Article Text

Abstract
Objective Conflicting microbiota data exist for primary sclerosing cholangitis (PSC) and experimental models. Goal: define the function of complex resident microbes and their association relevant to PSC patients by studying germ-free (GF) and antibiotic-treated specific pathogen-free (SPF) multidrug-resistant 2 deficient (mdr2−/− ) mice and microbial profiles in PSC patient cohorts.
Design We measured weights, liver enzymes, RNA expression, histological, immunohistochemical and fibrotic biochemical parameters, faecal 16S rRNA gene profiling and metabolomic endpoints in gnotobiotic and antibiotic-treated SPF mdr2−/− mice and targeted metagenomic analysis in PSC patients.
Results GF mdr2−/− mice had 100% mortality by 8 weeks with increasing hepatic bile acid (BA) accumulation and cholestasis. Early SPF autologous stool transplantation rescued liver-related mortality. Inhibition of ileal BA transport attenuated antibiotic-accelerated liver disease and decreased total serum and hepatic BAs. Depletion of vancomycin-sensitive microbiota exaggerated hepatobiliary disease. Vancomycin selectively decreased Lachnospiraceae and short-chain fatty acids (SCFAs) but expanded Enterococcus and Enterobacteriaceae. Antibiotics increased Enterococcus faecalis and Escherichia coli liver translocation. Colonisation of GF mdr2−/− mice with translocated E. faecalis and E. coli strains accelerated hepatobiliary inflammation and mortality. Lachnospiraceae colonisation of antibiotic pretreated mdr2−/− mice reduced liver fibrosis, inflammation and translocation of pathobionts, and SCFA-producing Lachnospiraceae and purified SCFA decreased fibrosis. Faecal Lachnospiraceae negatively associated, and E. faecalis/ Enterobacteriaceae positively associated, with PSC patients’ clinical severity by Mayo risk scores.
Conclusions We identified novel functionally protective and detrimental resident bacterial species in mdr2−/− mice and PSC patients with associated clinical risk score. These insights may guide personalised targeted therapeutic interventions in PSC patients.
- intestinal microbiology
- primary sclerosing cholangitis
- antibiotics
- short chain fatty acids
- cholestatic liver diseases
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- intestinal microbiology
- primary sclerosing cholangitis
- antibiotics
- short chain fatty acids
- cholestatic liver diseases
What is already known on this subject?
Microbiotal alterations are associated with idiopathic primary sclerosing cholangitis (PSC) but provide limited insights into functional activities and mechanisms of resident bacteria and their metabolites.
Germ-free mice and antibiotics lead to increased reuptake of bile acids, suggestive of microbial control of bile acid homeostasis.
Antibiotics have variable protective and detrimental outcomes in clinical and experimental studies but lack unified mechanistic explanations.
What are the new findings?
Germ-free mdr2−/− mice, a PSC murine model, exhibited 100% fatality by 8 weeks driven by lack of microbial modulation of toxic progressive hepatic and plasma bile accumulation unless rescued with faecal microbiota transfer from syngeneic specific pathogenic stool by 4 weeks of age, confirming an overall protective role of resident microbiota in this model.
Antibiotic-induced dysbiosis of specific pathogen-free (SPF) mdr2−/− potentiated hepatobiliary disease by non-bacteremic hepatic translocation of Enterococcus faecalis and Escherichia coli and resulted in accelerated detrimental hepatic bile acid accumulation without changing ileal FXR signalling. Inhibition of ileal bile salt transporter, Asbt, resulted in attenuated hepatic inflammation and fibrosis in antibiotic-treated SPF mdr2−/− mice, validating microbial modulation of bile acid homeostasis.
What are the new findings?
Antibiotic-induced dysbiosis of SPF mdr2−/− potentiated hepatobiliary disease with hepatic translocation of E. faecalis and E. coli and increased bile salt hydrolase activity.
Microbial manipulations and metabolomic analysis in mdr2-−/−- mice identified hepatoprotective Lachnospiraceae that inhibited pathobiont (E. faecalis and E. coli) enterohepatic translocation and mediated their anti-fibrotic effects by producing short-chain fatty acids.
In human cohorts, fecal E. faecalis/Enterobacteriaceae positively and Lachnospiraceae negatively associated with PSC clinical severity measured by Mayo risk score.
How might it impact on clinical practice in the foreseeable future?
These microbial insights have the potential to better predict clinical disease courses in individual patients by assessing abundance of detrimental versus protective resident microbial populations; improving PSC faecal transplant outcomes by matching donor and recipient selection; and guiding selective microbial manipulations for personalizsed therapeutic approaches.
Introduction
Chronic biliary inflammation, cirrhosis and variable progression to liver failure characterise primary sclerosing cholangitis (PSC).1 No current therapies exist to halt disease progression, because the mechanisms that underlie the onset and progression of PSC remain unclear. Several studies show dysbiotic microbiota in PSC patients with decreased microbial diversity and over-represented intestinal pathobionts like Enterobacteriaceae (Escherichia coli and Klebsiella pneumonia), Enterococcus (Enterococcus faecalis), Veillonella and biliary tract E. faecalis, compared with controls suggesting potential translocation.2–5 Promotion of interleukin (IL)-17-mediated mucosal immunity play a role in PSC pathophysiology.2 5–7 In parallel, PSC patients exhibit decreased abundance of presumed protective faecal Lachnospiraceae,4 8 which have bile acid (BA) metabolic activities and anticolitogenic properties9 but unclear functional hepatobiliary impact. Short-term traditional probiotic therapy (Lactobacillus and Bifidobacterium)10 or single donor faecal microbial transfer (FMT)11 are ineffective for PSC patients, possibly related to no native colonisation of these bacteria.3 4 8 These studies indicate the complexity and strong association of microbiota with PSC and illustrate the need for further understanding of pathogenic and protective roles of resident gut microbiota.
Preclinical studies document the protective role of unfractionated gut microbiota in experimental PSC. In both chemically induced12 and genetic (FVB/N mdr2−− )13 murine PSC models, germ-free (GF) mice exhibited accelerated liver disease compared with conventionally raised controls. GF conditions lead to extensive reuptake of intestinal BAs,12 emphasising the importance of microbial mediation of BA homeostasis. Microbial depletion expands hepatic non-micellar BA concentrations contributing to cholangiocyte inflammation, ductal disruption and parenchymal disease in mdr2−/− mice.13–15 Dysbiosis in PSC patients results in altered BA pools that potentially contribute to PSC pathophysiology.3 14 Furthermore, increased microbial bile salt hydrolase (BSH) activity was found in PSC patients’ bile,3 possibly related to the high BSH activity of E. faecalis, abundant in the faeces and bile of PSC patients.3 4 16 17 These observations suggest a potential pathogenic role for bacterial BSH activity in PSC. Functional microbial or metabolic studies are needed to further delineate mechanistic differences among intestinal bacteria that differentially influence hepatobiliary damage, inflammation and fibrosis in PSC.
In this study, we used gnotobiotic and antibiotic approaches in specific pathogen-free (SPF) mdr2−/− mice to study the functional roles of resident bacteria in cholestatic liver disease. We identified functionally protective (Lachnospiraceae) and pathogenic (E. faecalis and E. coli) resident bacteria and defined in vivo mechanisms through complementation studies. Conserved relative abundance of these bacteria was found in metagenomic analysis of PSC patient faeces with a moderate association with Mayo clinical risk score in PSC patients denoting parallel microbial roles in experimental and clinical disease. These insights could target donor selection in future PSC-FMT trials and guide selective faecal enrichment/depletion approaches to increase efficacy of future personalised microbial therapies.
Methods
Ethics statement/animal husbandry
The Popov lab generated C57Bl/6 SPF mdr2(abcb4) −/− mice.18 Mdr2−/− and WT SPF control C57BL/6 mice were housed in vivaria microisolator cages with no more than five mice/cage or in GF Trexlar isolators. The UNC National Gnotobiotic Rodent Resource Center derived GF mdr2−/− mice by caesarian section. We bred GF mdr2+/− ±mice due to early mortality of GF mdr2−/− mice. Age-matched and sex-matched mice had unrestricted access to autoclaved water and 5v0F Purina LabDiet chow with a 12-hour light/dark cycle. Human microbiome data were analysed from a previous study.4
Antibiotic treatments
Non-fasted 3–4 week old male and female mice received either combined or single antibiotics (0.5 mg/mL vancomycin (Hospira), 1 mg/mL neomycin (Medisca) and 50 mg/kg metronidazole (G.D. Searle)) in drinking water ad libitum, consuming 6–7 mL/mouse/day.19 20 The antibiotic mixture was diluted in deionised H2O, sterilised through a 0.2 µm filter and replaced twice weekly for 7–14 days.
Patient and public involvement
No involvement.
Statistical analysis
Murine data are shown as mean±SEM, with ‘n’ representing number of mice indicated in figure legends. One dot represents one mouse. Linear fitting and normalisation were performed in GraphPad Prism. Unless specified in the figure legends, statistical significance between two groups was determined by unpaired, two-tailed Student’s t-test; significance between >2 groups determined using one-way analysis of variance with Fisher’s LSD test using GraphPad Prism default settings. We compared CFU numbers by the Poisson generalised linear model, estimated with a Markov Chain Monte Carlo method robust for small sample sizes. Error bar represents mean±SEM with *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 and n.s. as no significance. Further details of histological staining, IHC staining and analysis, bacterial or metabolite inoculation, cultivation and translocation, biochemical, liver enzyme, metabolomic and 16S rRNA sequencing/analysis are presented in the online supplemental material and methods.
