10-4) and heterotrimeric G protein signalling (O/E: 3.22, p=8.97×10-4, table 2). Enrichment for WNT signalling in type-AB promoters was stronger than in type-A promoters (O/E 2.65 vs 2.09), indicating a selection for accelerating age-associated DNA methylation at WNT signalling pathway gene promoters following Braf mutation.
Type-AB DNA methylation at gene promoters targets specific signalling pathways
Intestinal epigenetic age is increased by prolonged exposure to oncogenic BRAF
Epigenetic age is the predicted biological age of a tissue based on the DNA methylation level of CpGs that are predictive of chronological age in normal tissues. As we identified a robust acceleration of age-associated DNA methylation in Braf mutant intestinal samples, we next assessed whether epigenetic age was similarly accelerated. We estimated epigenetic age using two previously validated epigenetic age prediction models (Stubbs et al33 and Meer et al34). As expected, both epigenetic age predictors yielded estimates of age in our wild-type animals that was strongly correlated with chronological age; however, these models lack precision (R2 (Stubbs) 0.91 (Meer) 0.97 figure 4A,B). The Stubbs model tended to underpredict epigenetic age of wild-type animals (ratio of chronological age to epigenetic age (CA/EA: 1.83, root mean square error (RSME): 184.54), in contrast the Meer model tended to overpredict the age of wild-type animals, but was generally more accurate (CA/EA: 0.76, RMSE: 117.02). Intestinal epigenetic ageing was significantly accelerated in Braf mutant mucosa using both the Stubbs model (CA/EA: 0.65 vs 1.83, p<2.2×10-16, figure 4A) and the Meer model (CA/EA: 0.39 vs 0.76, p<2.2×10-16, figure 4B). We also built an in-house, tissue-specific, epigenetic clock model, which was much more precise in estimating the epigenetic age of wild-type animals. Prediction of epigenetic age using this model showed the same marked increasing in epigenetic ageing revealed by the two published models (figure 4C, analysis of covariance p<2.2×10-16). Thus, constant exposure to oncogenic Braf induces a marked and sustained acceleration of epigenetic ageing.

Epigenetic age versus chronological age in the Braf mutant and wild-type (Wt) intestine. Epigenetic age is persistently accelerated by prolonged exposure to oncogenic Braf V637. Epigenetic age was estimated using models by Stubbs et al (A), Meer et al (B) and a model built by elastic net regression modelling of age using methylation data from the intestine of Wt animals (C). Analysis of covariance (ANCOVA) was performed to test whether the rate of epigenetic ageing differed between Braf V637 and Wt animals.
Braf mutation-specific DNA methylation alterations
Next we analysed the DNA methylation alterations that occurred over time but were exclusive to the Braf mutant small intestine. This represented 61% of all temporal DNA methylation alterations that occur in the presence of Braf mutation (105 561 CpGs, online supplemental figure 1). As these events are specific to oncogenic Braf, we term them type-B methylation events to discriminate them from age-associated methylation (type-A, type-AB). Type-B methylation is similarly distributed to type-A methylation (figures 1B and 3C). Of all type-B hypermethylation events, 17 919 mapped to the promoter region of 3984 genes. Type-B methylation is enriched at cancer-associated signalling pathways (table 3), including the WNT signalling pathway (O/E: 1.82, p=7.34×10-5), the angiogenesis pathway (O/E: 2.0, p=6.89×10-4), the TGF-Beta signalling cascade (O/E: 1.92, p=0.03) and EGF signalling (O/E: 1.76, p=0.04). We identified 310 tumour suppressor genes that were affected by type-B methylation, representing 17.26% of all tumour suppressor genes identified in the TSGene 2.0 database.
Type-B DNA methylation at gene promoters targets specific signalling pathways
Expression analysis of type-B genes that were hypermethylated revealed significant differential expression of 472 genes between 10-day and 14-month-old Braf mutant animals (online supplemental table 4), which corresponds to 11.8% of all type-B genes. This is consistent with our findings on the frequency of gene repression by methylation in human colon cancers.6 Of differentially expressed type-B loci, most were differentially downregulated in 14-month-old Braf mutant animals, suggesting methylation-induced gene silencing (online supplemental figure 3A). Online supplemental figure 3B,C are depictions of type-B genes Cstf2t and Fbn2 that were differentially expressed. Differentially expressed type-B genes included 21 WNT signalling pathway genes (table 4).
Supplemental material
Supplemental material
Differentially expressed type-B WNT signalling pathway genes between BrafW-10D and BrafW-14M animals
We assessed human CIMP panel genes for type-B methylation and report that Igf2 and Neurog1 undergo significant type-B methylation (online supplemental table 3). We did not observe evidence of type-B methylation in Cacna1g, or Socs1. Cacna1g, however, is an age-associated locus that is accelerated by oncogenic Braf (type-AB). We did not capture any CpGs in the Runx3 promoter. Of the five human CIMP panel genes, the expression of two genes, Igf2 and Runx3, was significantly downregulated versus wild-type samples (analysis of variance p value<0.0001, 6.59×10–6, respectively; online supplemental figure 4A).
Supplemental material
Supplemental material
The differential effects of Braf mutation in the aged intestine
We have previously shown that prolonged exposure to mutant Braf from wean can induce murine serrated lesions (mSLs).19 Based on our finding that extensive DNA methylation alterations accumulate with age (figure 1A), we hypothesised that the small intestine of an elderly mouse would rapidly develop serrated neoplasia following induction of Braf mutation. To test this hypothesis, we aged animals for 9 months prior to activation of mutant Braf for a period of 5 months (Braf9–14) and compared both histology and DNA methylation to animals with Braf activated at wean for an identical period of time (BrafW-5).
