Article Text

Abstract
Objectives Transcriptomic-based subtyping, consensus molecular subtyping (CMS) and colorectal cancer intrinsic subtyping (CRIS) identify a patient subpopulation with mesenchymal traits (CMS4/CRIS-B) and poorer outcome. Here, we investigated the relationship between prevalence of Fusobacterium nucleatum (Fn) and Fusobacteriales, CMS/CRIS subtyping, cell type composition, immune infiltrates and host contexture to refine patient stratification and to identify druggable context-specific vulnerabilities.
Design We coupled cell culture experiments with characterisation of Fn/Fusobacteriales prevalence and host biology/microenviroment in tumours from two independent colorectal cancer patient cohorts (Taxonomy: n=140, colon and rectal cases of The Cancer Genome Atlas (TCGA-COAD-READ) cohort: n=605).
Results In vitro, Fn infection induced inflammation via nuclear factor kappa-light-chain-enhancer of activated B cells/tumour necrosis factor alpha in HCT116 and HT29 cancer cell lines. In patients, high Fn/Fusobacteriales were found in CMS1, microsatellite unstable () tumours, with infiltration of M1 macrophages, reduced M2 macrophages, and high interleukin (IL)-6/IL-8/IL-1β signalling. Analysis of the Taxonomy cohort suggested that Fn was prognostic for CMS4/CRIS-B patients, despite having lower Fn load than CMS1 patients. In the TCGA-COAD-READ cohort, we likewise identified a differential association between Fusobacteriales relative abundance and outcome when stratifying patients in mesenchymal (either CMS4 and/or CRIS-B) versus non-mesenchymal (neither CMS4 nor CRIS-B). Patients with mesenchymal tumours and high Fusobacteriales had approximately twofold higher risk of worse outcome. These associations were null in non-mesenchymal patients. Modelling the three-way association between Fusobacteriales prevalence, molecular subtyping and host contexture with logistic models with an interaction term disentangled the pathogen–host signalling relationship and identified aberrations (including NOTCH, CSF1-3 and IL-6/IL-8) as candidate targets.
Conclusion This study identifies CMS4/CRIS-B patients with high Fn/Fusobacteriales prevalence as a high-risk subpopulation that may benefit from therapeutics targeting mesenchymal biology.
- colorectal cancer
- intestinal microbiology
- molecular mechanisms
- biostatistics
Data availability statement
Data are available in a public, open access repository. Processing and analysis code along with bacterium estimates with corresponding clinical and molecular datasets for the Taxonomy and TCGA-COAD-READ cohorts included in this study are publicly available and archived at Zenodo (https://10.5281/zenodo.4019142). Bacterium estimates include Fusobacterium nucleatum load (Taxonomy cohort) and Fusobacteriales relative abundance, along with higher resolution estimates at genus, family and species taxonomic ranks, (TCGA-COAD-READ cohort).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Fusobacterium nucleatum (Fn), a commensal Gram-negative anaerobe from the Fusobacteriales order, is an oncobacterium in colorectal cancer (CRC), and a causal relationship between Fn prevalence and CRC pathogenesis, progression and treatment response has been reported in vivo.
Broad-spectrum antibiotics have proven moderately successful in reducing tumour growth promoted by Fn in preclinical models. However, the use of antibiotics to treat bacterium-positive cases in the clinic is not a viable option as it may further alter the already dysbiotic gut microbiome of patients with CRC and may also have limited efficacy against Fn, which penetrates and embeds deeply within the tumour.
The highly heterogeneous population of patients with CRC can be classified into distinct molecular subtypes (consensus molecular subtyping (CMS) and colorectal cancer intrinsic subtyping (CRIS)) based on gene expression profiles mirroring the underlying transcriptional programmes. Patients classified as CMS4 and CRIS-B exhibit a mesenchymal phenotype and have poorer outcome.
Significance of this study
What are the new findings?
Fn/Fusobacteriales prevalence is associated with immune involvement (decrease in antitumour M1 macrophages and increase in protumour M2 macrophages) and activation of specific signalling programmes (inflammation, DNA damage, WNT, metastasis, proliferation and cell cycle) in the host–tumours.
The prevalence of bacteria from the Fusobacteriales order, largely driven by Fn species, plays an active or opportunistic role, depending on the underlying host–tumour biology and microenvironment.
Fn and other species of the Fusobacteriales order are enriched in CMS1 (immune-high, microsatellite unstable tumours) patients compared with CMS2–4 cases.
Fn/Fusobacteriales prevalence is associated with worse clinical outcome in patients with mesenchymal-rich CMS4/CRIS-B tumours but not in patients with other molecular subtypes.
How might it impact on clinical practice in the foreseeable future?
Fn/Fusobacteriales screening and transcriptomic-based molecular subtyping should be considered to identify patients with mesenchymal-rich tumours and high bacterium prevalence to inform disease management.
Fn/Fusobacteriales prevalence may need to be addressed exclusively in patients with mesenchymal-rich high-stromal infiltrating tumours rather than a blanket approach to treat all pathogen-positive patients.
Clinical management of the disease for this subpopulation of high-risk patients with unfavourable clinical outcome could be attained by administering agents currently in clinical trials that target aberrations in the host signalling pathways (NOTCH, WNT and epithelial-mesenchymal transition) and tumour microenviroment (inflammasome, activated T cells, complement system, and macrophage chemotaxis and activation).
Introduction
Colorectal cancer (CRC) has one of the highest morbidities and mortality rates among solid cancers, and its incidence is steadily on the rise, accounting for circa 10% of newly diagnosed cancer cases worldwide.1 Patients with CRC with similar macroscopic clinicopathological characteristics exhibit a high degree of heterogeneity at the molecular level, which translates into heterogeneous and often suboptimal response to treatment. Thus, research has focused on molecular subtyping strategies based on single or multiomics data from the host to categorise patients into subgroups to aid in risk stratification and disease management. Subtyping strategies such as the consensus molecular subtyping (CMS2) and the colorectal cancer intrinsic subtyping (CRIS3) classify patients into subgroups with more homogeneous signalling features based on key transcriptomic programmes. Among the four subtypes identified by the CMS classifier, CMS4 patients have high stroma infiltration along with upregulated angiogenesis and transforming growth factor-β (TGF-β) signalling and show poorer recurrence-free and overall survival.2 Similarly, CRIS-B patients feature mesenchymal traits and also exhibit poorer outcome compared with patients classified as CRIS-A and CRIS-C–E.3
Recent research has identified the microbiome as a key player in health and disease, including cancer.4 Several research groups, including ours, have shown that Fusobacteriales, largely from Fusobacterium nucleatum (Fn), are more abundant in tumour tissue compared with matched adjacent mucosa,5 suggesting a causative role in CRC progression.6 More advanced, right-sided, MSI tumours are typically enriched with Fn.7 Remarkably, antimicrobial treatment has been shown to reduce tumour burden in mouse xenograft models,8 corroborating the association between Fn-positive patients and poorer outcome observed in some studies.5 However, the prognostic value of Fn prevalence was not observed in other cohort studies (reviewed in Gethings-Behncke et al 9). Thus, we hypothesised that the impact of Fn/Fusobacteriales may differ according to the underlying tumour biology.