Supplemental material
Results
Autologous stool transplant alleviate the non-micellular hepatic BA accumulation, hepatobiliary inflammation, fibrosis and mortality in germ-free mdr2−/− mice
To evaluate resident microbiota’s role in this experimental cholestatic murine model, we generated and characterised GF C57BL/6 mdr2−/− mice compared with littermate C57BL/6 WT mice. Relative to SPF conditions, 6–7 week old GF mdr2−/− mice had decreased weight gain, 2× increased alkaline phosphatase (ALP) and 15× increased total bilirubin (TB) (figure 1A–C). GF mdr2−/− mice from weaning 5 weeks had histologically normal livers (data not shown) and normal TB (figure 1C). Rapid onset of cholestasis in GF mdr2−/− occurred beyond age 6 weeks, not seen in age-matched SPF mdr2−/− mice, in GF WT C57BL/6 or older GF mdr2+/− ± (≤70 weeks) (figure 1C, online supplemental figure S1A,D). Loss of microbiota in mdr2−/− mice resulted in >3–5 fold progressively increased hepatic and serum BAs strongly correlating with body mass wasting and biomarkers of cholestatic injury (figure 1C–E). Most hepatic BA were 1oBA (cholic and muricholic acids, S1B-C). GF mdr2−/− and wild-type (WT) C57BL/6 mice exhibited elevated hepatic BA, attenuated liver cyp7a1 gene expression (figure 1E, online supplemental figure S1H) despite no difference in ileal ASBT, FXR or FGF15 (figure 1G, online supplemental figure S1E–G) compared with SPF controls.
Supplemental material
Supplemental material
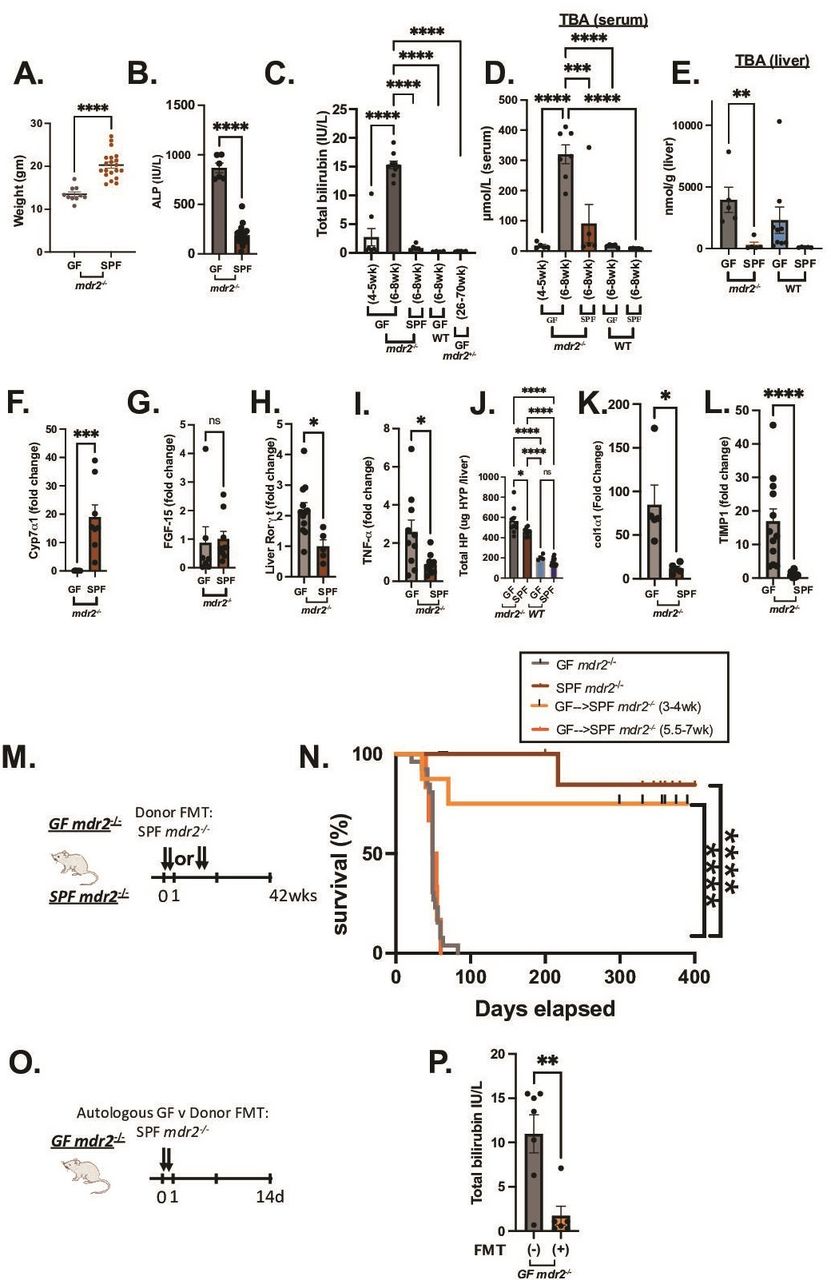
Clinical, biochemical and liver histological indicators of germ-free (GF) mdr2−/− mice with or without faecal microbiotal transplant compared with control mice. Mouse body weight (A) and serum alkaline phosphatase (AP), (B) of 6–8 week old GF (n=6–10) and SPF (n=20) mdr2−/− mice. Longitudinal total bilirubin (TB) (C) or total serum bile acids (TBA) (D) assessment in GF mdr2−/− mice from 4–5 week old (n=7) to 6–8 week old (n=8), compared 6–8 week old SPF mdr2−/− and C57/BL6 WT GF mice and older (26–70 week old) mdr2+/− ±. Total liver bile acids (Bas) in mdr2−/− or WT mice in GF or SPF environments (E, n=5–9/mice per group). Liver cyp7a1 expression (F) and liver FGF-15 expression (G). Relative expression (fold change) of liver cyp7a1, terminal Ileal FGF-15, Rorγt and tnf-α (F–I) of 6–8 week old GF (n=5–7) and SPF (n=5–7) mdr2−/− mice. Hepatic collagen deposition expressed as total hepatic hydroxyproline (HYP) expressed in ug HYP/whole liver, calculated by multiplying individual liver weight with relative HYP content (J). Relative expression (fold change) of collagen I alpha-1 (col1α1) and tissue inhibitor of metalloproteinases (TIMP) 1 (K–L) in livers of 6–8 week old GF (n=5–12) and SPF (n=5–8) mdr2−/− mice. Experimental design of autologous faecal microbiota transplant (FMT) study (M) Kaplan-Meyer survival curves of pooled SP mdr2−/ donor stool orally gavaged into GF mdr2−/− C57Bl/6 mice twice in the first 2 days at age 3–4 weeks (n=8) or 5.5–7 weeks (n=6) compared with untreated GF (n=23) or SPF (n=14) mdr2−/− mice monitored for survival for 42 weeks (N). Experimental design of GF mdr2−/− given autologous GF (n=7) or SPF donor (n=6) stool for 14 days (O) monitoring serum TB (P). Results are expressed as means±SEM. Survival data are analysed by log-rank (Mantel-Cox) test, group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. Welch correction was applied to histological scoring analysis. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. SPF, specific pathogen free.
GF mdr2−/− had increased hepatic RNA expression of IL-17/IL-22 master regulator, Rorγt and inflammatory cytokine TNF-α (figure 1H,I). Total hepatic hydroxyproline content, a collagen biochemical marker, was increased in GF mdr2−/− mice, along with RNA expression of fibrinogenic markers, co1α1 and timp-1 (figure 1J–L). GF and SPF mdr2−/− mice developed hepatomegaly versus WT controls (online supplemental figure S1F).
GF mdr2−/− mice had markedly decreased survival (median mortality 7.5 weeks and 100% mortality by 8 weeks) compared with SPF mdr2−/− mice (figure 1N). Faecal transplantation (FMT) of SPF mdr2−/− faeces to 3–4 week old GF mdr2−/− mice improved survival (75% at 400 days; figure 1N) and decreased TB (figure 1P) 2 weeks post-transplant. FMT later in life (5.5–7 weeks) provided no protection (figure 1N). Although both SPF and GF female mdr2−/− mice developed more prominent periportal inflammation relative to males, mortality was not sex related in GF mdr2−/− mice. FMT did not affect mortality in GF mdr2+ /−, SPF mdr2−/− or WT mice (online supplemental figure S1J).
GF mdr2−/− mice displayed heightened periportal inflammation, periductular macrophages, centrilobular necrosis and ductular proliferation (figure 2A,B and E,F) and more pronounced macrophage (figure 2C–D) than neutrophilic (S1K-L) infiltration versus SPF mdr2−/− mice. SPF mdr2−/− mice displayed more prominent sinusoidal macrophage distribution compared with periductular localisation in GF mice, whereas SPF donor FMT-treated GF mdr2−/− had a mixed phenotype. Notable hepatic lesions (figure 2A, E and G, arrows) range from hepatocytes with vacuolar and cystic degeneration to coagulation necrosis rimmed by inflammation. The cause of these lesions is not apparent, but a similar lesion has been reported in biliary ligation21 and antibiotic-induced cholestatic14 studies to be the result of bile salt leakage and described as bile infarcts. in GF mdr2−/− mice. SPF mdr2−/− mice developed periductal fibrosis with focal parenchymal extensions, but GF mdr2−/− mice had doubled histological fibrosis scores with extensive periductal fibrosis extending well into the parenchyma with frequent bridging to portal veins, resembling stage 3–4 Ishak fibrosis patterns (figure 2G). Early FMT attenuated periportal inflammation and fibrosis in GF mdr2−/− mice (figure 2B and H). No histological hepatic or ileocolonic inflammation occurred in GF or SPF WT or mdr2+/− ± (data not shown). Thus, early life exposure to resident microbiota in GF mdr2−/− mice promotes survival and decreased hepatic inflammation, ductular reaction and fibrosis.