Following the induction of mutant Braf for 5 months from wean, animals rarely develop mSLs (figure 5A). We compared these animals (n=24, BrafW-5) to animals aged for 9 months prior to induction for the mutation for the same 5-month time period (n=32, Braf9–14). The incidence of mSL in BrafW-5 animals was 4.2% and in Braf9–14 mice it was significantly higher at 43.8% (relative risk: 10.5, Fisher’s exact p<0.001, figure 5A), despite being exposed to oncogenic Braf for the same duration of time. An example of a representative mSL is shown in figure 5B. These data suggest that the age, rather than the length of exposure alone, influences the risk of Braf-induced transformation.

(A) The aged intestine is primed for Braf-induced spontaneous neoplastic transformation. Braf mutation rarely induces murine serrated lesions (mSL) in animals exposed for 5 months from wean. Mutation for the same interval after 9 months of ageing frequently induces murine serrated lesions. (B) Representative example of a murine serrated adenoma located in the intestine of a Braf9–14 mouse. This is a protuberant lesion which projects from the small intestinal surface, forming numerous club-shaped papillae. The papillae are lined by a variety of cell types. At the base the cells are cuboidal, contain clear cytoplasm and resemble the mucin-rich cells seen in human sessile serrated lesions. This transitions to slender cells with abundant pink cytoplasm, which resembles human traditional serrated adenomas. Finally, the tips of the papillae are lined by crowded and hyperchromatic cells with overt dysplastic cytology.
To determine if the increased risk of lesion development in the aged animals may be due to DNA methylation alterations, we performed genome-wide DNA methylation analysis on intestinal mucosa from a subset of these animals (BrafW-5 n=9, Braf9–14 n=12). 68 385 CpG sites were differentially methylated, of which 72.2% were more methylated in the Braf9–14 animals.
We next classified these loci as either type-A (age-associated, independent of Braf mutation), type-AB (age-associated, but the rate of methylation accumulation differs between Braf mutant and wild-type animals) or type-B (occurring exclusively in the setting of Braf mutation), if they were identified in earlier analysis, and ‘other’ if they were not. Forty-three per cent of differential methylation occurred at type-AB loci, 16% at type-A loci, 15% at type-B loci and 26% at unclassified loci (figure 6A).
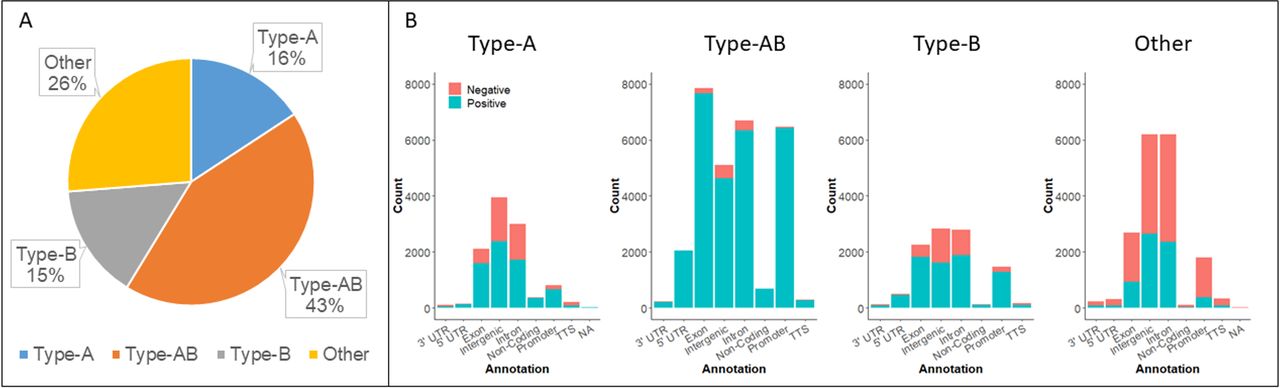
(A) The distribution of differentially methylated CpG sites by methylation type. (B) Methylation type and genomic context. Negative refers to differentially hypomethylated and positive to differentially hypermethylated CpGs. NA, not applicable; TTS, transcription start site; UTR, untranslated region.
Type-A, type-AB and type-B differential methylation was predominantly hypermethylation (65.4%, 96.3%, 72.1%, respectively). However, unclassified methylation events were more likely to be hypomethylation (37% hypermethylated). The distribution of methylation events was similar for type-A and type-B differential methylation (figure 6B). By contrast, type-AB methylation was predominantly hypermethylation across all genomic features and we note that type-AB hypomethylation at gene promoters was extremely rare.
We performed pathways analysis on differentially methylated genes that were classified as type-A, type-AB or type-B. We observed no significant pathway level enrichment for type-A loci that were differentially methylated between BrafW-5 and Braf9–14 animals (table 5). Type-B differentially methylated genes were enriched for G-protein signalling, glutamate receptor signalling and the cadherin signalling pathway (table 5). Similarly, type-AB differentially methylated genes were associated with cadherin and glutamate receptor signalling. Notably, type-AB differentially methylated genes were also strongly enriched for involvement in the WNT signalling cascade (table 5).
