Article Text

Abstract
Objective The transcriptional response in the liver during HCV infection is critical for determining clinical outcomes. This issue remains relatively unexplored as tissue access to address this at scale is usually limited. We aimed to profile the transcriptomics of HCV-infected livers to describe the expression networks involved and assess the effect on them of major predictors of clinical outcome such as IFNL4 (interferon lambda 4) host genotype and sex.
Design We took advantage of a large clinical study of HCV therapy accompanied by baseline liver biopsy to examine the drivers of transcription in tissue samples in 195 patients also genotyped genome-wide for host and viral single nucleotide polymorphisms. We addressed the role of host factors (disease status, sex, genotype, age) and viral factors (load, mutation) on transcriptional responses.
Results We observe key modules of transcription which can be impacted differentially by host and viral factors. Underlying cirrhotic state had the most substantial impact, even in a stable, compensated population. Notably, sex had a major impact on antiviral responses in concert with IL28B (interleukin 28B)/IFNL4 genotype, with stronger interferon and humoral responses in females. Males tended towards a dominant cellular immune response. In both sexes, there was a strong influence of the underlying host disease status and of specific viral mutations, and sex-specific expression quantitative trait loci were also observed.
Conclusion These features help define the major influences on tissue responses in HCV infection, impacting on the response to treatment and with broader implications for responses in other sex-biased infections.
- HEPATITIS C
- INFLAMMATION
- INFECTIOUS DISEASE
- GENE EXPRESSION
- BIOSTATISTICS
- Sex immunology
Data availability statement
Data are available in a public, open access repository. Data are publicly available and deposited on online repository Gene Expression Omnibus Database (GEO, GSE149601)
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
What is already known on this subject?
Spontaneous HCV clearance and successful antiviral therapy rates are largely in favour of women and/or in subjects with a specific interferon lambda 4 (IFNL4) genotype. The unfavourable IFNL4 genotype leads to an exaggerated interferon response eventually detrimental for the patient. However, women tend to have a more robust and prompt interferon activity. Beyond this, the causes of sex differences in infections are insufficiently understood.
What are the new findings?
We detected transcriptional networks in infected livers influenced by sex, host genotype and age. In females we observed a consistent interferon response largely enhanced by IFNL4 genotype. Males show overexpression of genes linked to cellular immune responses. Expression of other gene networks (eg, immunoglobulin genes) are sex biased and further influenced by the presence of viral variants. The evidence that clinically relevant genes can be differentially active between sexes was corroborated by the detection of sex-specific single nucleotide polymorphisms through expression quantitative trait loci analysis.
How might it impact on clinical practice in the foreseeable future?
These findings help define the patient-specific impacts of IFNL4 genotype. Beyond HCV, the combined impact of sex and genotype on transcriptional responses could have implications in personalised medicine, where combinations of factors must be considered to effectively treat a patient. In parallel, host genetic polymorphisms could have clinically different impacts between sexes, with broader implications for patient stratification.
Introduction
HCV is an enveloped, single-stranded, positive-sense RNA virus (genus Hepacivirus, family Flaviviridae).1 Based on the sequence variability of its 9.6 kbp genome, HCV is classified in seven genotypes (G1: 44%–46%, G3: 22%–25%; G2 and G4: 12%–15% of cases worldwide).1 2 The prevalence of persistent HCV infection has been estimated to be 1%–2% worldwide, corresponding to 72–149 million infected people.1 2 Following acute infection (which is mostly asymptomatic), around 20% of individuals clear the virus spontaneously, while the majority progress to chronic infection of the liver.1 As this state persists lifelong unless treated, patients are at risk of developing cirrhosis (15%–30%) and hepatocellular carcinoma (HCC) (2%–4% of HCV cirrhosis patients), leading to approximately 700 000 deaths per year.1 2 Factors identified to impact virological outcomes include sex, host human leukocytes antigen (HLA) genes and the host IL28B/IFLN4 genotype (linked to interferon lambda 3 and 4 expression).3–7 While female sex is associated with lower prevalence and higher rates of spontaneous viral clearance both in HCV and HBV, men are more prone to chronic infections and disease progression to cirrhosis and HCC.3–5 The favourable IL28B/IFNL4 genotype8 9 (described here as CC) is associated with higher rates of disease resolution and treatment response compared with the non-CC genotype. However, despite these clear-cut results, the mechanisms defining successful versus unsuccessful control of this virus (and their impacts on treatment responses) are still not understood.