Histological impact of absent and reconstitution of resident microbes in GF mdr2−/− mice. Representative photomicrographs of 6–8 week old GF mdr2−/− mice±faecal microbiota transplant at 3–4 week old compared with age-matched SPF mdr2−/− mice with blinded scoring for liver inflammation (A and B), macrophages (4-4/80 immunohistochemical staining) (C and D), ductular reaction (CK-19 immunohistochemical staining) (E and F) and fibrosis (G and H) stained by H&E (200×), antibodies to F4/80 (200×) and CK-19 (200×), and Sirius Red (100×), respectively. Arrows indicate foci of hepatocyte degeneration and necrosis bordered by inflammation (bile infarct). CK-19 staining in panels D and F count positive cells in five high powered fields/ slide. Results are expressed as means±SEM. Group or pairwise comparisons were performed by analysis of variance or Student’s t-test, respectively. Welch correction was applied to histological scoring analysis. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. CV, central vein; PV, portal vein.
mdr2−/− mice have dysbiotic microbial landscapes with altered metabolic pathways exacerbated by antibiotics
Resident faecal microbial community structure diverged in SPF mdr2−/− and WT mice by β-diversity analysis (figure 3A) with decreased putative protective Clostridial family abundance (figure 3B). To validate the protective role of host–microbiota and identified potential beneficial bacterial groups and metabolites, we developed an antibiotic depletion model in SPF mdr2−/− mice via administration of a broad-spectrum antibiotic cocktail (Abx) targeting resident Gram-positive, Gram-negative and anaerobic bacteria19 20 for 14 days (figure 3C). As predicted, Abx similarly depleted microbial abundance in mdr2−/− and WT control mice by α-diversity and faecal qPCR (figure 3D–G). β-diversity analysis demonstrated markedly different microbial communities in the two hosts following Abx (figure 3H,I).

Faecal microbiome and metabolite profiles of preantibiotic/postantibiotic treated mdr2−/− and wild-type (WT) mice on C57BL/6 background. Pooled baseline untreated SPF mdr2−/− and WT C57BL/6 16S sequencing faecal beta diversity (Permanova test) (A) and linear discriminant analysis plot showing differential enrichment of taxa (B). Experimental design of broad-spectrum antibiotic (Abx) study, 3–5 week old SPF mdr2−/− or WT mice exposed ad libitum to broad-spectrum antibiotics (metronidazole: 30 mg/mL; vancomycin: 0.5 mg/mL; and neomycin: 1 mg/mL) in drinking H2O or H2O alone (n=10 mice/group) for 14 days (pooled from three experiments) (C). Effect of 14 days of Abx assessing pooled baseline alpha diversity (Faith’s PD) and faecal universal 16S qCR (expressed in ΔΔCT differences) in mdr2−/− (D and E) and WT BL/6 (F and G) mice, respectively. Principal coordinates analysis (PcoA) plots assessing beta diversity (Permanova test) antibiotic exposure between genotypes (WT and mdr2−/− mice) (H) and antibiotic exposure in mdr2−/− mice (I). 3D-PCA plot shows the separation of cecal (J) and serum (K) metabolites from mdr2−/− with and without antibiotics. Mummichog pathway cloud plot of potential pathway differences with exposure of broad antibiotics in SPF mdr2−/− mice (L). The radius of each circle represents the number of metabolites relative to the number of metabolites represented by other circles. Darker circles mean more pathways are represented (mdr2−/− no Abx: n=13, mdr2−/− with Abx: n=25). Group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. SPF, specific pathogen free.
To identify potential metabolic profiles reflecting protective microbial effects in mdr2−/− mice, we analysed the metabolome of serum and cecal contents from Abx-treated versus untreated mdr2−/− mice by liquid chromatography-mass spectrometry (LC/MS) (figure 3J–L). Abx decreased multiple compounds, spanning BA, alcohols, indoles, oxosteroids, bilirubin and methyl-branched fatty acid pathways (figure 3L). We used these metabolomic profiles to identify putative functional properties of protective bacterial candidates, Clostridiaceae and Lachnospiraceae that might rescue inflammation/fibrosis driven by Abx depletion of protective microbial subsets.
Antibiotic depletion of resident microbiota augments intestinal permeability, liver inflammation, biliary epithelial reactivity and fibrosis and restricts weight gain in mdr2−/− mice
Abx markedly depleted putative protective Clostridiaceae and Lachnospiraceae families (figure 4B) and increased intestinal permeability versus WT mice (figure 4C). Weight loss in mdr2−/− mice limited Abx treatment to 14 days (figure 4D) with increased serum ALP and clinical illness (lethargy, hunching, unkempt fur and peritoneal jaundice), and minimal serum bilirubin elevation (figure 4E,F), not seen in Abx-WT mice (online supplemental figure S1M–O). Like GF mdr2−/− mice, Abx-induced inflammation increased hepatic Tnf-α expression (figure 4G). Histological evaluation of Abx-treated mdr2−/− displayed accelerated cholangiopathic injury similar to GF mdr2−/− mice with expanded portal/peribiliary inflammation and ductular reaction (figure 4K–N).
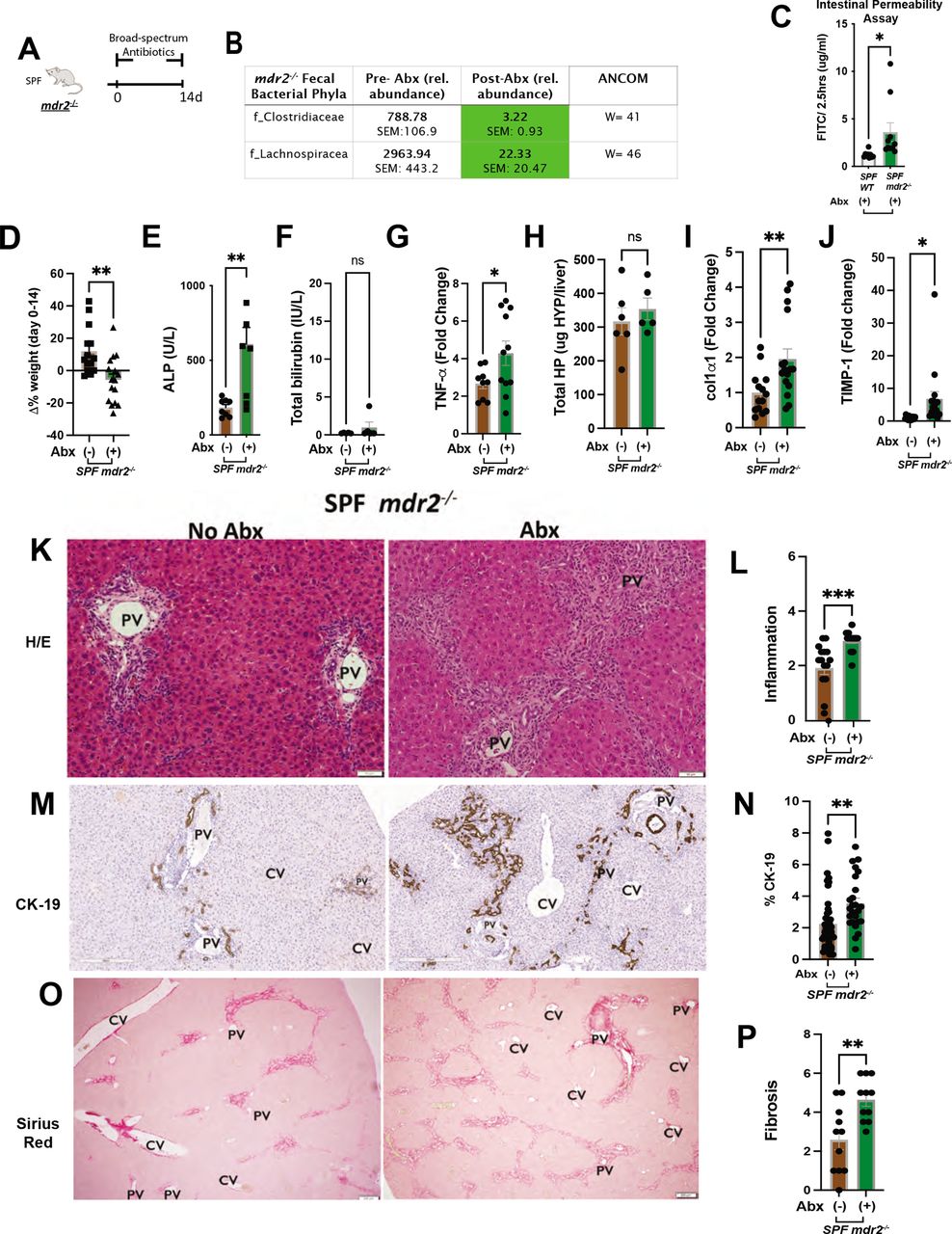
Clinical, biochemical and liver histological indicators of broad-spectrum antibiotic treated mdr2−/− mice. Experimental design of broad-spectrum antibiotic (Abx) study, 3–5 week old SPF mdr2−/− mice exposed ad libitum to broad-spectrum antibiotics in drinking H2O (n=15) or H2O alone (n=13) for 14 days (pooled from three experiments) (A). Effect of Abx exposure on various outcomes including relative abundance of selected bacterial phyla expressed as unadjusted raw average operational taxonomic unit (OTU) relative abundance (B), colonic/TI permeability (WT & mdr2−/− mice) (C), 14 days body weight change (D), serum ALP, TB (E and F), RNA expression of liver tnf-α (G), col1α1 (I) and timp-1 (J) and hepatic collagen deposition expressed as ug HYP/whole liver (H). Representative photomicrographs and blinded composite histological scoring murine liver stained with H&E (K–L), CK-19 (M–N) and Sirius Red (O–P) of untreated (n=15) versus Abx-treated SPF mdr2−/− mice (n=15). Results are expressed as means±SEM. Pairwise comparisons were performed by Student’s t-test for biochemical and molecular studies; Welch correction was applied to histological scoring analysis. Unadjusted raw average OTU relative abundance and SEs of bacterial groups against the variables detected with significant effects by analysis of composition of microbes). *P<0.05, **p<0.01, ***p<0.001. ALP, alkaline phosphatase; SPF, specific pathogen free; TB, total bilirubin.