PANTHER pathway enrichment for differentially methylated genes in BrafW-5 vs Braf9–14 animals
We also compared the methylation profile of Braf9–14 animals with mSLs to Braf9–14 animals that did not have mSLs (n=6 for both groups). On differential methylation analysis, we identified 2490 CpG sites that differed between these groups, 72.5% of which were significantly more methylated in Braf9–14 animals with mSLs (FDR<0.05, absolute methylation difference of >10%, online supplemental figure 5). One hundred seventy-seven of hypermethylated CpG sites mapped to 92 protein coding genes, and gene ontology enrichment analysis revealed an association with WNT signalling genes (p=0.000907).
Supplemental material
Expression profiling of Braf9–14 mice reveals repression of WNT in the intestinal epithelium
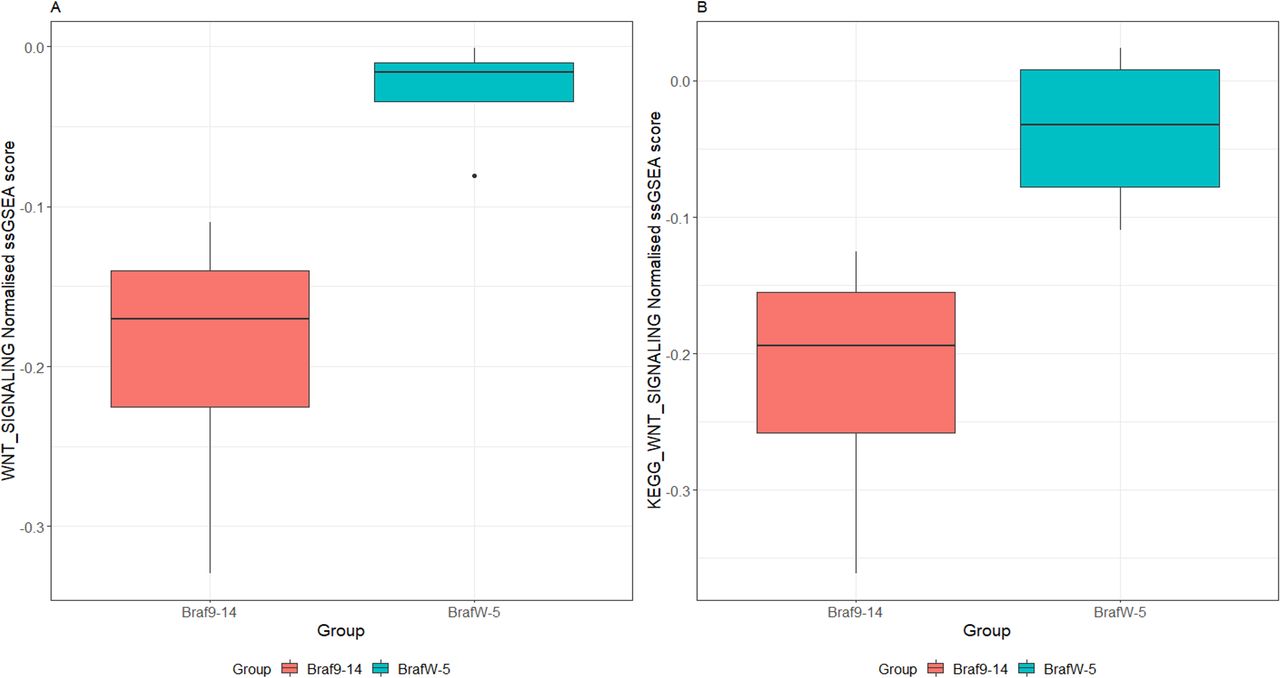
Gene expression profiling and analysis by single-sample gene set enrichment analyses revealed substantial downregulation of the WNT signalling cascade in Braf9–14 intestinal epithelium in comparison to BrafW-5 (p=0.02, figure 7A,B). Lower levels of basal WNT in the epithelium of older mice may increase the selective advantage afforded by (epi)-genetic alterations that increase WNT signal, which may manifest later in the neoplastic cascade. To confirm that WNT signalling is elevated in lesions, we stained sections for β-catenin. In keeping with our hypothesis, β-catenin was significantly elevated in lesions when compared with hyperplasia (p<0.0001, online supplemental figure 6).