In this study, we combined mechanistic in vitro experiments in colon cancer cells with an in-depth analysis in two independent CRC patient cohorts and a systematic multiomic characterisation of cell signalling and tumour microenvironment in n=745 patients to investigate the interaction between the dysregulation induced by Fusobacteriales prevalence (including Fn) on the human host and, conversely, the characteristics of the host microenvironment that allow pathogens to thrive. Here, we provide evidence that the prognostic value of Fn/Fusobacteriales strongly relates to the molecular subtype of the host–tumour and is confined to subtypes showing mesenchymal involvement.
Materials and methods
Detailed methods for the in vitro experiments and the patients’ study (design, cohorts’ description and analysis steps) are provided in the online supplemental materials and methods.
Supplemental material
Results
Fn infection induces inflammation mediated by tumour necrosis factor alpha (TNF-α) and NFκB in CRC cellular cultures
Due to the presence of Fn in CRC tumour tissue,5 8 a causative role for this bacterium in exacerbating tumourigenesis has been put forward. Infection of colon cells with Fn has previously been shown to induce inflammation, activate NFκB signalling and increase expression of the proinflammatory cytokine TNF-α10 11 (figure 1A). Hence, we infected HCT116 and HT29 colon cancer cell line cultures for 6 hours to assess epithelial cell response to increasing amounts of Fn (multiplicity of infection, (MOI), bacteria-to-cancer-cells: 10, 100 and 1000). We found that NFκB signalling was activated on infection with Fn in CRC cell lines, as evidenced by the degradation of IκBα (alpha nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor) (figure 1B), an increase in NFκB transcriptional activity (figure 1C) and a marked increase in mRNA expression of the NFκB target gene, TNF-α (figure 1D). Taken together, these results confirm that Fn coculture with human colon cancer epithelial cells promotes a proinflammatory response.

Fn infection induces inflammation mediated by TNF-α and NFκB in HCT116 and HT29 CRC cell lines. (A) Schematic representation of the experimental set-up to investigate how Fn may trigger inflammation via TNF-α and NFκB signalling pathways. (B) Western blot analysis of IκBα and β-actin in HT29 and HCT116 cell cultures following infection with Fn (MOI bacteria-to-cancer-cells 10, 100 and 1000). (C) NFκB transcriptional activity assay in HCT116 cells 6 hours following infection with Fn (MOI bacteria-to-cancer-cells 100 and 1000). (D) TNF-α mRNA expression relative to β-tubulin in HT29 cells 6 hours following infection with Fn (MOI bacteria-to-cancer-cells 100 and 1000). (B–D) Representative results from duplicate experiments. Fn, Fusobacterium nucleatum; TNF-α, tumour necrosis factor alpha.
Prevalence of Fn/Fusobacteriales in tumour resections
We sought to investigate the relationship between inflammation in the human host and prevalence of Fn and Fusobacteriales in tumour resections of patients with CRC. We selected an in-house multicentre stage II–III cohort (Taxonomy, n=14012 13) and the colon and rectal cases of The Cancer Genome Atlas (TCGA-COAD-READ cohort, n=605 patients; figure 2A) to encompass the heterogeneity of the CRC clinicopathological characteristics observed in the clinic. Demographic, clinicopathological characteristics for the Taxonomy and TCGA-COAD-READ cohorts are summarised in online supplemental table 1. We determined Fn load by a targeted quantitative real-time PCR in tumour resections of the Taxonomy cohort where we detected Fn in n=101 of 140 (72%) patients (figure 2B and online supplemental table 2). The distribution of Fn positivity levels (relative to the human PGT gene) was heterogeneous, and we categorised patients as Fn-high or Fn-low using the 75th percentile as cut-off (figure 2B). We estimated Fusobacteriales relative abundance (RA) in the TCGA-COAD-READ cohort from RNA sequencing data by mapping non-human reads to microbial reference databases and retaining only high-quality matches (see the Materials and methods section) with a PathSeq analysis14 15 (figure 2A and online supplemental table 2). For downstream analyses, we reported the RA at the order, family, genus and species taxonomic rank, and expressed it as percentage of the total bacterial abundance. We detected Fusobacteriales (defined as RA over zero, at the order level) in n=558 of 605 (92%) of the TCGA-COAD-READ patients (figure 2D). Fn was the most abundant species and was detected in 82% of the TCGA-COAD-READ patients (compared with 72% in the Taxonomy cohort), accounting on average for approximately 45% of total Fusobacteriales RA and accounting for over 75% of total Fusobacteriales RA in 16% of cases (figure 2C). Analogous to the Taxonomy cohort, we categorised patients as Fusobacteriales-high or Fusobacteriales-low using the 75th percentile as cut-off.
Supplemental material
Supplemental material
Supplemental material

Fn/Fusobacteriales prevalence is associated with inflammation and immunosuppression in patients with CRC of the Taxonomy and TCGA-COAD-READ CRC cohorts. (A) Schematic representation of the cohorts included in the study and methods to estimate Fn load and Fusobacteriales (order) RA in the Taxonomy and TCGA-COAD-READ cohorts, respectively. (B–D) Per-patient (waterfall plot, 1, left) and distribution (violin plot with overlaid data-points, 2, right) of bacterium prevalence in tumour resections of the Taxonomy (n=140, B) and TCGA-COAD-READ (n=605, D). In B,D 1, patients are sorted in ascending order of either Fn load (Taxonomy cohort, B) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-READ cohort, D). Cut-off of 75th percentile used for patients’ stratification in downstream analysis is also indicated (black dotted line). (C) Corresponding per-patient fraction of Fn species to total Fusobacteriales order RA detected for the TCGA-COAD-READ cohort . (E,F) Violin plots depicting the expression distribution of key genes or signatures involved in inflammation and immunosuppression grouped by patients with low (in green) or high (in orange) either Fn load (Taxonomy cohort, E) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-READ cohort, F). Median and lower (25th) and upper (75th) percentiles are indicated by white solid or dashed lines, respectively. Statistical significance was evaluated using Kruskal-Wallis tests and p values are reported. (G,H) Stacked bar plots indicating cell type composition per patient estimated from gene expression by quanTIseq in tumours with low versus high either Fn load (Taxonomy cohort, G) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-cohort, H). Cell type composition is shown sorted in ascending order of tumour and stromal content (1 and 3) and aggregated (by mean, 2 across the low and high subgroups). (I,J) Distribution of specific tumour/stroma and immune cell types determined as indicated by either quanTIseq or MCP-counter grouped by either Fn load (Taxonomy cohort, I) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-READ cohort, J). Median and lower (25th) and upper (75th) percentiles are indicated by white solid or dashed lines, respectively. Statistical significance was evaluated using Kruskal-Wallis tests and p values are reported. CRC, colorectal cancer; Fn, Fusobacterium nucleatum; NK, natural killer cells; RA, relative abundance; TCGA-COAD-READ, colon and rectal cases of The Cancer Genome Atlas; Treg, regulatory T cell.