Abx-treated mdr2−/− mice developed increased bridging fibrosis compared with untreated SPF mdr2−/− mice (figure 4O–P). Unchanged hydroxyproline content (figure 4H) was likely due to the short study duration, but increased liver RNA expression of co1α1 and timp-1 occurred in Abx-treated mdr2−/− mice versus untreated controls (figure 4I,J). This Abx depletion validated our GF mdr2−/− findings, confirming that decreased protective resident bacteria potentiates disease in this cholestatic model.
Ileal BA transport inhibition attenuates hepatic BA pool size and hepatobiliary inflammation/cholestasis in antibiotic treated mdr2−/− mice
To test mechanistic interventions, we identified the shortest duration of Abx therapy that accelerated hepatobiliary inflammation/fibrosis but maintained a 2-week experimental window for microbial transfer experiments. Interestingly, the shorter 7-day treatment enhanced hepatic inflammation and ALP elevation (online supplemental figure S2B, C) versus 14-day treatment, without significantly altering fibrosis scores (online supplemental figure S2D,E).
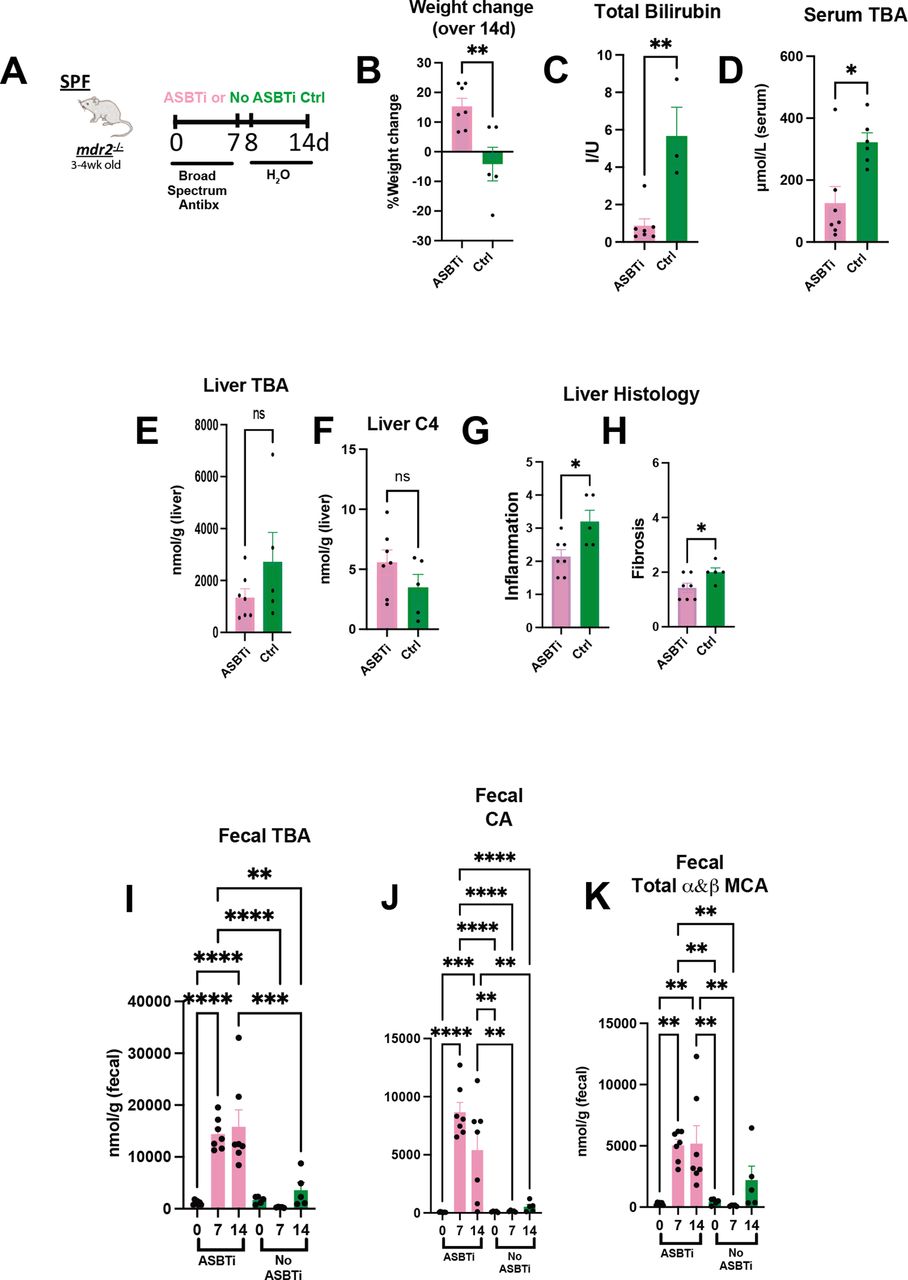
To further investigate the role of microbial-mediated altered BA pool size and hepatobiliary disease in this model, we identified effects of an apical sodium-dependent bile acid inhibitor (ASBTi) in our accelerated antibiotic model (figure 5A). ASBTi restored age-appropriate weight gain and attenuated cholestasis, inflammation and fibrosis (figure 5B,C and G,H, online supplemental figure S2F). Abx increased serum TBA hepatic shunting, like GF mice, that was attenuated by ASBTi (figure 5D). The predicted ASBTi-induced increased faecal BA was sustained for the 2-week treatment (figure 5I). Compensatory induction of the cyp7a1 metabolite, hepatic C4 (figure 5E,F), supported the trending decreased hepatic BA pool. Additionally, we found increased faecal primary unconjugated BAs (cholic acid (CA) and muricholic acid (MCA) (figure 5I–K). These studies indicate that resident bacterial depletion elevates non-micellular BA hepatobiliary toxicity in susceptible mice, mediated by enterohepatic BA transport.