Supplemental material
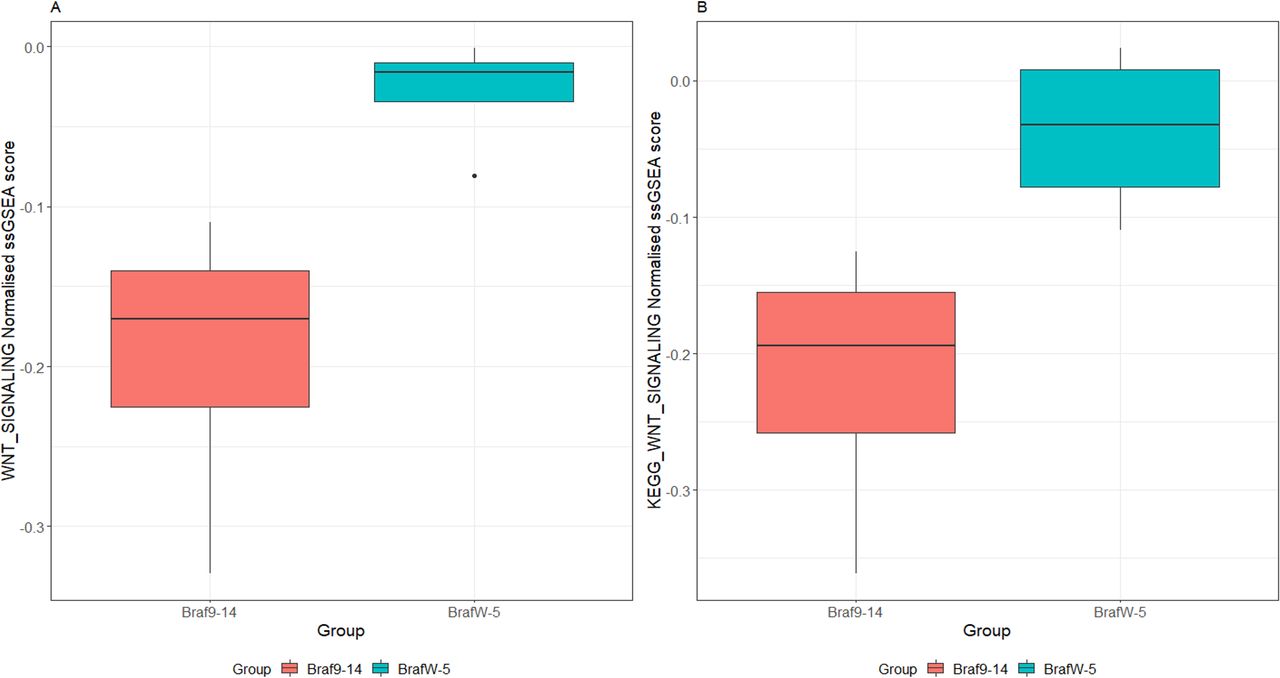
The WNT signalling pathway is repressed in the hyperplastic epithelium of Braf9–14 mice. Pathway activity was assessed by single sample gene set enrichment analysis of transcript expression data obtained via RNA-Seq. The genesets used for this analysis were the WNT_SIGNALING and KEGG_WNT_SIGNALLING gene sets. Data presented is the normalised single sample gene set enrichment score (ssGSEA score).
Cancer associated signaling in murine serrated lesions
To investigate whether WNT is altered in the context of mSLs, we microdissected and extracted RNA from formalin fixed paraffin embedded mSLs (n=3 mSLs and matched mucosal tissue). Using the Nanostring nCounter assay, we profiled the gene expression of 750 cancer associated genes (Extended methods). We identified 173 significantly differentially expressed genes (p<0.05, figure 8A,B). Gene set analysis implicated eight key signalling pathways that were deregulated, including JAK/STAT (GSA score: 4.16), PI3K (GSA score: 4.08), Cell cycle (GSA score: 3.39), TGF-Beta (GSA score 3.06), Wnt (GSA score: 3.03), MAPK (GSA score: 3.03), Apoptosis (GSA score: 2.97), and RAS (GSA score 2.89). We identified several Wnt signalling genes that were significantly differentially expressed in mSLs (figure 8D), including receptors (Fzd3, Fzd9), Wnt ligands (Wnt11), transcription factors (Lef1), and negative regulators (Axin2, Nkd1). These data are in keeping with the upregulation of Wnt observed via β-catenin staining and supports the hypothesis that molecular alterations increasing Wnt signalling may be selected for during tumour formation, and that the selection pressure may be heightened in the aged small intestine with repressed basal Wnt signalling.

(A) Gene expression profiling of 750 cancer-associated genes in murine serrated lesions (mSLs) and hyperplastic mucosa (n=3 mSLs and matched hyperplasia) identifies 173 significantly differentially regulated genes (p<0.05). The horizontal broken line represents p=0.05 and the vertical broken lines represent a log2 fold change of 0.5. (B) Hierarchical clusterings of differentially regulated genes. Hyperplastic samples are denoted by the blue sample bar and mSLs by the green. (C) Top 20 differentially expressed genes and their pathway involvement. (D) Log2 fold change of WNT signalling pathway genes.
Inhibition of DNA methylation attenuates viability of aged intestinal organoids following activation of oncogenic Braf
To link DNA methylation of the aged small intestine to the propensity for lesions to develop, we harvested intestinal tissue from a mouse aged to 10 months. This animal carried the requisite genotype for conversion of the oncogenic BrafV637 allele, but was not yet exposed to tamoxifen. Organoids were established and passaged three times in complete media (see the Extended methods section). Twenty-four hours after the third passage, media was changed to complete media containing 250 nM azacitidine (or diluent control). Organoids were exposed to azacitidine for 48 hours, with drug and media replaced 24 hours after initial exposure. Organoids were passaged, and 24 hours later, 1 μM 4-Hydroxytamoxifen (or diluent control) was spiked into media to induce recombination of the Braf V637E allele.