Higher Fn/Fusobacteriales prevalence correlates with inflammation and immune involvement
We examined the association between host gene expression profiles of key inflammatory markers and either Fn load or Fusobacteriales RA in the Taxonomy and TCGA-COAD-READ cohorts, respectively. In line with the in vitro experiments (figure 1), we detected an increase in NFKB1 and a trend in TNF-α gene expression, recapitulated by transcriptomic-based signatures for an overall inflammation status mediated by the cytolytic and interferon gamma (IFN-γ) pathways in the Taxonomy cohort (figure 2E). When investigating further key inflammation players, we observed a marked increase in proinflammatory interleukins (ILs) (IL-6, IL-8, IL-10, IL-1β and IL-13), cytokines/chemokines (CCL8, CSF1 and ICAM1), metalloproteins (MMP1, MMP3 and MMP9), NOS2, the inflammasome complex (NLRP3) and decrease in COX2 in Fn-high versus Fn-low Taxonomy patients (figure 2E and online supplemental figure 1).
Supplemental material
We sought to validate and build on our findings from the in-house Taxonomy cohort by analysing the TCGA-COAD-READ cohort (figure 2F). At the transcription level, we confirmed an exacerbated inflammatory state when comparing Fusobacteriales-high and Fusobacteriales-low patients mediated by the NFκB–TNF-α axis and IFN-γ with cytolytic involvement. Fusobacteriales-high patients overexpressed proinflammatory ILs (IL-6, IL-8, IL-10 and IL-1β), cytokines/chemokines (CCL8 and ICAM1), metalloproteinases (MMP1 and MMP3), NOS2 and inflammasome markers (NLRP3) (figure 2F).
As inflammation is strongly tied to immune cell migration and activity, we investigated whether there was a link between immune cell composition and either Fn load (taxonomy) or Fusobacteriales RA (TCGA-COAD-READ). Cell composition was computationally deconvoluted from gene expression profiles with quanTiseq16 and microenvironment cell populations (MCP)-counter17 (figure 2G,H). Despite observing high interpatient heterogeneity in cell composition within the Taxonomy and TCGA-COAD-READ cohorts, we detected higher immune cell activation and polarisation when comparing patients with high versus low Fn load (Taxonomy) or Fusobacteriales RA (TCGA-COAD-READ). Patients with high Fn load (Taxonomy) or Fusobacteriales (TCGA-COAD-READ) showed higher predicted abundance of regulatory T cells coupled with an increase in M1 macrophages and a decrease in M2 macrophages (figure 2I,J). MCP-counter identified a strong positive association between neutrophil infiltration and either Fn load (Taxonomy) or Fusobacteriales RA (TCGA-COAD-READ). However, no difference in predicted neutrophils abundance was detected by quanTIseq. Importantly, no difference in fibroblasts and endothelial cells was observed by Fn/Fusobacteriales in either cohort by either method (figure 2I,J).
Multiomic characterisation of the association between Fusobacteriales RA and human host–tumour microenvironment in the TCGA-COAD-READ cohort
We leveraged the rich molecular characterisation of the TCGA-COAD-READ cohort to perform a systematic and unbiased characterisation of the association between Fusobacteriales RA and patient clinical and molecular features to identify human host vulnerabilities that may be conducive for tumour development (figure 3).

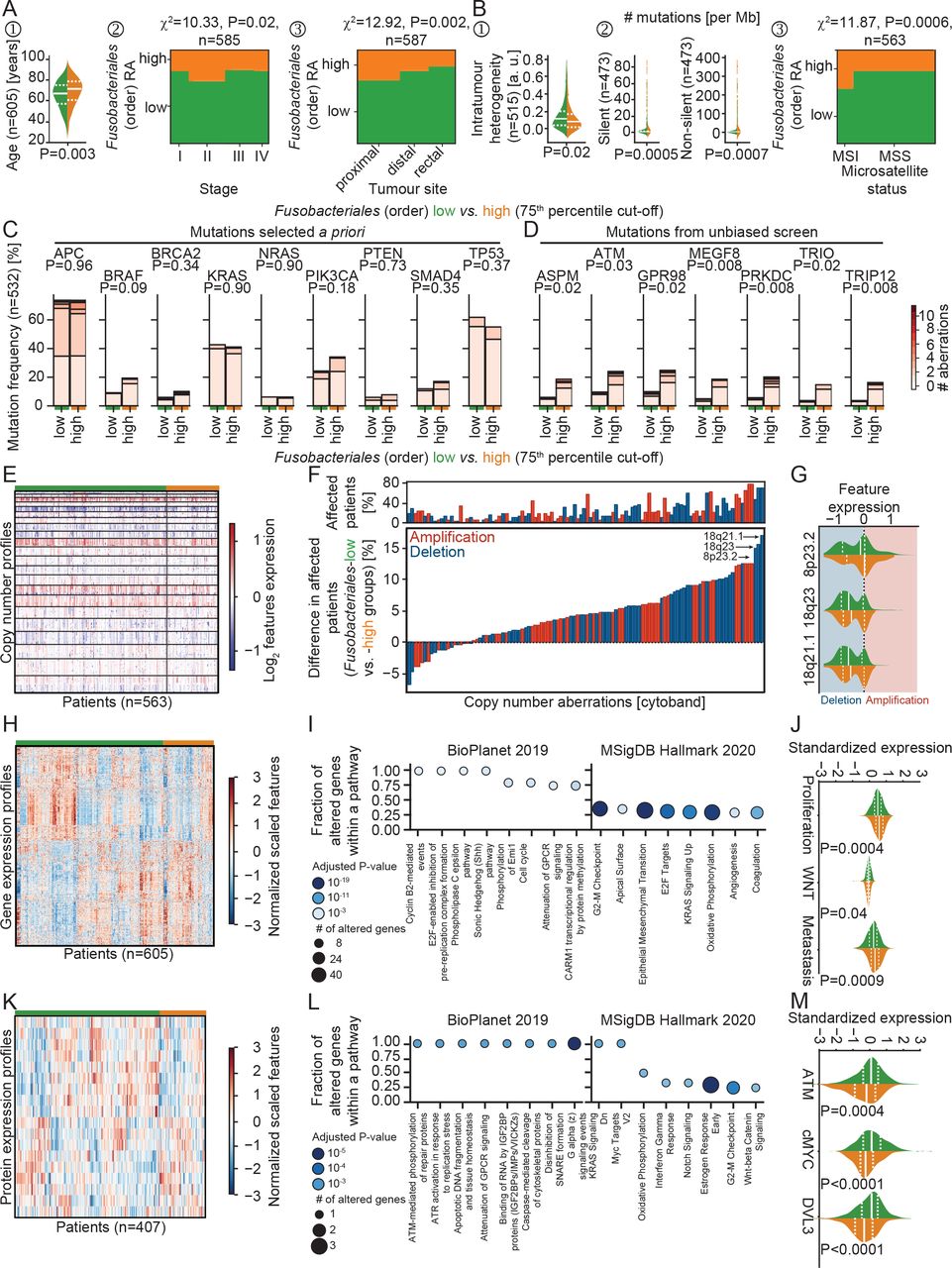
Multiomic characterisation of the association between Fusobacteriales RA and human host–tumour microenvironment in the TCGA-COAD-READ cohort. (A,B). Association between Fusobacteriales at the order taxonomic rank binned into low versus high (cut-off 75th percentile) and clinicopathological (A) and mutational (B) characteristics of the human host. (C,D) Comparison of frequency of occurrence of mutations selected a priori (C) or identified by an unbiased scan (D) in Fusobacteriales-low versus Fusobacteriales-high patients. Colour bar indicates number of detected aberrations among frame shift deletions and insertions, in frame deletions and insertions, missense and nonsense mutations, and splice sites. P values were computed with χ2 independence tests and adjusted for multiple comparisons (Benjamini-Hochberg false discovery rate). (E–G) Heatmap (E) displaying copy number alterations grouped by Fusobacteriales-low (in green) and Fusobacteriales-high (in orange) RA. Waterfall plot (F) displaying differences in recurrent copy number aberrations detected in patients with low Fusobacteriales versus high Fusobacteriales. Top panel (F) reports percentage of patients affected by recurrent copy number aberrations. Distribution of top 3 deletions, the frequency of occurrence of which differs between Fusobacteriales-low and Fusobacteriales-high patients (G). Red and blue shadings indicate amplification and deletions, respectively. (H–J) Heatmap (H) displaying expression of genes differentially expressed when comparing Fusobacteriales-low versus Fusobacteriales-high patients and corresponding pathway enrichment analysis (I). Expression distribution grouped by Fusobacteriales RA (low, in green, vs high, in orange) for selected gene expression signatures (J). (K–M) Heatmap (K) displaying expression of proteins differentially expressed when comparing Fusobacteriales-low versus Fusobacteriales-high patients and corresponding pathway enrichment analysis (L). Expression distribution grouped by Fusobacteriales RA (low, in green, vs high, in orange) for key proteins (M). In violin plots, the median and lower (25th) and upper (75th) percentiles are indicated by white solid or dashed lines, respectively. Green and orange annotation bars denote patients with low versus high Fusobacteriales RA (75th percentile cut-off). (Unadjusted) P values (J,M) were determined by Kruskal-Wallis tests. MSS, patients with microsatellite stable tumours; RA, relative abundance; TCGA-COAD-READ, colon and rectal cases of The Cancer Genome Atlas.