While the overall concept of sex differences in immune responses are well described, the underlying mechanisms of the sexual dimorphisms in infectious disease, and in HCV in particular, are poorly understood. Studies suggest that the higher rates of spontaneous clearance in women are determined by stronger, more efficient innate inflammatory responses, potentially triggered by an increased toll-like receptor 7 (TLR7) response.4 Treatment with TLR7 agonists (SM360320/isatoribine) both in vitro and in vivo led to a decrease in HCV RNA plasma levels and more potent IFN-α response.3 10 Other theories include the direct effects of oestrogens, shown to decrease the production of HCV virions and the expression of hepatic viral entry receptors.4 11
To address this knowledge gap, we designed a transcriptomic analysis of liver biopsies obtained from 195 chronic patients infected with HCV (BOSON trial).12 This carefully curated cohort allowed parallel investigation of the contextual effects of underlying disease status, sex, host genotype and viral genotype. In this study, we aimed to define the key drivers of transcriptional variability in human tissue biopsies during HCV infection.
In addition to impacts of cirrhosis and the role of IL28B genotype, we find a strong and consistent impact of sex differences in immune response to chronic HCV infection, with evidence for different pathways of response in males and females. The results of this study not only facilitate novel insights into the pathogenesis of chronic hepatitis, the immune response to and treatment of HCV, but also have implications for other RNA virus infections.
Results
Defining transcriptional variability in the sample set
We performed bulk RNA-Seq on 195 liver biopsy samples taken pretreatment in patients from the previously described BOSON HCV cohort.12 We first explored this large dataset through an unsupervised sample clustering analysis. It was first observed that underlying liver cirrhosis was the major source of gene expression variability (figure 1A) driving these severe patient samples to cluster together. Of note, study entry required evidence of no current or prior decompensation.
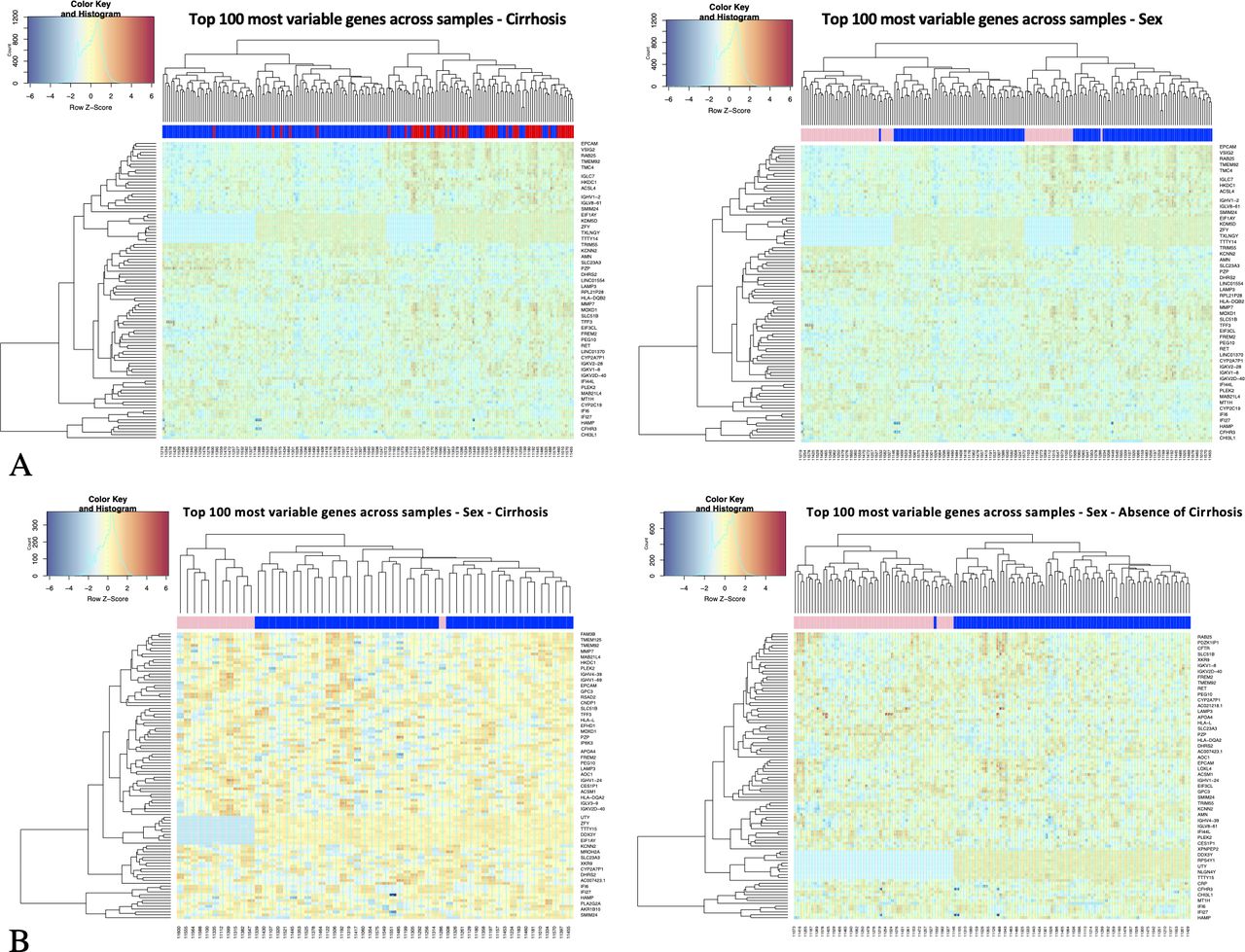
Unsupervised sample clustering analysis of HCV-Liver transcriptomics. Hierarchical clustering of transcription profiles using the top 100 most variable genes across all samples; (A) all sample clustering with sample class labels colour coded by cirrhotic status (left plot, red=cirrhosis, blue=absence of cirrhosis) and by sex (right plot, pink=female, blue=male). (B) Cirrhotic (left plot) and non-cirrhotic (right plot) sample clustering with sample class labels colour coded by sex. Gene counts are log transformed (logCPM), centralised across rows (z-scores); Euclidian distance as metric of similarity.