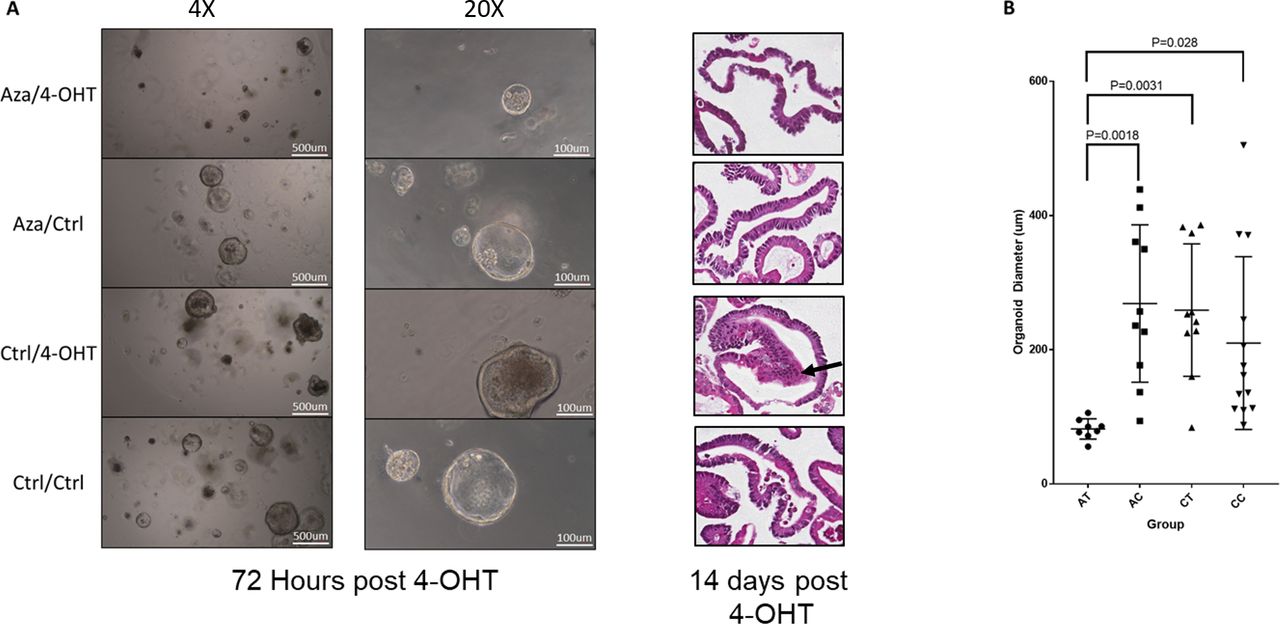
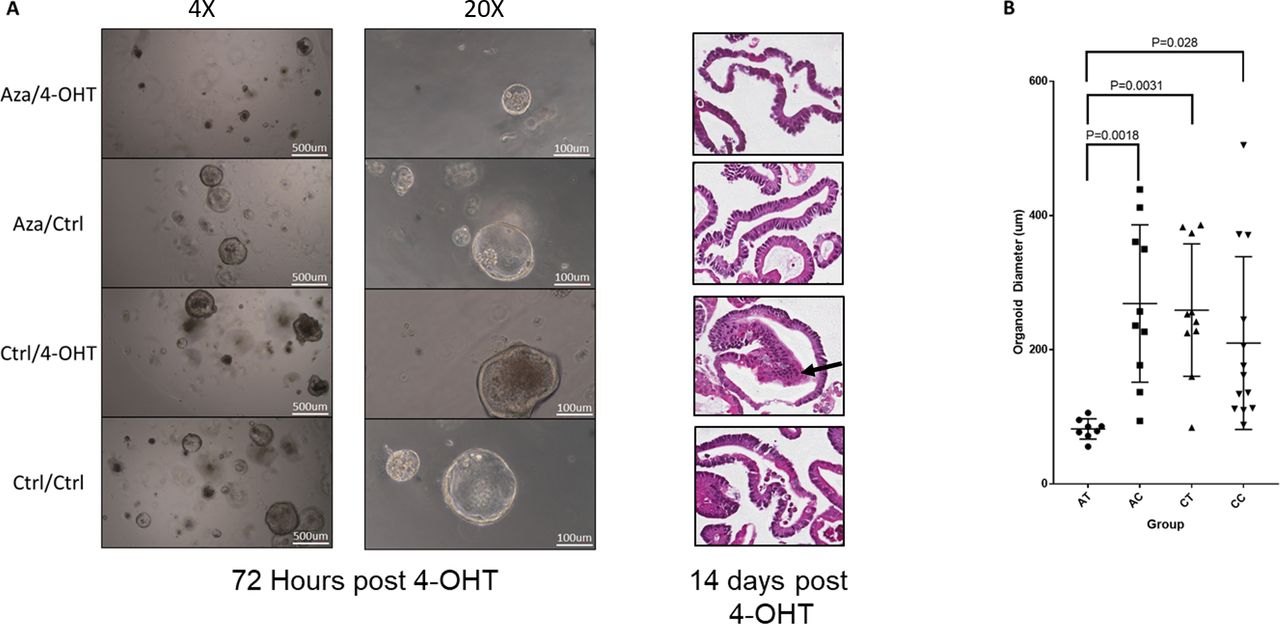
We observed a reduction in organoid growth at 72 hours post-Braf induction in azacitidine-treated organoids (Aza-4OHT, figure 9A), with organoids the average diameter of exposed to azacitidine and 4-OHT being threefold lower than those exposed to azacitidine alone (p=0.0018), 4-OHT alone (p=0.0031) or control (p=0.029) (figure 9B). There was no difference in the average diameter of organoids between the remaining groups (Aza-Ctrl, Ctrl-4OHT, Ctrl-Ctrl; figure 9B). All organoids were observed to maintain a predominantly cystic appearance. This is atypical of non-transformed small intestinal organoids,35 which normally develop complexing branching structures. However, the cystic phenotype has been noted to occur in organoids derived from aged animals36 and may be attributed to dysregulated Wnt signal.36
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Organoids were derived from the intestine of a mouse aged for 10 months. Organoids were passaged three times in vitro, and then treated with 250 nM of azacitidine or diluent control for 48 hours, with drug and media changed at 24-hour intervals. Organoids were passaged and treated with either 4-OHT to induce recombination of the BrafV637 allele, or diluent control. (A) Micrographs of organoids at ×4 and ×20 magnification at 72 hours post 4-OHT and H&E-stained sections at 14 days post 4-OHT. The Ctrl-4OHT organoids demonstrated morphological changes of dysplasia, with cells showing high nuclear to cytoplasmic ratios and atypical nuclear appearance accompanied by multilayering of the epithelium (arrow). This was not identified in the other three groups. (B) Quantification of average organoid diameter. Organoid diameter was measured in a subset of randomly selected organoids from each treatment group.