We observed higher Fusobacteriales in patients of older age, diagnosed with more advanced disease stage and tumours located in the colon, particularly in proximal site (figure 3A) cohorts. In contrast, we found no statistically significant differences in Fusobacteriales RA by sex, body mass index and either lymphovascular or perineural invasion (online supplemental figure 2). We observed similar patterns and a slightly higher prevalence in women (Taxonomy cohort, p=0.049), when assessing Fn in both the TCGA-COAD-READ and Taxonomy cohorts (online supplemental figure 3A), corroborating previous studies.18
Patients harbouring higher Fusobacteriales showed lower genomic intratumour heterogeneity, had higher silent and non-silent mutational burden and were enriched in microsatellite unstable cases (figure 3B and online supplemental figure 3B). Fusobacteriales-high patients had an increase in transitions, defined as the exchange of two-ring purines (A↔G) or of a one-ring pyrimidines (C↔T), coupled with a decrease in transversions, a substitution of purine for pyrimidine bases (online supplemental figure 4A) as evidenced by a decrease in conversion changes of C>G and T>A (online supplemental figure 4B). We found no difference in prevalence of common mutations in CRC by Fusobacteriales (low vs high) except for BRAF (figure 3C). BRAF mutations trended to be more common among Fusobacteriales-high and Fn-high patients, as previously reported when assessing Fn 18 (figure 3C and online supplemental figure 3B). A comprehensive screen revealed that mutations in cell cycle (ATM), Hedgehog signalling (MEGF8), DNA damage/repair (TRIP12 and PRKDC), mitotic spindle (ASPM) and migration/adhesion (TRIO, GPR98) were more prevalent in Fusobacteriales-high patients (figure 3D) (online supplemental table 3).
Supplemental material
We set out to investigate the relationship between copy number alterations (CNAs) and Fusobacteriales presence in the TCGA-COAD-READ cohort (figure 3E–G). We determined recurrent CNA amplifications and deletions across the whole cohort by applying the genomic identification of significant targets in cancer (GISTIC) algorithm19 (online supplemental figures 5 and 6 and online supplemental table 4). Fusobacteriales-high cases showed lower chromosomal instability with a lower fraction of the genome affected by recurrent CNAs, in line with the increased incidence of MSI. We identified CNA amplifications or deletions, the frequency of occurrence of which differed between Fusobacteriales-high versus Fusobacteriales-low patients and, thus, may be specifically associated with the bacterium presence (figure 3F). CNAs more frequently (>15%) observed in Fusobacteriales-high versus Fusobacteriales-low cases included deletions in 8p23.2 (tumour suppressor CSMD1 and LOC100287015), 18q21.1 (MIR4743 and RNA binding by CTIF) and 18q23, which impact the regulation of IL-6 and chemokine secretion, cell–cell adhesion and host response of viral transcription (figure 3G).
Supplemental material
We then focused on the transcriptional level and combined enrichment analyses with pathway-activity signatures to compare the impact of Fusobacteriales RA on cellular processes (figure 3H–J). Transcriptional profiles that differed included mTORC1 and cMYC signalling, cell cycle (G2-M checkpoint), mitotic spindle, epithelial-to-mesenchymal transition, TGF-β and IL-1 regulation of extracellular matrix, matrix remodelling including focal adhesion, cytoskeleton and contractile actin filament bundle, mitochondrial translational elongation/termination, and protein complex assembly and stromal estimates (figure 3H,I, online supplemental figure 7 and supplemental table 5). We corroborated these findings by comparing the activation of signalling pathways estimated by gene set signatures identified in the literature (see the Materials and methods section) in Fusobacteriales-low versus Fusobacteriales-high patients. Fusobacteriales RA was inversely linked to WNT signalling and positively associated with proliferation, metastasis (figure 3J) and DNA damage.
Supplemental material
We sought to investigate whether the findings at the genomic and transcriptional levels were also observed in protein profiles determined by reverse phase protein array. We found that Fusobacteriales RA correlated with differential expression of proteins involved in microenvironment composition (Claudin7), cell cycle (Cyclin1), cMYC, apoptosis (cleaved Caspase7), proliferation (DLV3), Hippo pathway (Yap), DNA damage (Chk1 and ATM), receptor and mitogen-activated protein(MAP) kinases and PI3K signalling (figure 3K–M, online supplemental figure 8 and supplemental table 6).