In a first analysis, when samples are colour labelled by sex, they show sex segregation, clear in both cirrhotic and non-cirrhotic samples. This implies that sex clustering is not due to the greater incidence of cirrhosis in males than females (figure 1B). This initial analysis, filtering active genes (sufficiently expressed and most variable across samples) chosen for clustering analysis, includes features mapping in X and Y chromosomes.
Variance partitioning analysis of autosomal gene expression
We next assessed whether the variability of expression of genes mapping in autosomal chromosomes can be also accounted for by sex. The sources of expression variation in a complex dataset required the use of mixed linear model to quantify contribution of known and relevant covariates (figure 2A). Using the variancePartition13 statistical framework, we determined that within the subset of most variable genes (IQR >0.95, n=2934) the main sources of variability were attributable, as expected, to the cirrhotic status and IL28B/IFNL4 genotype; nevertheless the analysis showed that the expression of a significant proportion of genes is driven by other factors, for example, age and sex. Of note, genes located in sex chromosomes were excluded from this analysis to highlight sex-driven variation not explained by heteromorphic sex chromosomes.14
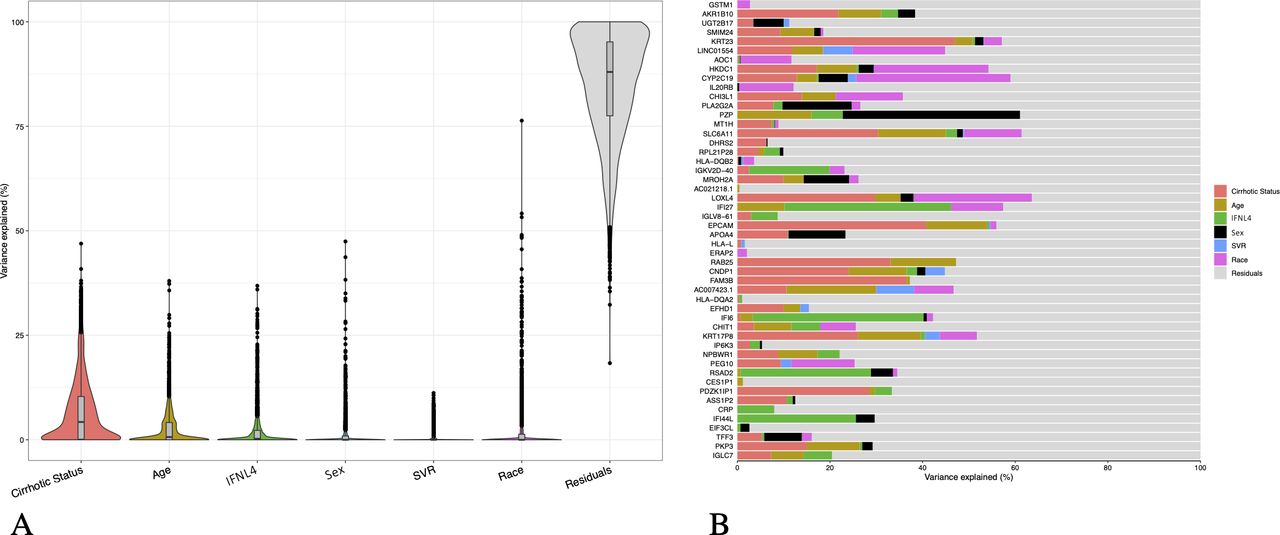
Variance partition analysis of main drivers of expression variability in autosomal genes. (A) Violin plots of variance fraction for each gene in each variable ranked by total contribution of variability for each variable (IQR >0.95, n=2934) (B) Bar plot of variance fractions for the subset of the top 50 most variable genes. Genes mapping in sex chromosomes were excluded from the analysis. SVR, sustained virologic response.