Organoids were passaged for an additional 11 days to obtain sufficient material for histological processing and analysis. On reviewing the morphology of each group, we observed overt dysplastic changes only in the aged organoids following Braf mutation (Ctrl-4OHT organoids). These organoids showed high nuclear to cytoplasmic ratios and an atypical nuclear appearance, accompanied by multilayering of the epithelium. These alterations were similar to what was observed by Tao et al18 in organoids where Braf was mutated following 10 months of organoid culture. This indicates that inhibition of DNA methylation reduces the capacity for oncogenic Braf to induce neoplastic transformation and provides specific evidence of a functional role for DNA methylation as compared with other age-associated maladies.
Epigenetic drift is accelerated in human SSLs
We also examined the effects of BRAF mutation on epigenetic drift and epigenetic ageing in human samples. To achieve this, we combined three publicly available DNA methylation datasets. The first dataset contained 232 normal colonic mucosal samples (GSE11390437) and was used to train a model to predict epigenetic age. The second dataset contained 149 colonic mucosal samples (GSE10176438) used to test the accuracy of the model. The third dataset consisted of 80 BRAF mutant SSLs (E-MTAB785439). Forty of these SSLs had a focus of dysplasia which was macrodissected and discarded, the remaining 40 had no evidence of dysplasia but were nonetheless CIMP-high lesions. We regard the former as ‘high risk’ SSLs, and the later as ‘moderate risk’ SSLs.
Our human model for intestinal epigenetic age was constructed using elastic net regression on probes that were shared by all three cohorts (n=376 280). The model identified 238 CpG sites that were predictive of age. In our training dataset, the mean error was 0.34 years. In the test dataset, the mean error rate was 2.94 years. When the model was applied to the DNA methylation data from SSLs, we observed a marked increase in epigenetic age of 11.0 years greater than the expected chronological age (p<0.0001, online supplemental figure 7A). This analysis did not include high-risk SSLs; however, when we apply our model to these lesions (n=40), we report a further elevation in epigenetic age compared with moderate-risk SSLs (p=0.08, mean difference: 4.76 years, online supplemental figure 7B). This difference, although not at the threshold for significance, provides evidence that epigenetic age may inform the transformational risk of SSLs.
Supplemental material
Discussion
This study shows that prolonged exposure to mutant Braf markedly increases age-associated epigenetic drift. In the context of BRAF mutant human colorectal cancer, these findings shed light on why the risk of neoplastic transformation increases dramatically with age. The conventional model for colorectal tumourigenesis describes a multistep process whereby somatic mutations sequentially accumulate to promote tumour development over time. Data presented here support a new paradigm pertinent to serrated neoplasia, whereby risk of progression scales with increasing patient age, rather than the length of time since polyp initiation. We have shown that a milieu of epigenetic alterations occur in the ageing intestinal cells, and hypothesise that some of these alterations may contribute to the heightened risk of transformation in the aged intestine. These findings have implications for understanding the natural history of serrated neoplasia, for informing SSL surveillance strategies relative to age and for the rational design of chemopreventive strategies that target age-associated molecular alterations.
Recently, using the same model presented in the current study, we showed that induction of the Braf mutation results in consistent DNA methylation changes analogous to CIMP in human colorectal cancer.19 Here, we have examined both normal and Braf mutant intestinal mucosa in much greater depth. Profiling of the murine intestinal methylome revealed age-associated DNA methylation changes in 8.0% of the >1 million CpGs examined. Most of the loci that were associated with epigenetic drift were also associated with Braf mutation; however, the rate these loci accumulate DNA methylation is significantly faster in the Braf mutant intestine. DNA hypomethylation is also common during both ageing and tumourigenesis.40 It is important to note that the technology used in this study (RRBS) enriches for CpG islands, and that CpGs residing outside of these regions tend to become demethylated with age.40
We report that age-associated loci overlap significantly with the WNT signalling pathway, and genes associated with development and differentiation. Moreover, we identified >100 promoters of tumour suppressor genes that accumulate DNA methylation in a temporal manner. As WNT signalling is crucial in serrated colorectal carcinogenesis41 and the silencing of tumour suppressor genes by DNA methylation is a well-established means of escaping normal cellular regulation, we hypothesise that age-associated DNA methylation at the promoter region of these genes primes the aged intestine for a more rapid neoplastic transformation once a Braf mutation is acquired and an SSL is formed. To test for the involvement of age-associated methylation in determining the risk of Braf-induced neoplasia, we conducted an in vitro organoid experiment. Here, treating organoids established from aged mice, with azacitidine, a DNA methyltransferase inhibitor, greatly reduced the ability for oncogenic Braf mutation to induce growth and neoplastic transformation when activated. Additional studies are necessary to form a definitive causal link between these DNA methylation alterations and risk.