Supplemental material
Fn/Fusobacteriales prevalence differs by transcriptomic-based molecular subtype
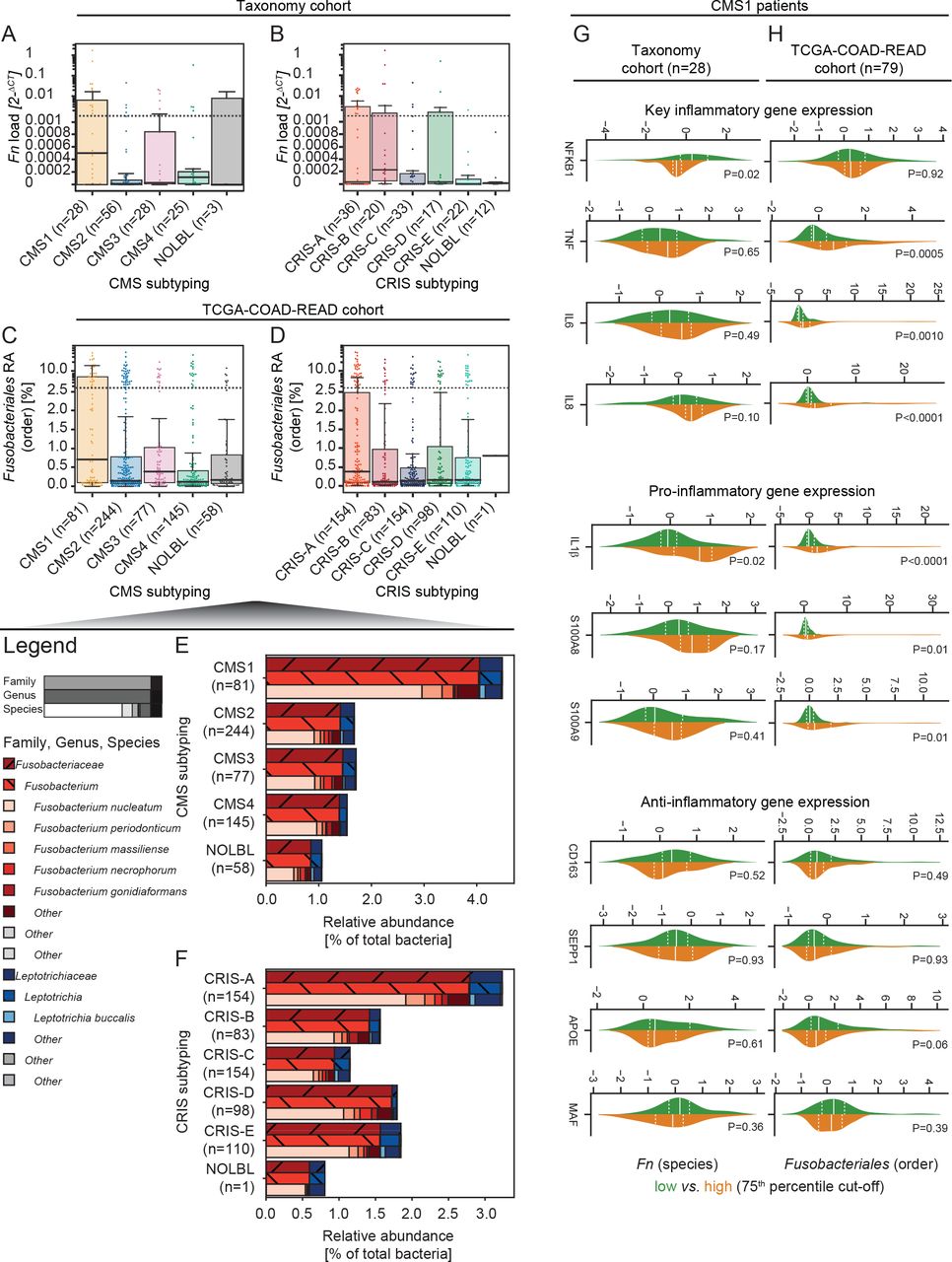
The aforementioned systematic screen pinpointed host aberrations associated with Fusobacteriales hallmarked by transcriptomics-based molecular subtypes. Hence, we classified patients in the study by CMS2 and CRIS3 subtyping. We observed higher Fn load (Taxonomy, figure 4A) and Fusobacteriales RA (TCGA-COAD-READ, figure 4C) in immune-high CMS1 tumours, corroborating the link between pathogen prevalence and host immunity. We observed higher Fn load in CRIS-B tumours (figure 4B) and Fusobacteriales RA in CRIS-A cases (figure 4D) of the Taxonomy and TCGA-COAD-READ cohorts, respectively. At the family rank, Fusobacteriaceae were more abundant than Leptotrichiaceae, accounting for 77% and 23% of total Fusobacteriales RA and ~2% and ~<1% of the total bacteria RA, respectively. In line with the findings at the order level, we observed an increase in Fn, the most abundant Fusobacterium species, in CMS1 and CRIS-A cases (figure 4E,F). In line with the findings at the order level, we observed an approximately threefold increase when comparing patients classified as CMS1 versus the rest (figure 4E). Fn, the most abundant Fusobacterium species, was enriched in CMS1 and CRIS-A cases (figure 4E,F). We examined whether the positive association between inflammation and immune involvement by Fn/Fusobacteriales presence could be ascribed to the host CMS1 milieu or whether there was an additional pathogen-induced component. When restricting the analysis to CMS1 cases, we observed higher expression of proinflammatory markers in Fusobacteriales-high patients of the TCGA-COAD-READ cohort. We detected no association between pathogen prevalence and expression of anti-inflammatory markers or inflammation signatures in either CRC cohort (figure 4G,H). Taken together, these results suggest that Fn/Fusobacteriales may play an active role in mediating inflammation in the host.

Prevalence of Fn/Fusobacteriales by transcriptomic-based molecular subtypes of the host. (A–D) Boxplot with overlaid dot plots displaying the dependency by CMS (A,C) and CRIS (B,D) molecular subtyping by either Fn load (Taxonomy cohort; A,B) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-READ cohort; C,D). (E,F) RA (to total bacterial kingdom) of Fusobacteriales reported at increasing resolution of taxonomic rank (family, genus and species) by CMS (E) and CRIS (F) subtypes (aggregated by mean). Genuses/species with an average RA lower than 0.05 were aggregated as ‘other’. (G,H) Distribution of key (pro-)/(anti-)inflammatory genes in CMS1 patients classified as -low (in green) or -high (in orange) using the 75th percentile as cut-off. Patients’ stratification was based on either Fn load (Taxonomy cohort (G) or Fusobacteriales RA at the order taxonomic rank (TCGA-COAD-READ cohort, H). Median and lower (25th) and upper (75th) percentiles are indicated by white solid or dashed lines, respectively. (Unadjusted) P values were determined by Kruskal-Wallis tests. CMS, consensus molecular subtyping; CRIS, colorectal cancer intrinsic subtyping; Fn, Fusobacterium nucleatum; RA, relative abundance; TCGA-COAD-READ, colon and rectal cases of The Cancer Genome Atlas.