The bar plot in figure 2B shows the variance fractions of the top 50 most variable genes, highlighting for each feature the main sources of variation: other than unknown causes of variability (residuals), the main sources of variation for gene expressions can be attributed to cirrhosis status, although genes driven by sex or age are noted. For instance, expression variability is mainly accounted by sex for PZP (chromosome 12), reported to play a role in immunity-modulating T-helper cells.15 16 Other genes of interest driven by sex are PLA2G2A (with an antiviral function in HCV),17 MROH2A (associated with high level of bilirubin),18 APOA4 (functional in HCV assembly and cell entry),19 EIF3CL (a translation initiation factor)20 21 and TFF3 (overexpressed in HCCs).22
Unsupervised gene–gene correlation analysis
Next, exploiting the notion that genetic programmes are implemented through gene coexpression networks fitting a scale-free topology,23 24 we attempted to detect modules of highly correlated genes likely to represent networks that best characterise the main active transcriptional engines of inflamed livers. Weighted gene coexpression network analysis (WGCNA)23 was performed including all samples, filtering genes by overall variance (IQR >0.75, n=6997), and estimating the most suitable beta parameter (β=9) to fit a scale-free network. The dendograms in figure 3A show gene clustering revealing seven gene modules detected with WGCNA. Over-representation analysis of gene ontology (GO) categories (figure 3B, online supplemental table S1) and Reactome pathways25 (online supplemental figure S1) was carried out to evaluate the homogeneity and biological meaning of the gene content of each module. In each module, significant categories appear distinct from each other. The heatmap in figure 3C shows how modules’ eigengenes relate to relevant traits (including viral load and alanine transaminase (ALT) test). An expected module enriched in interferon pathways showed highest correlation with IFNL4 genotype (positive correlation with non-CC set colour coded in red, p value=4e-12) along with a significant contribution of age (p value=0.05) and sex (p value=0.01). Cirrhotic status does not have an effect on this module, while the CC genotype has a significant impact (correlation coded in green) in other modules enriched with immunological pathways. Sex shows also significant correlation to different modules (correlation to females in red, to males in green) although this analysis does not adjust for covariates.
Supplemental material
Supplemental material

Weighted gene coexpression network analysis (WGCNA) on HCV-liver transcriptomics. (A) Hierarchical clustering dendograms showing gene modules (m1–m7) detected with WGCNA in the set of most variable genes (IQR >0.75, n=6997). (B) Dot plot showing and comparing over-representation analysis of GO term categories in detected gene modules. (C) Heatmap showing bivariate correlations between module genes to sample traits with p values (in heatmap cells); highlighted (dashed red line) the trait correlations with the module enriched with interferon stimulated genes (ISGs) (m6) and module correlations with sex (dashed light blue).
PCA with reduced dimensionality
We next exploited this information to reduce the dimensionality of a complex dataset and conduct a more focused clustering analysis. Principal component analysis (PCA) of samples using genes in a module enriched with ‘extracellular matrix organisation’ and ‘degradation’ pathways (module=2) shows that samples segregate according to cirrhotic status rather than sex or IL28B/IFNL4 genotype (online supplemental figure S2a). In contrast, a sample PCA using exclusively the genes in a module enriched with interferon-related pathways (module=6) depicts segregation guided by IL28B/IFNL4 genotype, and clearly not driven by cirrhosis (figure 4A, online supplemental figure S3a). Preranked gene set enrichment analysis26 (GSEA) highlighted the significant overexpression of the interferon module in females versus males (figure 4B, online supplemental figure S3a). Linear modelling and data stratification were used to control for covariates and confirm that these genes are differentially expressed between sexes (figure 4B) and are not only explained by the prevalence of non-CC genotype (n=121) over CC (n=74) samples in the dataset.27 Preranked GSEA in figure 4B was performed ranking t-tests from linear modelling (adjusted for covariates).