We identified a strong enrichment for immune-related signalling among genes with altered expression in the aged intestine. These genes were not type-A methylation genes and are likely regulated by some other cellular process. Alternatively, the microenvironment of the aged intestine may be comprised of different immune cells, leading to the altered gene expression signal that we observed. Immunosensence is associated with cancer,42 and identifying whether this may be contributing to the elevated risk of neoplasia in the context of Braf mutation remains an important area for future studies.
Here, we have identified three distinct patterns of temporal DNA methylation in the intestine in response to Braf mutation and in the wild-type setting. Type-A DNA methylation alterations occur with age in the normal intestine. There exists a subset of age-associated loci that are prone to a change in the rate of DNA methylation accumulation in response to oncogenic Braf, we have termed these loci type-AB. Type-B DNA methylation alterations occurred with age, but were exclusive to animals with Braf mutation. Both type-A and type-AB loci were enriched for pathways that are relevant to colorectal carcinogenesis, and many of these loci have been reported to be methylated in colon cancers, although to a much greater extent. Thus, we hypothesise that in humans, given an unlimited lifespan, many of the DNA methylation alterations that are observed in BRAF mutant cancers may arise in the absence of BRAF mutation, although over a significantly longer period. The functional consequences of type-A and type-AB methylation in the setting of serrated neoplasia remain unclear, and further study is warranted to elucidate their role in priming the intestinal epithelia for neoplastic transformation.
We next examined tumour burden in animals exposed to mutant Braf for 5 months, either from wean or in animals aged to 9 months. In the aged mice, there was a 10-fold increased relative risk of developing a mSL compared with mice exposed to mutant Braf from wean. We did not observe invasive cancer in our study, and thus it remains possible that ageing facilitates the acquisition of overt cytological dysplasia in serrated lesions, but that some dwell time is still required for malignant progression. In humans, dysplastic SSLs terminate to malignancy in ~12 months.11
Comparisons of the DNA methylation landscape of these groups revealed highly divergent epigenetic profiles. Braf9–14 animals displayed extensive hypermethylation, predominantly confined to type-AB loci. We observed some hypermethylation of type-B loci, suggesting that certain genomic regions become predisposed through ageing to rapid methylation acquisition following activation of oncogenic Braf. Pathways analysis of the affected genes did not reveal enrichments for cancer-associated pathway, and thus we believe it is improbable that type-B methylation differences explain the increased risk of neoplasia in the aged intestine; however, given the correlative nature of our study, it remains possible that some methylation alterations in type-B loci might contribute to the overall increased risk. In contrast, type-AB methylation differences were enriched for the WNT signalling pathway, and the enrichment for WNT signalling among these specific type-AB loci was stronger than the overall fold enrichment of type-AB loci. When we examined Braf9–14 mice that had mSLs, in comparison to those that did not, we observed significant hypermethylation at gene promoters and especially those encoding WNT signalling regulators. Many of the loci that were hypermethylated with age, either in the wild-type or Braf mutant setting, were lowly expressed. Methylation was not correlated with expression in many of these genes. Tao et al18 reported a similar finding in their in vitro system. They note that although lowly expressed in all conditions, DNA methylation appeared to restrain the activation of affected genes, and thus transient, context-dependent expression was prevented. We hypothesis that this phenomena is also occurring in our in vivo model. Transcriptomic profiling of the intestine of Braf9–14 (in comparison to BrafW-5 animals), revealed significant repression of WNT signalling. The repression of WNT signalling in the ageing intestine is consistent with previous studies.43 The repression of WNT signalling in the aged epithelium may heighten the selective pressure for pro-transformation (epi)genetic alterations in WNT signalling, which may ultimately result in neoplastic transformation and the formation of an mSL. This is consistent with our earlier finding that mSLs44 (and growth factor independent Braf mutant organoids18) frequently harbour Ctnnb1 mutations, and our observation here of increased Wnt signalling activity in mSLs.
In vitro assessment of colonic organoids has also shown that DNA methylation changes accumulate over time and may create an environment more permissive to oncogenic transformation.18 The elegant experiments of Tao et al18 showed that organoids continuously cultured for 12 months were signficiantly more predisposed to Braf-induced transformation than those that were cultured for shorter periods. This finding implied that ageing may modify the risk of Braf-induced transformation. The in vivo study we present here provides an important validation of these findings and resolves a key limitation in this earlier work, being the possibility that the culture environment may have contributed to the effects Tao et al18 observe. Our study shows this not to be the case, and lays forth a foundation for clinical studies in this area. On the mechanism of DNA methylation acceleration of by Braf, we identify three hypotheses. The first, as proposed by Tao et al,18 is that cells harbouring DNA methylation alterations that have occurred during ageing have a growth advantage, and continue to be selected for in the presence of Braf mutation, thus increasing the overall level of DNA methylation in the sample. The second, that Braf mutation induces a process that directly influences DNA methylation (such as oxidative stress, or via MAFG as suggested by Fang et al16), and the third that age-associated DNA methylation alterations are accelerated by Braf as a consequence of oncogene-induced proliferation and mitosis, and thus the tissue ‘ages’ relatively more rapidly compared with the wild-type intestine. Discerning between these competing (or potentially co-occuring) hypotheses is an important area of future research.