Patients with high Fn/Fusobacteriales have worse outcome in CMS4/CRIS-B
We sought to investigate whether bacterium presence correlated with patient clinical outcome assessed by overall survival (OS), disease-specific survival (DSS) and disease-free survival (DFS) endpoints (figure 5 and online supplemental figures 9 and 10).

High Fn/Fusobacteriales prevalence is associated with negative clinical outcome in patients with mesenchymal-like tumours. (A–L) Kaplan-Meier estimates comparing survival curves in patients of the Taxonomy (OS, A–D) and TCGA-COAD-READ (DSS and DFS cohorts, E–L). Patients across the whole cohort were grouped by bacterium subgroup (low, in green, vs high, in orange; A,E,I) or mesenchymal status (CMS4 and/or CRIS-B, in light blue, vs remaining cases, in dark blue; B,F,J). Patients were grouped by bacterium group and further stratified by mesenchymal status (C,D,G,H,K,L). Patients were binned into a bacterium group (low vs high) using the 75th percentile as cut-off and based on either Fn load (Taxonomy cohort; A,C–D) or Fusobacteriales RA at the order level (TCGA-COAD-READ cohort; E,G–I,K,L). (M) Cox regression models fitted on bacterium RA reported at the order, family, genus and species taxonomic ranks. for each taxonomic rank, patients were classified as low or high subgroup using the corresponding 75th percentile RA abundance as cut-off. Univariate Cox regression models were fitted when evaluating the association between bacterium subgroup (high vs low, reference low) at each taxonomic rank and either DSS or DFS in the whole unselected patient population (left panel). Cox regression models with an interaction term between bacterium subgroup (high vs low; reference low) and mesenchymal status (mesenchymal, ie, either CMS4 and/or CRIS-B, vs non-mesenchymal, ie, neither CMS4 nor CRIS-B) at each taxonomic rank and either DSS or DFS was fitted to evaluate differential impact of bacterium on clinical outcome by tumour biology (right panels). CMS, consensus molecular subtyping; CRIS, colorectal cancer intrinsic subtyping; DFS, disease-free survival; DSS, disease-specific survival; Fn, Fusobacterium nucleatum; OS, overall survival; TCGA-COAD-READ, colon and rectal cases of The Cancer Genome Atlas.
We found no statistically significant differences in either cohort when comparing survival curves from patients grouped by either Fn load or Fusobacteriales RA (figure 5A,E,I and online supplemental figure 9-10). We hypothesised that Fn/Fusobacteriales may result in poorer outcome in a subtype-dependent context (ie, mesenchymal status; figure 5B,F,J). Indeed, we identified a differential association between Fusobacteriales RA and clinical outcome of the TCGA-COAD-READ cohort in mesenchymal (either CMS4 and/or CRIS-B) versus non-mesenchymal (neither CMS4 nor CRIS-B) tumours (figure 5G,H,K,L and online supplemental figure 10). Fusobacteriales-high mesenchymal patients had approximately twofold higher risk of worse outcome, whereas these associations were null in non-mesenchymal patients (figure 5G,H,K,L and online supplemental figure 10). Importantly, these findings held true when accounting for key (adjusted model 1) and more extensive (adjusted model 2) clinical–pathological characteristics that may represent confounders or disease modifiers (online supplemental table 7). We fitted two additional Cox regression models where, in addition to the interaction term between Fusobacteriales and mesenchymal status, we included adjustment covariates. In adjusted model 1, we included age, stage, tumour location and sex as key clinicopathological and demographic covariates. In adjusted model 2, we expanded on adjusted model 1 by also including history of colon polyps and history of other malignancy as comorbidities. We found that the risk of unfavourable outcome (HRs) and statistical significance were minimally impacted by accounting for potential disease modifiers in adjusted models 1 and 2, confirming the robustness of our findings (online supplemental table 7).
Supplemental material
Although numbers in the Taxonomy cohort are more limited, when restricting the analysis to CMS4 and/or CRIS-B cases, we observed a trend in which Fn-high patients had shorter OS than those with low Fn load. Again, no difference in survival according to Fn load was observed in non-mesenchymal Taxonomy patients (figure 5C and online supplemental figure 9).
Exploratory analyses examining the association between clinical outcome and pathogen prevalence at taxonomic ranks of increasing resolution (order, family, genus and species) in the TCGA-COAD-READ cohort by fitting Cox regression models on the whole unselected population and in mesenchymal versus non-mesenchymal settings revealed that the prognostic impact stems primarily from, but is not limited to, species, including Fn, from the Fusobacterium genus from the Fusobacteriaceae family (figure 5M and online supplemental figure 11).
Putative mechanisms underlying selective Fusobacteriales virulence in mesenchymal tumours
Having identified a patient subpopulation that has an unfavourable clinical outcome when their tumours exhibit mesenchymal traits and are highly positive with Fn/Fusobacteriales, we reasoned that intervening by either clearing Fn/Fusobacteriales with broad-spectrum antibiotics or targeting the host–tumour biology could ameliorate clinical outcome for this subpopulation of patients. Given that broad-spectrum antibiotics may not represent a viable avenue in the clinic and narrow-spectrum antibiotics currently do not exist, we set out to identify clinically actionable host-specific vulnerabilities that could be exploited. We examined the host signalling pathways and microenvironment to identify alterations that may be mediated by and/or exacerbated by Fusobacteriales (ie, interact) and, thus, may promote virulence and, ultimately, result in an unfavourable clinical outcome. To disentangle the three-way association between Fusobacteriales RA, gene/signature and molecular subtyping, we fitted two distinct logistic regression models for each feature of interest in the TCGA-COAD-READ cohort. The selection of features was hypothesis-driven and included key host signalling pathways and immunomodulators (figure 6A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