Supplemental material
Supplemental material
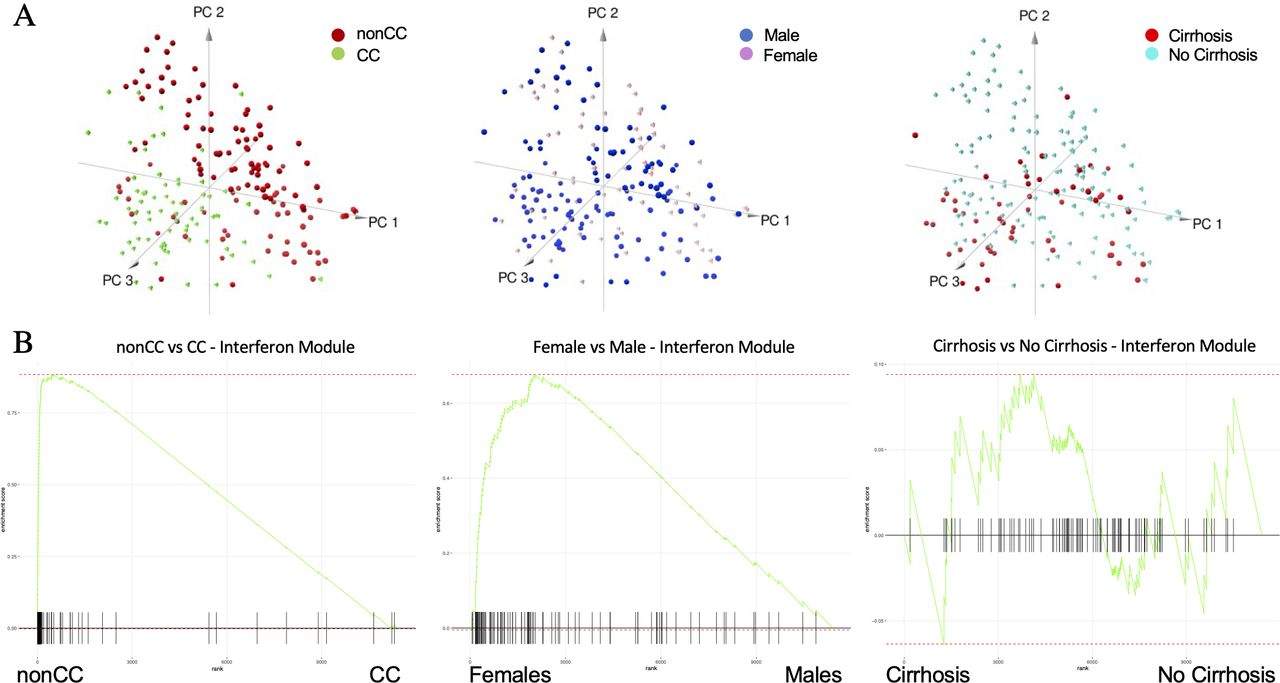
Principal component analysis (PCA) and preranked gene set enrichment analysis (GSEA) of interferon module genes. (A) Samples’ PCA using as dimension gene expression in interferon module and sample labels colour coded by IFNL4 (interferon lambda 4) genotype (left plot), sex (middle plot) and cirrhotic status (right plot). (B) GSEAs to assess enrichment of interferon module genes (m6) between non-CC versus CC (left plot), female versus male (middle plot) and cirrhosis versus non-cirrhosis (right plot) using genome-wide preranked t-tests after controlling for all covariates.
Network gene module enrichment between sexes
We next analysed sex effects in each gene set in the network modules detected with WGCNA using a preranked GSEA. As in figure 4B, t-test scores (females versus males) are obtained from a linear model controlling for cirrhotic status, IFNL4 genotype, age and race covariates. The GSEA table in figure 5 shows that the module associated with interferon signalling GO categories (m6) is the highest enriched in females over males while a module enriched in T-cell GO terms (m4) appears to be robustly enriched in males. Modules of less evident immunological relevance, related to cell RAS signalling transduction and ubiquitination process (m1), and carboxylic acid metabolism (m3), are still significantly enriched in females, whereas modules associated with cirrhosis status with genes implicated in vesicle secretion and extracellular matrix organisation pathways (m2) and catalytic process (m5) are more enriched in male patients. These analyses shows clear evidence that genes involved in regulatory networks associated with immune responses or characteristic of a disease outcome can be divergently expressed between sexes.

Weighted gene coexpression network analysis (WGCNA) module enrichment between sexes. Preranked (after covariates adjustment) gene set enrichment analysis (GSEA) table showing enrichments between sexes (F=female; M=male) of all WGCNA modules and ranked by adjusted p values. Enrichment score curves (left) showing divergent enrichment in sexes of interferon module (m6 in females) and T-cell module (m4 in males).
Impact of sex on specific host genotypes
Since non-CC and CC samples could be considered two different patient populations, with distinct phenotypes and clinical features, a regression-based differential expression (DE) analysis was performed separately for these two categories of individuals (CC, expressing only IFNL3 protein isoform, non-CC, able to express also IFNL4) to get a better insight on their defining characteristics and observe any specific sex effects.