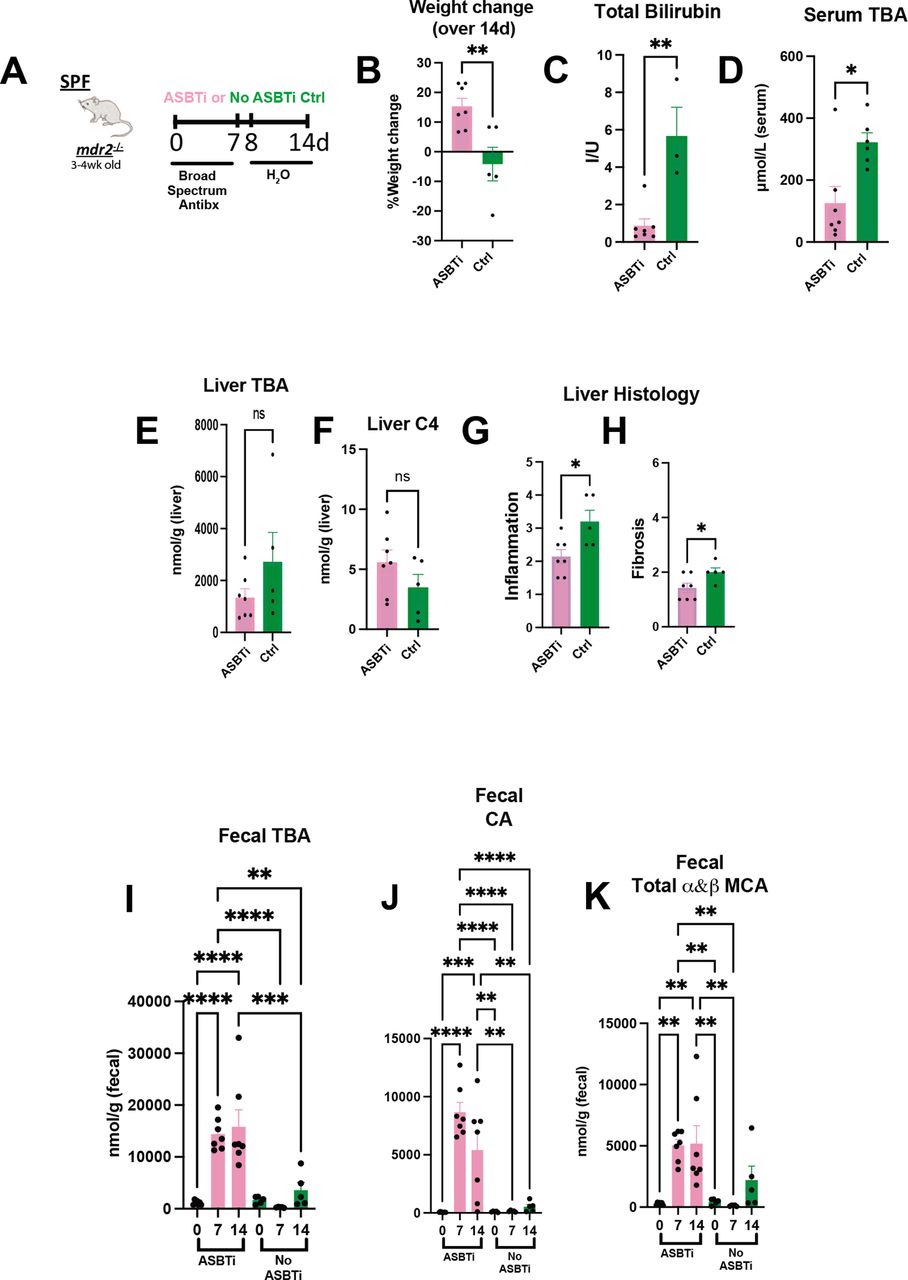
Bile acid homeostasis and hepatobiliary injury assessed in Abx treated mdr2−/− mice following ASBT blockage. Experimental design of ad libitum exposure in drinking H2O of ASBT inhibitor (GSK23306, 10 mg/kg) for 14-day concomitant with 7-day broad antibiotic pretreatment in 4–5 week old SPF mdr2−/− mice versus no ASBTi (water only) group (n=5–7 mice/group) (A). Effect of ASBT inhibition on: weight change (B), TB (C), serum and liver total BA (D–E), liver C4 (F, metabolite of Cyp7a1 activation), histological liver inflammation and fibrosis (G and H). Assessment of longitudinal faecal bile acid homeostasis at times 0, 7 and 14 days following initiation of ASBT inhibition evaluating TBA, cholic acid (CA) and α+β MCA (I–K). Group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ASBTi, apical sodium-dependent bile acid inhibitor; SPF, specific pathogen free; TB, total bilirubin.
Selective antibiotics have differential effects on mdr2−/− hepatobiliary disease
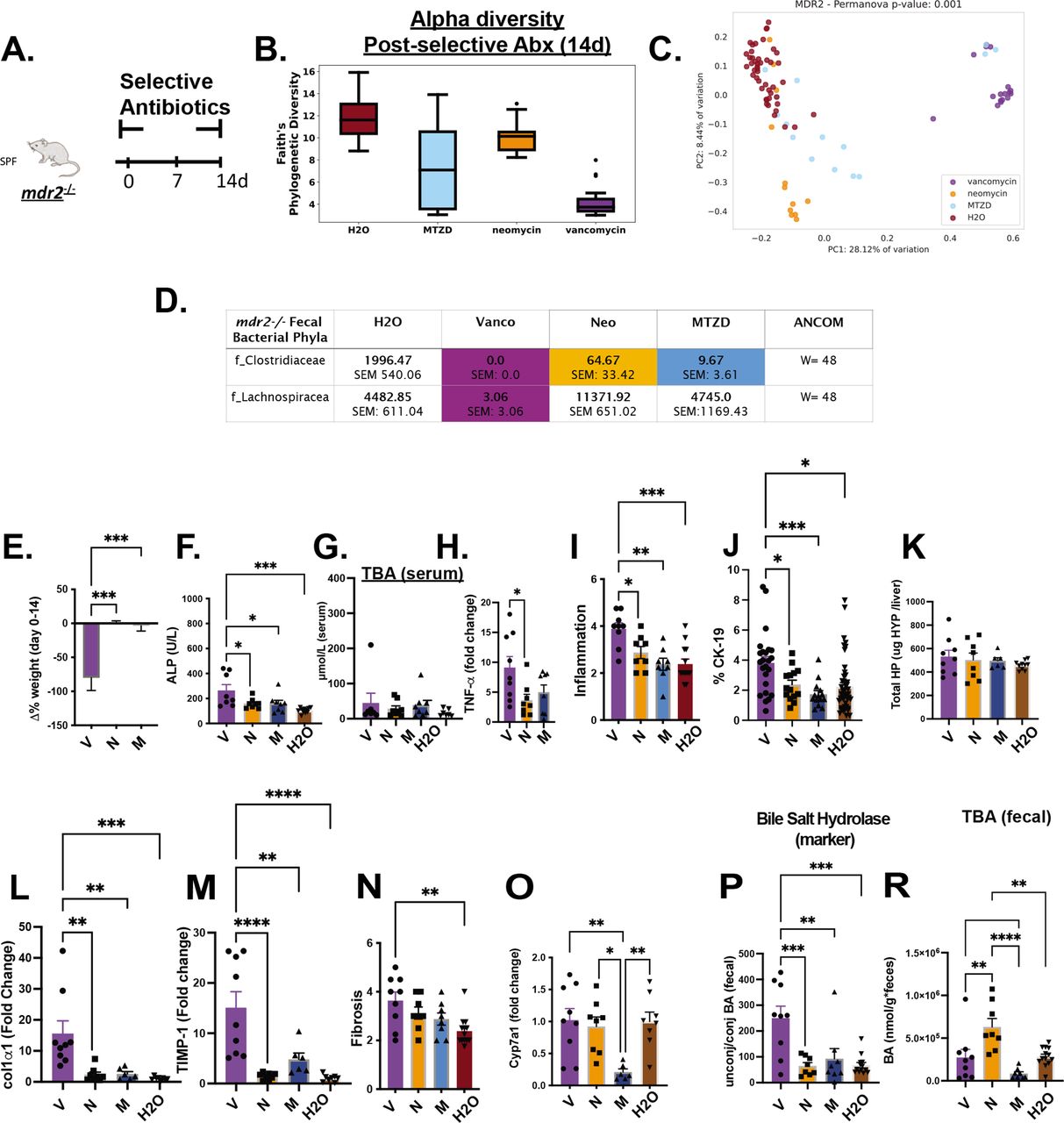
To identify microbial populations responsible for protecting mdr2−/− mice, we tested individual Abx components: vancomycin, neomycin and metronidazole that target Gram-positive, Gram-negative and anaerobic bacteria, respectively (figure 6A). Vancomycin most significantly decreased faecal bacterial α-diversity (figure 6B) and maintained most divergent bacterial populations with maximum β-diversity separation from untreated controls (figure 6C). Vancomycin and metronidazole decreased putative protective Clostridiaceae, but only vancomycin attenuated Lachnospiraceae. Neomycin did not significantly affect either family (figure 6D). Vancomycin treatment did not affect total serum BA levels (figure 6G), similar to Abx, but restricted weight gain and elevated serum ALP, hepatic TNF-αexpression, histological inflammation and ductal reaction (figure 6E,F and H–J).

Differential effects of selective antibiotics on mdr2−/− microbial, clinical, biochemical and histological outcomes. Experimental design of selective antibiotic treatment (A): SPF mdr2−/− mice were given individual antibiotics (vancomycin (V; 0.5 mg/mL, n=16), neomycin (N; 1 mg/mL, n=12) or metronidazole (M; 30 mg/mL, n=16) in autoclaved drinking water ad libitum and control mdr2−/− mice received autoclaved water alone (n=16) for 14 days. Alpha diversity, beta diversity (Permanova test) and differential abundance (centred log-ratio (CLR) transformation from CoDA methods) of Clostridiaceae and Lachnospiraceae from 14d faecal samples (B–D). Measured parameters include: 14-day weight change (E), serum ALP (F) and serumtotal bile acids (TBA) (G), and liver RNA expression of tnf-α (H); along with blinded composite histologic scoring murine liver stained with H&E (I) and CK-19 (J). Fibrosis readouts include hepatic collagen deposition (K), RNA expression of col1α1 (L) and timp-1 (M) along with Sirius Red composite staining (N) cyp7a1 liver expression (O) as well as pooled cecal content bile acids (BAs) differences in surrogate bile salt hydrolase activity indicator based on ratios of total unconjugated/conjugated BA (P) and total BA (R) of selective antibiotic treated mdr2−/− compared with H2O control. (V=9, N=8, M=8, H2O=8). CK-19+ represented of 5HPF/liver/mouse. Group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. Welch correction was applied to histological scoring analysis. Unadjusted raw average OTU relative abundance and SEs of bacterial groups against the variables detected with significant effects by analysis of composition of microbes. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ALP, alkaline phosphatase; SPF, specific pathogen free.
Concurrent with hepatobiliary injury and inflammation, vancomycin exacerbated hepatic fibrogenesis. Relative to control mice, only vancomycin significantly increased histological liver fibrosis scores (figure 6N), hepatic profibrotic co1α1 and timp-1 expression (figure 6L,M) but not hepatic hydroxyproline (figure 6K).