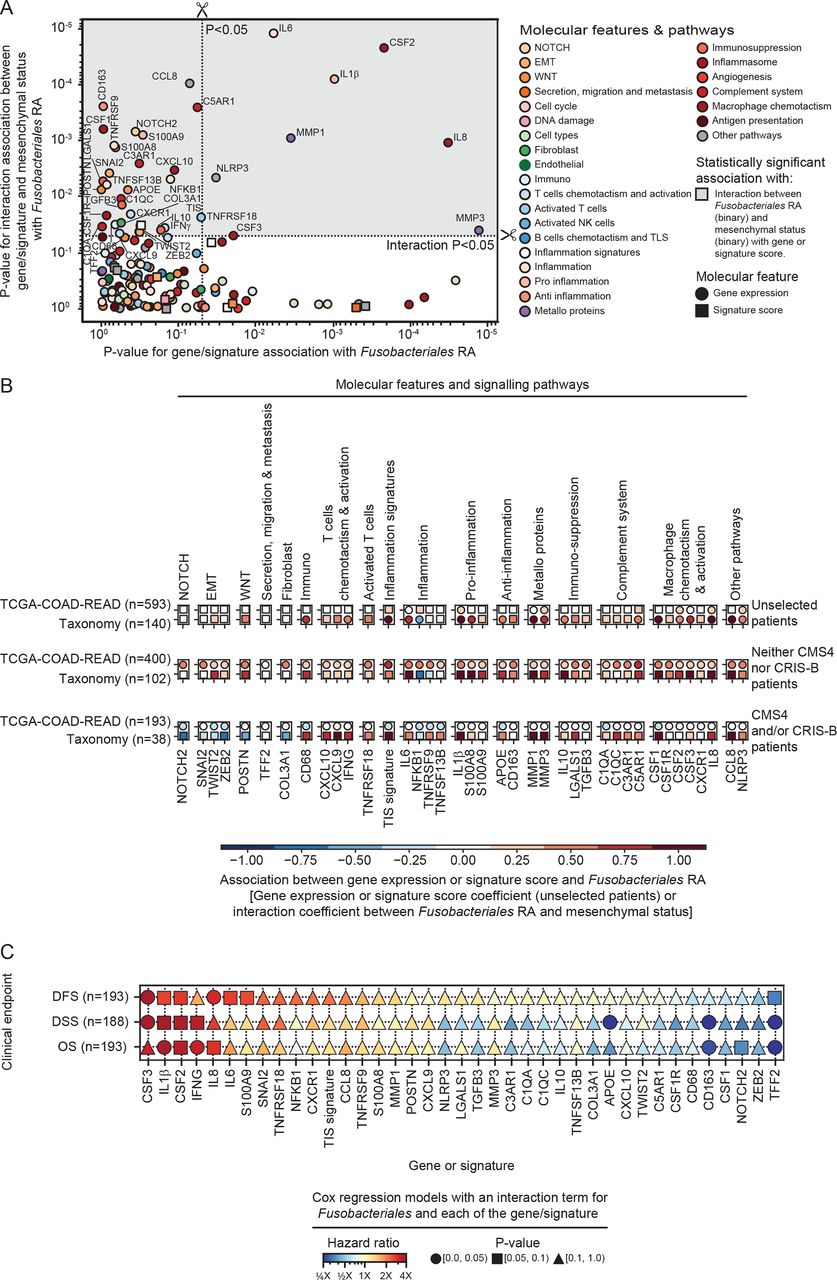
Exploration of mechanism underlying differential impact of Fn/Fusobacteriales in mesenchymal versus non-mesenchymal tumours. (A) Scatterplot depicting p values derived by assessing with logistic regression models the relationship between genes/signatures associated with Fusobacteriales RA in univariate analysis (model 1, x-axis) or the interaction with mesenchymal status (model 2, y-axis). Gene/signature with statistically significant p values from model 2 are highlighted by a grey shaded area. (B) Breakdown of association including direction and effect size, in the unselected patients’ population and within mesenchymal versus non-mesenchymal cases. Only gene/signatures with significant interaction between Fusobacteriales RA and the gene/signature interaction with the molecular subtype (model 2, top quadrant, grey-shaded area) in the TCGA-COAD-READ cohort are included. Associations for both the TCGA-COAD-READ (Fusobacteriales RA) and Taxonomy (Fn load) cohorts are shown. Statistically significant associations are represented with circle markers, whereas non-significant associations are indicated by squared markers. (C) Association between gene/signature identified as candidate targets, A) and clinical outcome in patients of the TCGA-COAD-READ cohort with mesenchymal tumours. HRs and p values are derived from Cox regression models with an interaction term for Fusobacteriales relative abundance (low vs high) and each of the gene/signature (low vs high) being evaluated. CMS, consensus molecular subtyping; CRIS, colorectal cancer intrinsic subtyping; DFS, disease-free survival; DSS, disease-specific survival; Fn, Fusobacterium nucleatum; OS, overall survival; TCGA-COAD-READ, colon and rectal cases of The Cancer Genome Atlas.
Figure 6A reports p values from the two models capturing the association between Fusobacteriales RA (high vs low) and either each gene/signature (model 1: Fusobacteriales~gene/signature, x-axis) or the interaction between each gene/signature with the molecular subtype (model 2: Fusobacteriales~gene/signature×molecular subtype, y-axis). The top half quadrant (darker grey shaded area) identifies a set of genes/signatures whose expression patterns differ by molecular subtype (statistically significant interaction p value in model 2) and thus may be mediating the signalling impact of Fusobacteriales and were prioritised for downstream analyses (figure 6B).
We tested whether the gene/signature we identified as candidate targets are indeed related to clinical outcome in patients of the TCGA-COAD-READ cohort with mesenchymal tumours and high Fusobacteriales. We restricted our analysis to patients with mesenchymal tumours, and for each clinical endpoint of interest, namely, OS, DSS or DFS, we fitted Cox regression models with an interaction term for Fusobacteriales RA (low vs high) and each of the gene/signature (low vs high) identified as statistically significant in the analysis presented in figure 6A. We reasoned that a gene/signature could be considered a candidate target with both specific and translatable impact on clinical outcome for patients with mesenchymal tumours if its association with unfavourable clinical outcome differed by Fusobacteriales. This analysis identified CSF1-3, IL-1β, IFN-γ, IL-8, IL-6, CD163, NOTCH2, ZEB2 and TFF2 as potential targets for patients with mesenchymal tumours and high Fusobacteriales (figure 6C and online supplemental figures 12–14).
Discussion
Fusobacteriales, predominantly Fn, have been associated5 6 8 11 20–26 with pathogenesis, progression and treatment response in CRC. We coupled mechanistic studies in cell cultures with hypothesis-driven and unbiased screening in clinically relevant and omics-rich CRC cohorts to examine the cross-talk between pathogen–host and pathogen–tumour microenvironment. We demonstrate relationships between Fn/Fusobacteriales prevalence and host immunity, signalling and transcriptomic-based molecular subtypes. Our findings suggest that host–pathogen interactions can define patient subpopulations where Fn/Fusobacteriales play an active or opportunistic role, depending on the underlying host–tumour biology and microenvironment and identify putative druggable and clinically actionable vulnerabilities.
We observed higher Fn/Fusobacteriales prevalence in CMS1 patients, corroborating findings by Purcell et al.27 Interestingly, we found that overall, higher pathogen prevalence did not correlate with poorer disease outcome. In contrast, high Fn/Fusobacteriales levels were associated with poor prognosis in the CMS4/CRIS-B patient subset, suggesting that the presence of Fn/Fusobacteriales has a specific clinical impact in mesenchymal-rich, high-stromal infiltrated tumours; this argues against a blanket approach for treating patients with Fn/Fusobacteriales-high tumours. Treatment with wide spectrum antibiotics reduces the growth of Fn-positive tumours in vivo.8 However, the use of antibiotics to treat Fn-positive CRC tumours may be limited as Fn penetrate deeply within tumour, immune and endothelial cells where they internalise with endosomes and lysosomes,28 adapt29 and persist.8 In addition, long-term use of antibiotics can cause gut dysbiosis, which may impact disease progression and outcome.