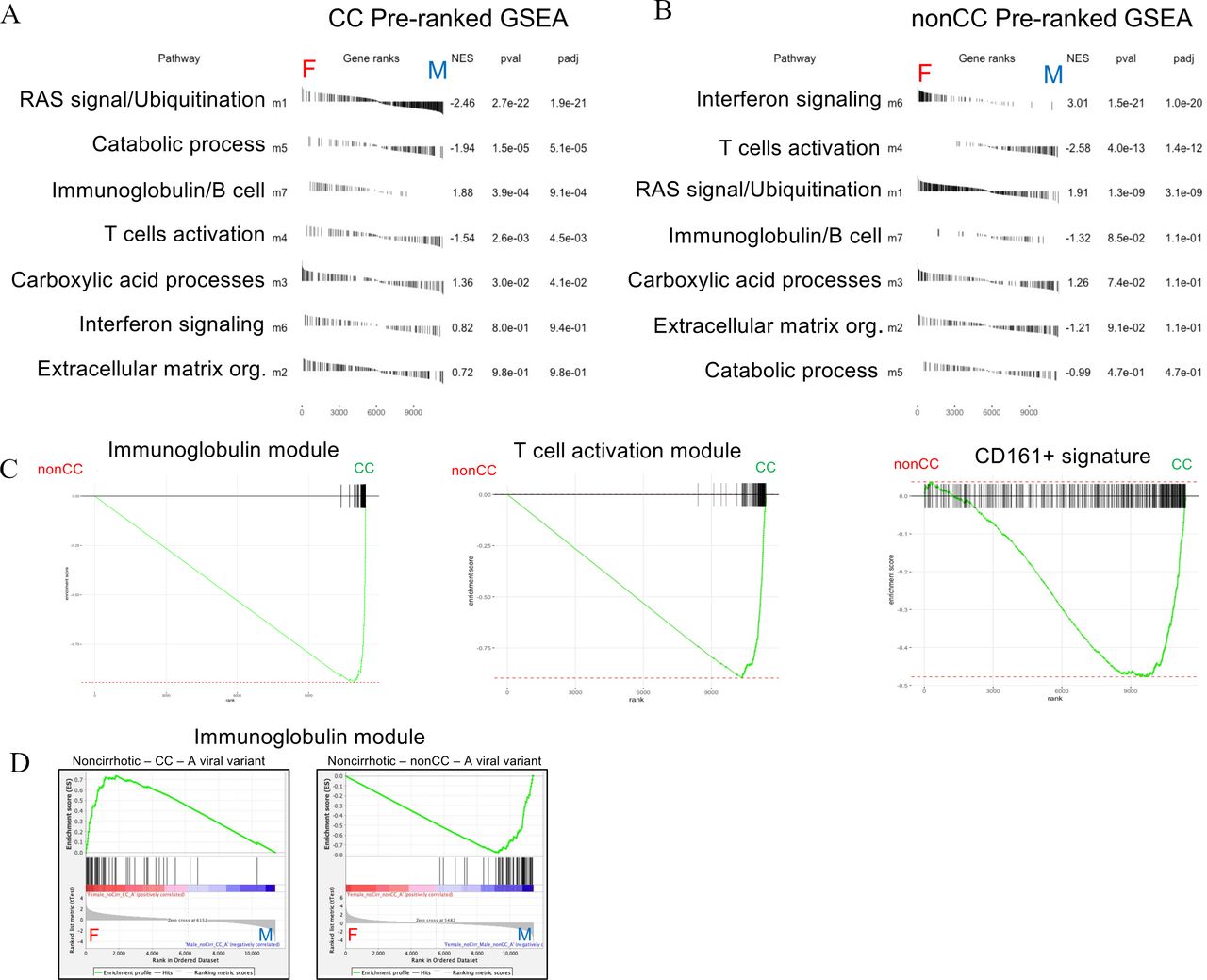
Tables in figure 6A,B show the enrichments of network modules between sexes in CC and in non-CC samples separately, describing how these genes can be divergent in sexes. The interferon module (m6) appears most significantly overexpressed in females in the non-CC group. In contrast, the T-cell activation module (m4) is more expressed in males both in CC and non-CC settings. Of note, the immunoglobulin module appears highly enriched in females in CC group.

Gene module enrichment between sexes in CC and non-CC genotypes. Preranked (after covariate adjustments) gene set enrichment analyses (GSEAs) showing sex enrichments of all weighted gene coexpression network analysis (WGCNA) modules (m1–m7) in CC (A) and non-CC (B) samples. Module enrichments are ranked by statistical significance (adjusted p values). (C) Preranked GSEAs with enrichment curves for immunoglobulin module (m7, left plot, false discovery rate (FDR) <<0.01), T-cell module (m4, middle plot, FDR<<0.01) and Mucosal Associated Invariant T (MAIT) cell signature (CD161+, right plot, FDR=0.003) between IFNL4 (interferon lambda 4) genotypes (non-CC versus CC) in overall analysis (all covariates control). Standard GSEAs (D) showing sex enrichments of immunoglobulin module in subsets of non-cirrhotic-CC with A viral variant in pos2570 (CC-A) (left plot, FDR<<0.01) and non-cirrhotic-non-CC with same viral variant (non-CC-A, FDR<<0.01) (right plot).
Although the interferon module was found significantly enriched in females from non-CC cohort, an equivalent analysis (preranked t-test from regressive model) performed only in less severe samples (non-cirrhotic) assessing enrichments of hallmark gene sets28 showed a conserved enrichment for interferon alpha response in CC females (online supplemental figure S4a). In less severe non-CC samples, a strong enrichment in females of interferon alpha/gamma (not WGCNA derived) genes indicates further that IFNL4 genotype could exaggerate constitutional sex differences in ISG (online supplemental figure S4b).