We also examined the impact of epigenetic drift in human samples. We developed an epigenetic clock model for human colon using two independent datasets. We then applied this to a series of SSL and observed an acceleration of epigenetic age of 11 years. Moreover, we report that DNA extracted from the non-dysplastic compartment of human SSLs with a focus of dysplasia has an elevated epigenetic age when compared with age-matched human SSLs with no evidence of cytological dysplasia. These data indicate that advanced epigenetic age may indicate risk of neoplastic progression.
In this study, we aged mice to 9 months prior to activating oncogenic Braf, equivalent to middle age in humans. Future studies could age animals for much longer to mimic elderly humans, before inducing the Braf mutation for even shorter periods of time. We also obtained samples from the small intestine, as this is where most of the phenotype associated with this model develop. This is consistent with prior works in models of colorectal cancer. Future work could develop models that develop phenotypes in the large bowel to maintain consistency with human colorectal cancer. In addition, further validation in human specimens is required, especially in polyps where the dwell time can be calculated from a previous clearing colonoscopy. As the CIMP is an indelible component of human BRAF mutant cancers, this was the focus of our investigations. Recently, Lee-Six and colleagues45 showed the pervasive accumulation of genetic alterations that scales with age, in the colonic crypts of healthy participants. Lee-Six et al45 reported that ~1% of colonic crypts of middle-aged participants contained driver mutations. In this study, we preserved the mSLs in formalin for histopathological analysis, as such we were unable to assess the mutational status of lesions. It is possible that age-associated risk of serrated neoplasia is also influenced by the accumulation of genetic lesions with age, and that some of these genetic alterations may subvert the cellular processes restraining transformation. It is likely that a combination of age-associated events contributes to the risk we have observed in the present study.
Conclusion
Here, we have comprehensively evaluated epigenetic drift in the murine intestine and shown that prolonged exposure to mutant Braf rapidly accelerates the rate of epigenetic drift. We have provided evidence that the WNT signalling pathway is a putative target of epigenetic drift. We have demonstrated in vivo that mutation of Braf in an aged intestine dramatically increases the rate of developing neoplasia compared with the same mutation occurring in young intestine, and show striking methylation differences between the intestinal epithelium of animals induced at a young age compared with those at an older age. Our in vitro organoid studies show that DNA demethylation via DNMT1 inhibition in aged organoids prevents Braf-induced cytological dysplasia and inhibits proliferation. These findings may have implications for our understanding of the development of serrated neoplasia across the age spectrum in humans and may influence the rational development of personalised surveillance guidelines according to the age of the individual. The ability to reverse epigenetic alterations also provides an impetus for investigating chemopreventive strategies to slow epigenetic drift to attenuate cancer development.
Methods
Murine model of serrated neoplasia
We recapitulated Braf mutant serrated neoplasia in vivo by crossing animals with the conditionally activated BrafV637E allele with VillinCreERT2 animals as previously reported.19 Intraperitoneal administration (75 mg/kg) of tamoxifen at 14 days post birth directs intestinally specific recombination of the BrafV637E allele, which is analogous to the human BRAFV600E mutation. Animals were genotyped using the method reported by Bond et al.19 Braf mutation was activated at wean (W) and animals were sacrificed at a range of timepoints across the murine lifespan (referred to as BrafW-X, where X = the number of months post induction of the Braf mutation) (table 6). We also examined the morphological implications of ageing following Braf mutation by activating oncogenic Braf at 9 months of age for a period of 5 months. We assessed the histology, DNA methylation and gene expression of the intestinal epithelium of these animals (table 6). All molecular analyses, with the exemption of the nanostring assay were performed on intestinal epithelial samples, and not mSLs, which were preserved for histological analyses. Extended methods are available as online supplemental materials. We confirmed efficient recombination of the mutant Braf allele by polyacrylamide gel electrophoresis of DNA extracted from intestinal samples (online supplemental figure 8).
Supplemental material
Summary of timepoints and analysis performed on the respective samples
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Acknowledgments
We acknowledge the work of the staff at the Australian Genome Research Facility and the QIMR Berghofer Sequencing Facility for their work on generating the sequence data analysed in this study, and the work of the QIMR Berghofer Histology department in preparing histological specimens. We acknowledge the contributions of the reviewers in enhancing the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors LF: Conceptualisation, data curation, formal analysis, investigation, software, methodology, writing—original draft, writing—review and editing. AK: Methodology, investigation, project administration, writing—review and editing. CL: Investigation, data curation, methodology, writing—review and editing. DM: Investigation, project administration, writing—review and editing. GH: Formal analysis, statistical analysis, writing—review and editing. CS: Investigation, writing—review and editing. CB: Conceptualisation, investigation, project administration, writing – review and editing. MB: Conceptualisation, funding acquisition, methodology, writing—review and editing. BL: Conceptualisation, funding acquisition, project administration, supervision, writing—review and editing. VW: Conceptualisation, funding acquisition, project administration, supervision, writing—original draft, writing—review and editing.
Funding This work was supported through funding from the National Health and Medical Research Council (Grant No: 1050455, 1063105), the Cancer Council Queensland (1160923), the RBWH Research Foundation and Pathology Queensland. VW is the recipient of a Senior Research Fellowship from the Gastroenterological Society of Australia. LF was supported by a Research Training Program Living Scholarship from the Australia Government, a Top-Up award from QIMR Berghofer and Australian Rotary Health.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.