Given that ‘it takes two to tango’, namely, a high pathogen prevalence and a conducive host milieu, we further examined this interdependence to identify druggable aberrations in the host signalling pathways and microenvironment. We identified putative targets related to (pro-)inflammation, inflammasome, activated T cells, complement system, metalloproteins and macrophage chemotaxis and activation. Fusobacteriales induce a constitutively activated NFκB-TNF-α-IL-6 state which results in activation of metalloproteins and inflammatory cytokines (CSF1-3) which mediate macrophage differentiation, inhibit cytotoxic immune cells and promote proliferation of myeloid-derived-suppressor (MDSC) cells. We observed an increase in inflammation and M1 macrophages and a decrease in M2 macrophages in patients with higher Fn/Fusobacteriales prevalence. We envisage that therapeutic options, such as NLRP3/AIM2 inflammasome suppression,30 IL-1β blockade,31 TNF-α32 or IL-6 inhibition,33 which have been approved for treatment of chronic inflammation and cytokine storm syndrome in multiple cancers, rheumatoid arthritis and COVID-19 may ameliorate the immunosuppressive microenvironment induced by Fn/Fusobacteriales. Importantly, these targets are involved in not only promoting an immunosuppressive microenvironment by recruiting tissue-associated macrophages (TAMs) and MDSCs, but also in orchestrating invasion, angiogenesis, epithelial-to-mesenchymal transition and, ultimately, metastasis. The prometastatic impact of Fn/Fusobacteriales is further corroborated by findings in the literature linking higher pathogen prevalence in more advanced disease stage and metastasis in clinical specimens5 and higher metastatic burden in mice inoculated with Fn.34
Cancer cells with an EMT phenotype secrete cytokines such as IL-10 and TGF-β that can further promote an immunosuppressive microenvironment. Additionally, secretion of IL-6 and IL-8 from stroma cells can further foster an EMT phenotype, activate primary fibroblasts (cancer-associated fibroblast (CAFs)) which, in turn, may promote angiogenesis and invasion.35 Taken together, these aberrations may result in a self-reinforcing mechanism that confers on cancer cells the ability to migrate, invade the extracellular matrix, extravasate and seed metastasis. When comparing the transcriptomic profiles by Fusobacteriales RA in the TCGA-COAD-READ cohort, we identified dysregulation affecting cell architecture involving apical surface dynamics and Aurora A kinase signalling, which regulate cMYC, DNA repair, cell motility/migration and induce EMT transition via β-catenin and TGF-β, leading to metastasis and resistance to treatment in multiple cancer types.36 Small molecule inhibitors against Aurora A have shown encouraging results in preclinical studies and clinical trials in CRC37 and other cancers.38 Cytoskeleton shape, filopodium protrusions and alterations in cell adhesion and structure are hallmarks of extracellular matrix invasion. EMT key effectors, SNAIL and ZEB1, alter apical surface dynamics by inhibiting scaffolding proteins and by inducing expression of matrix metalloproteins (MMP3 and MMP9), resulting in loosened tight junctions, altered cell polarity and increased plasticity which, in turn, enable cell invasion.39 Dysregulations in MMP expression may aid cancer cells that have reached the bloodstream to extravasate to distant tissues40 by priming the vascular endothelium via upregulation of VEGF-A41 and by increasing permeability via COX2 upregulation.42 Our analyses in the TCGA-COAD-READ cohort identified higher expression of vascular endothelial growth factor (VEGF) as well as an angiogenesis signature in patients with higher Fusobacteriales RA. Indeed, a new generation of selective and highly penetrative MMP inhibitors43 is being trialled in GI cancers,44 and Mehta et al reported lower Fusobacteriales RA in subjects treated with aspirin, a COX2 inhibitor.45
Green et al 46 demonstrated that MAPK7 is a master regulator of MMP9 and promotes the formation of metastasis. We observed a dysregulation in MAPK signalling at the protein level when comparing Fusobacteriales-high versus Fusobacteriales-low patients of the TCGA-COAD-READ cohort. MAPK7 induces EMT transition, cell migration and regulates TAM polarisation in a metalloprotein-dependent manner,46 rendering it an appealing upstream therapeutic target. IL-6 orchestrates MAPK-STAT3 signalling, which in turn regulates the dynamic transition between two CAFs subpopulations, EMT-CAFs and proliferation-CAFs,47 rendering the IL-6-TGF-β-EMT-CAFs cross-talk potentially a further therapeutic target. While directly targeting EMT via NOTCH or WNT has shown limited success in the clinic,48 microenvironment remodelling to reverse immunosuppression by inhibiting CXCL1249 or promoting T-cell infiltration50 or function via engineered oncolytic adenovirus,51 has shown promising results in reducing metastasis formation.52 Additionally, we observed a positive correlation between gene expression of IL-8, CXCL8, CXCR1 and CXCL10 and Fn/Fusobacteriales prevalence, corroborating findings from Casasanta et al. assessing Fn in HCT116 CRC cells.53
In conclusion, our analyses have identified a patient subpopulation that has an unfavourable clinical outcome when their tumours exhibit mesenchymal traits and are highly positive with Fn/Fusobacteriales and pinpointed clinically actionable host-specific vulnerabilities that suggest new treatments for these patients that extend beyond broad-spectrum antibiotics.
Supplemental material
Data availability statement
Data are available in a public, open access repository. Processing and analysis code along with bacterium estimates with corresponding clinical and molecular datasets for the Taxonomy and TCGA-COAD-READ cohorts included in this study are publicly available and archived at Zenodo (https://10.5281/zenodo.4019142). Bacterium estimates include Fusobacterium nucleatum load (Taxonomy cohort) and Fusobacteriales relative abundance, along with higher resolution estimates at genus, family and species taxonomic ranks, (TCGA-COAD-READ cohort).
Ethics statements
Patient consent for publication
Ethics approval
The Taxonomy cohort collection was approved by the Medicine, Dentistry, and Biomedical Sciences School Ethics Committee (ref: 12/12v4), as previously described (Allen et al, JCO Precision Oncology, 2018; PMID: 30088816). Approval for the cohort of patients with colon (COAD) and rectal (READ) cases denotated as TCGA-COAD-READ in this manuscript was acquired by the original investigators of The Cancer Genome Atlas consortium.
Acknowledgments
We thank the patients who kindly donated their samples and made this study possible.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MS, DL and JHMP conceptualised and designed the study. MS, NC, KS, SB, DL and JHMP were involved in collection, preparation, interpretation, validation and critical review of the data. NC and KS performed the cell culture experiments and quantified bacterium load in tumour samples of the patients of the Taxonomy cohort. MS performed formal analysis including bioinformatics and statistical analyses. MS created the manuscript figures and supplementary materials. MS and JHMP drafted the manuscript. All authors edited, reviewed, revised and approved the manuscript text. DL and JHMP acquired funding for the study.
Funding This study was supported by a grant from the Health Research Board and Science Foundation Ireland to JHMP (16/US/3301), a studentship to KS sponsored by The Northern Ireland Department for the Economy (NI DfE), and by funding from NI DfE (SFI-DEL 14/1A/2582, STL/5715/15). The results included here are in part based on data generated by the TCGA Research Network (https://wwwcancergov/tcga). We wish to acknowledge the information technology department at the Royal College of Surgeons in Ireland and the Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.