Supplemental material
The regressive analysis would also suggest that immunoglobulin and T-cell modules are generally upregulated in CC over non-CC cohorts, regardless of sex (figure 6C). Higher T-cell activity in CC patients is confirmed by a significant enrichment of a MAIT cell signature (CD161+ upregulated genes from our published study29 (figure 6C).
Effect of viral variants on host transcriptomics assessed by data stratification
We previously30 demonstrated that variants in viral sequences are associated with IL28B/IFNL4 host genotype. In particular, a variant at position 2570 in viral genome, causing an amino acid substitution in protein NS5B, is the most significantly associated. Viral variants in this region are reported to give resistance to treatments.31 Therefore, we next assessed the impact of this site on transcription, taking into account host IL28B/IFNL4 genotype and cirrhosis status. In the subcohort of non-cirrhotic patients with favourable genotype CC, the presence/absence of a Valine in viral site 2570 was used as discriminant to group patients and assess DE levels associated with the substitution. GSEA performed between samples from patients infected with a virus carrying a Valine (V) and not carrying a Valine (non-V) at this site showed significant enrichments of diverse and biologically interpretable GO categories. Genes in immune relevant pathways were found enriched in V patients when compared with non-V (online supplemental figure S5a). Furthermore, the V variant was found associated with WGCNA modules; interferon, T- and B-cell-related modules were significantly enriched in non-CC patients carrying the V viral variant (online supplemental figure S5b,c).
Supplemental material
Interaction of sex and viral variants in immune-relevant modules
Taking together all these results that indicate that interferon type I signalling is exacerbated in non-CC subjects and also boosted by the presence of a single amino acid residue change in NS5B, we were able to test further a sex effect in ISG expression, using data stratification to control for covariates. A standard GSEA was performed between sexes in the subcohort of non-cirrhotic, non-CC, and with the valine at viral site 2570 (non-CC-V) samples, revealing a significant upregulation of the interferon module genes in females (online supplemental figure S6a, left plot). However, in the presence of an alanine (V to A) in the same site (again in the non-cirrhotic/non-CC subgroup: non-CC-A), the interferon module is no longer biased toward women (online supplemental figure S6a, central plot). This suggests that sex effects on ISG are impacted also by viral variants.
Supplemental material
This impact is not restricted to interferon. An equivalent analysis using the same subset of patients to test coexpression of the two other immune-associated modules (m4, m7) shows an enrichment of immunoglobulin response genes in non-CC-V female subjects (non-CC-V) (online supplemental figure S6b, left). In non-CC-A patients, we observed a strong enrichment in males for both m4 and m7 modules (online supplemental figure S6b, figure 6D right plot). Conversely, in CC-A patients we observed significant enrichment for both T cells and immunoglobulin genes in females (online supplemental figure S6b, figure 6D left plot). These results suggest that not only innate but also sex-related adaptive immune responses can be influenced by a combinatorial effect of host and viral genotypes.