Exploration of selective Abx on BA metabolism demonstrated isolated metronidazole attenuated hepatic cyp7a1 expression in SPF mdr2− /− mice (figure 6O). Vancomycin profoundly increased BSH activity, measured by the unconjugated/conjugated BA ratio (figure 6P), despite not changing total faecal BAs versus control (figure 6R). These results suggest that depletion of vancomycin-sensitive bacterial populations, including Clostridiaceae and Lachnospiraceae, results in aggressive hepatobiliary responses.
Lachnospiraceae administration attenuates phenotypic effects of antibiotic-treated SPF mdr2−/− mice and inhibits hepatic bacterial translocation
We reconstituted the putative protective bacteria, Clostridiaceae and Lachnospiraceae, in the accelerated 7d Abx model (online supplemental figure S2A–E). Because Abx depleted protective faecal Clostridium clusters IV and XIVa22 23 in both SPF WT and mdr2−/− mice (online supplemental figure S2K,L), we initially tested a 17-strain consortium of protective human Clostridia.22 Despite efficacy of these Clostridium strains in multiple colitis models,22 their repletion in antibiotic-pretreated mdr2−/− mice did not alter body weight or liver enzymes (online supplemental figure S2M–P). Therefore, we assessed the biological activity of potentially protective Lachnospiraceae also depleted by Abx and vancomycin (figure 6D). Serial treatment with a protective colonic 23 strain Lachnospiraceae consortium (Lachno)24 (figure 7A) restored weight gain and reduced histological liver inflammation, ductular reaction and fibrosis (figure 7B and E–G) but did not decrease serum biochemistries (figure 7C, online supplemental figure S2Q). Lachno exposure minimally affected total faecal and serum BA, liver cyp7a1 and ileal FGF15 gene expression (online supplemental figure S2R–U). Reconsituting Lachnospiraceae in GF mdr2−/− mice resulted in reduced liver fibrosis, without affecting mortality or liver inflammation compared with non-colonised GF mdr2−/− mice (online supplemental figure S3F–H).
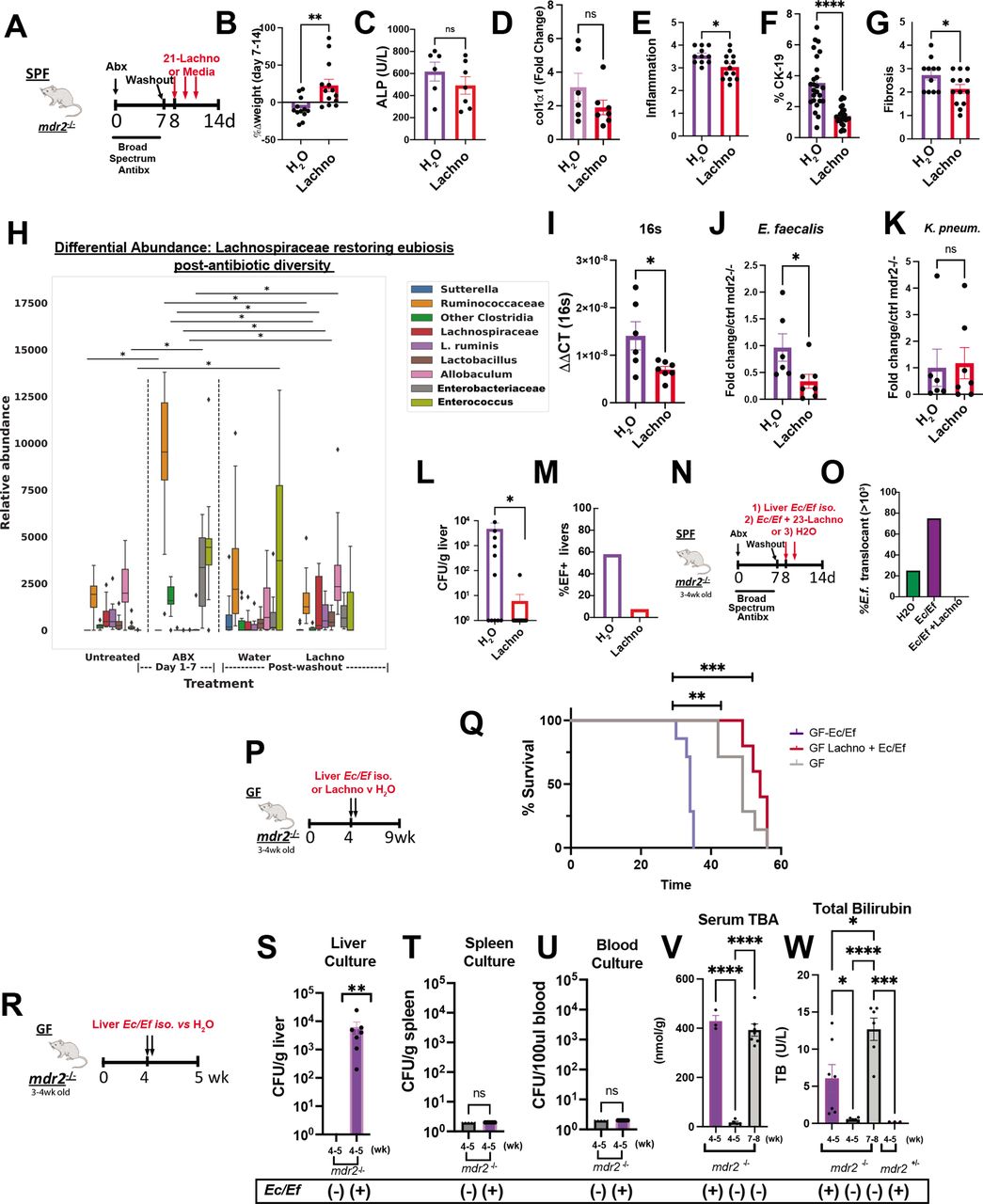
Lachnospiraceae phenotypic and metabolic effects on dysbiosis in mdr2−/− mice. Using the 7-day broad-spectrum antibiotic pretreatment model, followed by a 1-day washout, 21 Lachnospiraceae strains (Lachno) versus H2O were administered to SPF mdr2−/− mice on days 8, 10 and 12 (A). Changes following Lachno treatment included Δweight over 7 days (B), serum ALP (C), liver RNA col1α1 expression (D), blinded histological inflammation, ductal reaction and hepatic fibrosis scoring (E–G). Differential abundance expression of 16S rRNA analysis (H) (N, mdr2−/− untreated=24, Abx=36, H2O=11, Lachnospiraceae=13, pooled from two experiments. Presence of universal 16S, Enterococcus and Klebsiella pneumoniae (K. pneum) faecal DNA by qPCR (I–K) (one of two representative experiment, n=6 mice/group). Hepatic bacterial translocation (L), % of mice with Enterococcus faecalis liver translocation (M). Experimental design of treatment of 3–4 week old SPF mdr2−/− mice treated with broad-spectrum antibiotic cocktail (vancomycin, neomycin and metronidazole) for 7 days followed by a 1-day washout, then inoculated with mdr2− /− resident Ec/Ef isolates, versus Ec/Ef+23-Lachno combination or water only controls (N) assessing translocated bacteria cultured from homogenised liver (O) (n=4 mice/group). Experiment design (P) and Kaplan-Meyer survival curves (Q) of orally inoculated 108 pooled Ec/Ef hepatic isolates±Lachnospiraceae (Lachno) or H2O controls in GF mdr2−/− mice (H2O: n=12; Ec/Ef: n=7; Lachno: n=6). GF mdr2−/− mice were administered 108 pooled Ec/Ef hepatic isolates and euthanized at 5 weeks in order to measure translocated bacteria to liver, spleen or blood (S–U). Longitudinal measurements of serum TBA and bilirubin conducted in GF mdr2−/− ±Ec/Ef hepatic isolates (V and W). Group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. GF, germ free; SPF, specific pathogen free.
Abx altered faecal microbial communities 7 days after treatment with expanded Ruminococcaceae, Enterobacteriaceae and Enterococcus (figure 7H). Lachno supplementation resembled the pre-Abx state and particularly repressed Enterococcus expansion versus controls (figure 7H), confirmed by qPCR of faecal samples (figure 7I,J). Lachno treatment did not decrease faecal Klebsiella pneumoniae 2 (figure 7K). Importantly, Lachnospiraceae treatment reduced post-ABX culturable hepatic translocated bacteria (figure 7L). Sanger sequencing identified the translocating strains as E. faecalis (Ef) and E. coli (Ec), with Ef composing 60% of translocants in Abx-treated SPF mdr2−/− mice (figure 7M); all hepatic Ef isolates expressed the exotoxin cytolysin (online supplemental figure S3A–D).25
Antibiotics increase hepatic translocation of E. faecalis and E. coli, which accelerate mortality without sepsis
To more directly assess hepatic changes caused by Ef and Ec, 3–4 week old SPF-antibiotic pretreated or GF mdr2−/− mice were colonised with selected Ec/Ef hepatic isolate strains or H2O and euthanised at 5 weeks of age (figure 7N). Hepatic translocated Ec/Ef were isolated in both antibiotic pretreated and gnotobiotic conditions (figure 7O and S) versus untreated controls. Lachno treatment prevented Ec/Ef liver translocation (figure 5O) and Ef faecal concentrations (figure 7J and H). We assessed lethality of these bacteria in gnotobiotic mdr2−/− mice by orally inoculating Ec/Ef isolates with/without Lachno (figure 7P,Q). The Ec/Ef dual-associated mice exhibited earlier mortality relative to controls (figure 7Q), but survival improved with cocolonisation with Lachno relative to control GF mdr2−/− mice. Ec/Ef isolates colonised GF mdr2−/− mice had no evidence of bacteremia or splenic translocation (figure 7T,U). Additionally, Ec/Ef dual-association accelerated serum BA shunting and associated cholestasis in mdr2−/− mice (figure 7V,W). These results demonstrate that Ec/Ef translocation induces hepatobiliary injury and mortality driven by mechanisms independent of sepsis and systemic bacterial dissemination, and Lachnospiraceae protects from Ec/Ef hepatic translocation and mortality.
Short-chain fatty acids (SCFAs) produced by Lachnospiraceae have hepatic antifibrogenic effects
We postulate these anti-inflammatory and antifibrotic effects are mediated by Lachnospiraceae metabolic products, including SCFAs that are depleted by selective antibiotics (figure 8b). Vancomycin most effectively decreased SCFA (figure 8b), suggesting vancomycin-sensitive, resident bacteria (like lachnospiraceae) are predominant SCFA producers. Oral supplementation of SCFAs (acetate, propionate and butyrate) in vancomycin-treated mdr2−/− mice attenuated hepatic fibrosis (figure 8d–g). The 23 Lachno strains with variable SCFA production were divided into three strains with relatively low (8, 9 and 21-lo) or high (52, 60 and 70-hi) SCFA production as determined by mass spectrometry of supernatants (figure 8h,i). We serially gavaged Hi or Lo SCFA-producing Lachno to SPF mdr2−/− mice following antibiotic pretreatment and washout (figure 8i). Hi SCFA-producing Lachno attenuated histological hepatic fibrosis (figure 8j) but not hepatic inflammation (figure 8k) compared with Lo SCFA-producing Lachno. These results provide a mechanism for protection by our Lachnospiraceae consortium.

Effect of short-chain fatty acids (SCFAs) on mdr2−/− antibiotic model. Experimental design of 14-day selective antibiotic treatment (A) with measurement of cecal content SCFA in SPF mdr2−/− mice (B). Design of SPF mdr2−/− mice treated with vancomycin (0.5 mg/mL in drinking water, n=10) with or and without SCFA (67.5 mM acetate, 25.9 mM propionate, 40 mM butyrate and 3% sucrose, n=13) ad libitum (C), pooled from two experiments with representative Sirius Red photomicrographs of vancomycin versus vancomycin+SCFA SPF mdr2−/− mice, arrows highlight bridging fibrosis (D), blinded histologic fibrosis scoring (E) along with liver RNA col1α1 and timp1 expression (F–G). Three relatively low and high SCFA-producing (Lo strain #s: 8, 9 and 21; Hi #s: 52, 60 and 70) Lachnospiraceae strains selected from 21 consortium strains were identified by mass spectrometry (H). Experimental design of accelerated antibiotic pretreatment of SPF mdr2−/− mice for 7 days and following a 1 day washout, administering Hi or Lo SCFA-producing Lachnospiraceae strains by gavage on days 8, 9 and 10, then followed for 6 days (I) with measurement of histologic hepatic fibrosis (J) and inflammation (K) in Hi compared with Lo SCFA-producing strains. Group or pairwise comparisons performed by analysis of variance or Student’s t-test, respectively. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
E. faecalis and Lachnospiraceae are associated with divergent PSC clinical disease activity
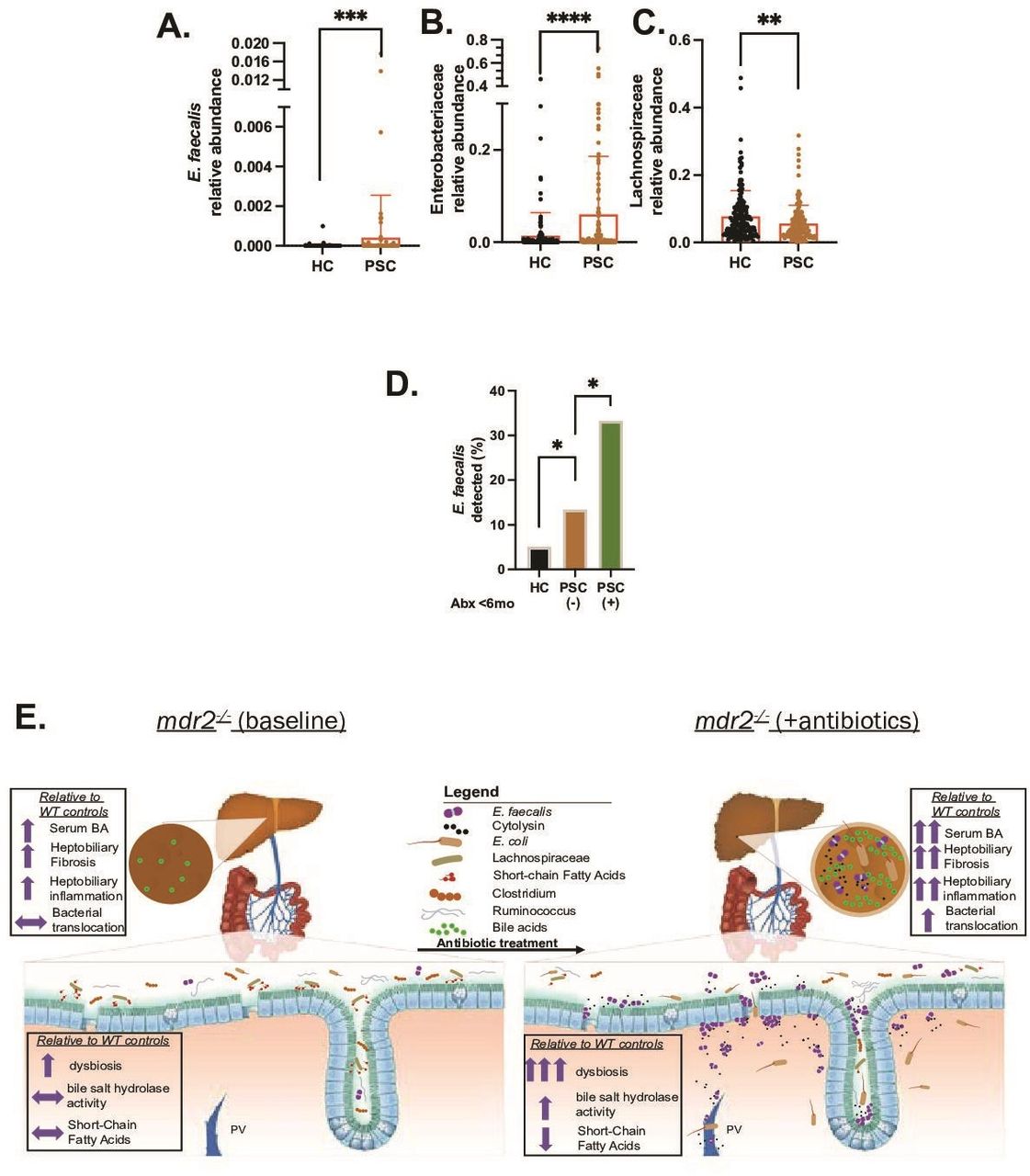
Analysis of metagenomic data from multinational repositories (Norway and Germany)4 of PSC faecal samples identified enrichment of E. faecalis and Enterobacteriaceae (figure 9A,B) and depletion of Lachnospiraceae (figure 9C) in PSC patients compared with healthy controls. Both E. faecalis (species) and Enterobacteriaceae (family) positively correlated with Mayo risk score (n=51, rho 0.28 for both, p<0.05) in patients with PSC, and Lachnospiraceae Blautia (genus), Lachnospiraceae bacterium 1_4_56FAA negatively correlated with Mayo risk score (rho=−0.34 for both, p<0.05) with only a trend at the family level (rho −0.2). E. faecalis was more commonly detected in patients with PSC than controls (17% vs 5%) (figure 9D), which was amplified in patients who received antibiotics the last 6 months (figure 9D). Trends existed towards increased Enterobacteriaceae and reduced Lachnospiraceae in patients with antibiotics compared with those without. These clinical results mirror observations in our mdr2−/− mice, providing clinical relevance to our murine results.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Selective faecal bacterial profile in PSC patients and effect of antibiotic exposure. Relative abundance of faecal Enterococcus faecalis (E. faecalis) (A), Enterobacteriaceae (B) and Lachnospiraceae (C) in heathy controls (HCs; n=158) versus PSC patients (n=136). (D) Prevalence of faecal E. faecalis in HC (irrespective of antibiotics use) versus PSC patients exposed to antibiotics (n=24) and no antibiotics exposure (n=112) the last 6 months. Data in figure parts A–C compared with Mann–Whitney U test, (D) with Fisher’s exact test. Graphical summary depicting the effect of antibiotic-induced dysbiosis of SPF mdr2−/− mice to potentiate hepatobiliary disease with hepatic translocation of E. faecalis and Escherichia coli, increased bile salt hydrolase activity (conjugated/unconjugated BA), hepatic bile acid pool size, and decreased SCFA production (E). *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. BA, bile acid; PSC, primary sclerosing cholangitis; SCFA, short-chain fatty acid.
Discussion
We identified functional subsets of resident luminal bacteria that either potentiate liver inflammation and fibrosis (E. faecalis and E. coli) or protect (Lachnospiraceae) through distinct mechanisms (figure 7E). We confirmed the overall protection of resident microbiota in GF mdr2−/− mice14 26 27 in a more aggressive genetic background (C57BL/6).28 We observed early lethality and severe cholestasis in GF mdr2−/− mice (median survival: 7.5 weeks) driven by loss of microbial-mediated hepatic accumulation of toxic non-micellular bile salts,12 29 30 stimulating robust hepatobiliary macrophage recruitment, inflammation, ductular reaction and fibrosis. Supporting progressive hepatic BA accumulation-induced toxic cholangiopathy, ASBTi treatment resulted in faecal BA loss, attenuated serum TBA, improved cholestatic liver and clinical endpoints independent of ileal FGF-15 expression in mdr2−/− antibiotic pretreatment model. Early (<5 weeks) but not later (>6 weeks old) autologous SPF FMT rescued this lethal phenotype and cholestasis, validating a strong protective role for SPF mdr2−/− microbiota within a short therapeutic window. This brief therapeutic window for effective microbial-mediated protection precludes the likely longer microbial engraftment required to mediate downstream events and mandates intervention at an early inflammatory stage. These preclinical studies imply the need for early-stage clinical microbial therapeutic interventions.
We used broad-spectrum and selective antibiotic treatment to validate protection by host microbiota and, more importantly, to delineate functional therapeutic and detrimental microbial subsets in this model system with shotgun faecal metagenomic and metabolomic analyses. We narrowed the protective microbial community to vancomycin-sensitive populations and metabolites: Lachnospiraceae,9 Clostridaeceae,22 BA and SCFA. Selective colonisation of GF and depleted mice with a human Clostridiaceae consortium with protective benefits in colitis models22 did not confer protection in our model. However, a Lachnospiraceae consortium24 attenuated hepatic inflammation and fibrosis in antibiotic pretreated mice without changing total faecal BA or FXR/Cyp7a1 signalling, suggesting alternate non-BA mechanisms. Direct supplementation with SCFAsdecreased hepatic fibrogenesis in our vancomycin-Lachnospiraceae depleted SPF model. Antifibrogenic effects of high-SCFA-producing Lachnospiraceae strains suggest that SCFAs partly mediate the protective effects of Lachnospiraceae. Our observations that Lachnospiraceae monoassociation of GF mdr2 −/− mice decreased hepatic fibrosis but not inflammation or extended lifespan compared with GF mice, yet decreased inflammation, decreased E. coli and E. faecalis luminal concentrations and hepatic bacterial translocation in antibiotic treateed SPF mice suggest a direct antifibrotic effect of Lachnospiraceae but that their anti-inflammatory benefits are mediated by suppressing resident aggressive species. Metagenomic analysis of feces4 from a PSC patient cohort demonstrated clinical relevance of our findings and showed decreased Lachnospiraceae species in PSC patients and Lachnospiraceae presence associated with lower Mayo risk scores.
Other studies show divergent hepatobiliary outcomes of FXR agonism in mdr2−/− mice.14 31 These differences are likely driven by the presence or absence of microbial depletion-mediated expansion of hepatic BA accumulation in each murine model. We found 20× attenuated liver cyp7a1 gene expression and increased hepatic BA accumulation in GF compared with SPF mdr2−/− mice, despite no difference in ileal FGF15 expression (a key downstream FXR target). Despite increased total serum BA, no difference in ileal FGF-15 expression was similarly reported recently by Schneider et al 14 between antibiotic treated vs untreated mdr2−/− mice, suggesting a microbial based, FXR-independent driver of hepatic BA accumulation in this model.12
We demonstrated clinical evidence of positive and negative correlations with Mayo risk score between both resident pathobionts (E. faecalis and Enterobacteriaceae) and protective Lachnospiraceae, respectively. In addition, E. faecalis expand in PSC patients exposed to antibiotics. Unfortunately, the antibiotic type and spectrum were unavailable in our clinical data set. Vancomycin clinical studies show heterogenous biochemical responses in PSC patients,32 suggesting individual vancomycin therapeutic effects on protective and pathogenic bacteria. These studies lack baseline and postintervention microbial profile assessments necessary to assess antibiotic microbial targets. Our mdr2−/− results suggest that broad-spectrum antibiotics should be administered with caution in PSC patients due to potential for exacerbating disease and contributing to recurrent cholangitis by promoting enterohepatic bacterial translocation.
This study has several limitations. Early lethality of our GF and broad-spectrum antibiotic-depletion mdr2−/− mice precluded longitudinal studies to assess delayed effects of Lachnospiraceae or SCFA on chronic disease progression. Second, we acknowledge the ongoing controversy regarding suitability of murine AP to monitor cholestasis.33 34 Therefore, we confirmed our findings with TB and bile duct reactivity as additional markers of cholestatic injury. Finally, because GF mdr2−/− mice expire before becoming fertile, our heterozygous mdr+/− breeding strategy with small litter sizes delayed experiments and limited more ± depth mechanistic studies but permitted littermate control comparisons.
Our mechanistic studies have direct translational implications for future personalised bacterial manipulation in PSC patients. For example, carefully screened cohorts of PSC patients may optimally respond to selective depletion of disease-inducing subsets and/or augmentation of protective bacteria or secreted metabolites. Additionally, our gnotobiotic model can explore hepatobiliary, microbial and metabolomic impacts of humanised faecal transfer and selective colonisation with WT and genetically engineered bacterial strains to dissect microbial protective and pathogenic mechanisms. This study also highlights specific in vivo roles for bacteria, such as Lachnospiraceae, play require additional protective resident bacteria to recapitulate various protective effects transferred by faecal microbiota transplantation. Future directions will determine the influence of macrophage phenotypes,35 downstream SCFA targets and activities of other putative protective bacteria on experimental hepatobiliary inflammation and fibrosis. In conclusion, our gnotobiotic and antibiotic mdr2−/− PSC murine models identified complex interactions between novel hepatoprotective resident bacteria balanced against pathobionts, replicated in PSC patient cohorts. Resident bacterial subsets exert both positive and negative influences partly through antimicrobial activities and altering bacterial metabolites.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Ethical approval was obtained from the respective Local Ethics Committees (Norway: Regional Committee for Medical and Health Research Ethics in South-Eastern Norway (reference number 2015/2140); Germany: Hamburg (reference number MC-111/15) and Kiel (reference numbers A148/14, A117-13 and A156-03)). We conducted mouse studies under the NIH guide for Care and Use of Laboratory Animals, approved and overseen by the UNC-Chapel Hill Institutional Animal Care and Use Committee (Protocol ID 18–266). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
Bo Liu, DVM, PhD, and Akihiko Oka, MD, PhD, provided technical assistance; Josh Frost, UNC Gnotobiotic Facility Manager for gnotobiotic research support; Fengling Li, MD, PhD, and Allison Rogala, DVM, for germ-free derivation of mdr2−/− mice; Lisa Holt for laboratory management and Cary Cotton for statistical support. Special appreciation to Dr Vincent Young, University of Michigan, for providing the original Lachnospiraceae stains and to Dr Kenya Honda, Rikken Institute, Tokyo, for providing the Clostridium consortium species. Special thanks to UNC Clinical Chemistry and CGIBD Pathology Cores for assistance with liver enzyme analysis and histologic preparation and staining.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @hov_jer
Contributors MA and RBS are guarantors of the paper. MA and RBS conceptualised the study. MA, BN, AV and MF contributed to the collection of samples. JT and JPYT grew and contributed Lachnospiraceae strains. KL and YL were responsible for metabolomics. JW performed the postsequencing data processing. SM performed blinded H&E and Sirius Red histological scoring. VM and HLF performed IHC staining along with image analysis. HZ, PBH, JSB and MA performed bile acid analysis. MA executed statistical analysis and data organization. MK, LT, SC, CB, AF and JRH collected and analysed human faecal metagenomic data. MA, VM, JW, JT, MK, YL, KL, JPYT, HLF and RBS performed data curation. YVP provided murine resources and technical advice. MA, JW and RBS planned the analysis reviewed data and edited the results. MA, JW, MK, JRH and RBS performed formal analysis. MA and RBS interpreted data. RBS supervised the study, and MA was responsible for project administration. MA and RBS wrote the original draft. MA, JW and RBS contributed to the writing of the manuscript. All authors read, critically revised for important intellectual content, and approved the final manuscript. RBS and MA acquired funding.
Funding National Institute Health (NIH) grants P01DK094779, P40OD10995 and P30DK034987 (to RBS), K01DK119582 (to JW); T32DK07737 and T322A1007273 (to MA), The Crohn's and Colitis Foundation #2434 (to RBS); CA232109, DK094779 and AI067798 (to JT); VA 1I01BX003031 and IK6BX005226; DK108959 and DK119421 (to HLF). JRH is funded by the European Research Council, grant no. 802544. CS is supported by the DFG, CRU306, and by the Helmut and Hannelore Greve Foundation and grant support from BiomX and Galapagos. VA Merit Award I01BX004033; Research Career Scientist Award (IK6BX004477); VA ShEEP Grant (1 IS1 BX004777); NIH Grants R01 DK104893, R01DK-057543 (to HZ). VA Merit Review 2I0CX001076, R21TR003095 (NCATS) (to JSB). VA Merit Award BX001328 (to PBH).
Competing interests RBS has consulted for and received grant support from Takeda, Janssen, Second Genome, Vedanta, BiomX, Biomica, SERES and Artizan; JRH has served on advisory boards and/or given lectures for Orkla Health, Novartis, Amgen and Roche, and received research support from Biogen, all unrelated to the present study.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.