We validated results of enrichment of immunoglobulin genes in non-CC-V females assessing CibertsortX32 imputed signatures of 22 immune cell types (LM2233) in stratified data (non-cirrhotic, non-CC-V, n=39, females=16, males=23) and using a preranked GSEA (online supplemental figure S6c). We found consistent results, that is, B-cell/plasma cell signature enrichment in non-CC-V females.
Expression quantitative trait loci (eQTL) analysis by sex for the impact of sex-specific single nucleotide polymorphisms
Given that sexes have a different responsiveness to the infection, regardless of genotype, we next focused on exclusively upregulated genes in females or males, for instance genes that are not concomitantly upregulated in non-CC versus CC (at lfc >1, padj <0.05). As shown schematically in online supplemental figure S7a,b, we selected the genes in the left set of the Venn diagram, the upregulated genes in females (in pink), not intersecting with the set of upregulated genes between non-CC and CC.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
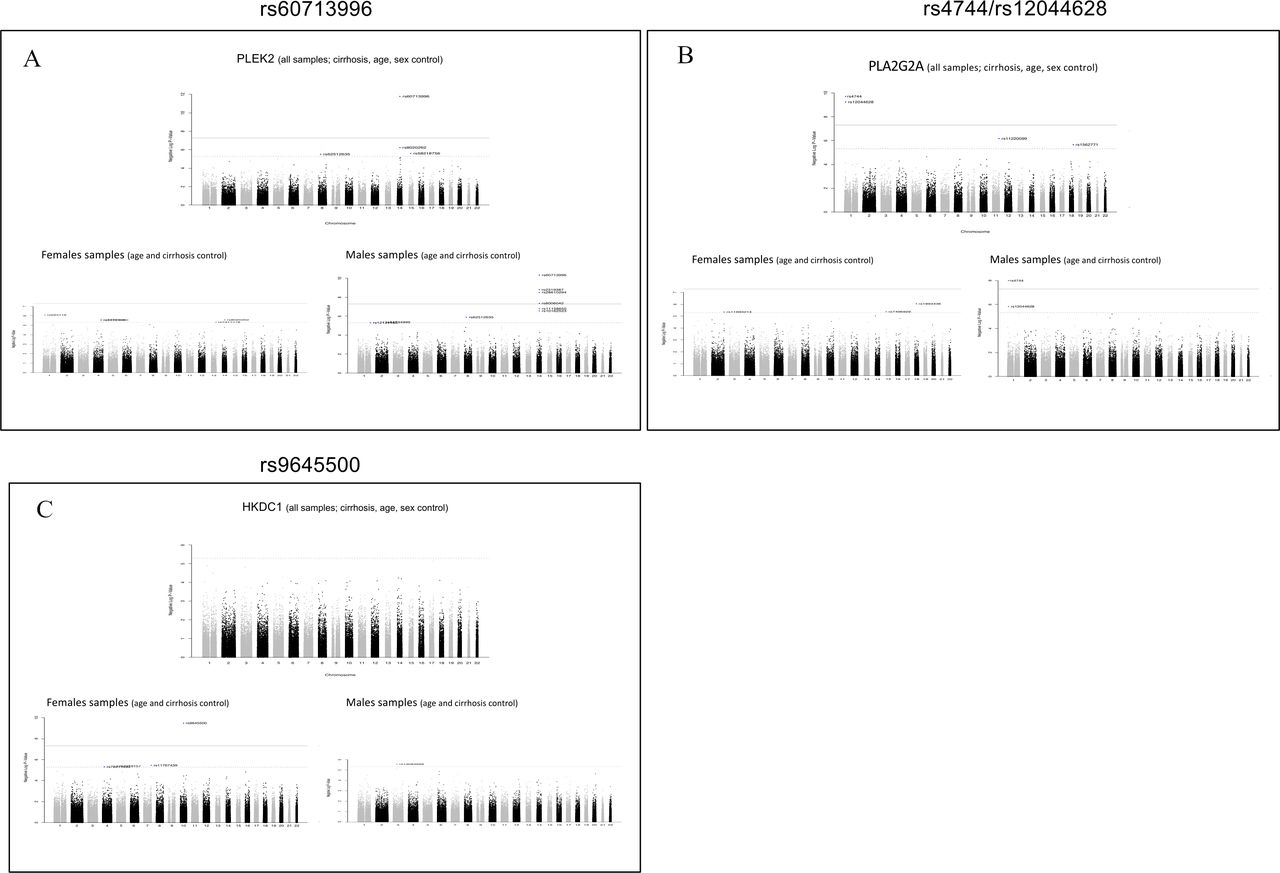
Manhattan plots for expression quantitative trait loci (eQTL) analysis results of significant loci in all samples and disaggregated by sex. Visualisation of eQTL analysis results using all samples (upper Manhattan plots) compared with results obtained separately in male and female subsets (lower Manhattan plots). PLEK2 (A) and PLA2G2A (b) are exclusively upregulated in females (and not in non-CC) and significant single nucleotide polymorphisms (SNPs) are detected only in male samples analysis. (C) HKDC1 is a gene upregulated only in cirrhotic males and a significant SNP is detected only in female sample eQTLs.
The uniquely and most significantly upregulated genes in females are GYG2, SLC23A3, PZP, PCP4L1, PLEK2, PFN2, SLC3A1, TMEM154, VIL1, H19, PLA2G2A, MROH2A, APOA4, GPC3 and XPNPEP2. Of note, PZP is reported to be an immune modulator and was previously detected by variance partition analysis (figure 1B).
eQTL analysis was executed for these sex-specific gene expressions, separately for each sex group and in combined set (adjusting for main covariates); Manhattan plots were generated and results from overall and separate sexes analysis were compared. We did not find sites associated with PZP expression, supporting that this immune modulator is driven by sex and not by a frequent mutation or polymorphism in this cohort. In contrast, PLEK2 and PLA2G2A have sites (rs60713996 and rs4744/rs12044628, respectively) detectable in males (and in combined samples, which could be expected since male patients are dominant in this cohort study) but not in females. The SNP map in the same chromosome of the genes providing further support of significant effect or association to the expression level of the features (figure 7A,B).
Another important example is the eQTL analysis detecting a site associated with HKDC1 expression (figure 7c) in female samples only. HKDC1 is a gene overall more expressed in women (adjusting for all covariates, workflow in online supplemental figure S7a) with an adjusted p-value=0.19. In cirrhosis, the gene is significantly upregulated only in males (p<<0.01, workflow in online supplemental figure S7b), hence the SNP genotype could impact responsiveness of HKDC1 expression in cirrhotic females. The female-specific site (rs9645500) associated with HKDC1 expression also maps in the same chromosome of the gene locus. These data provide evidence that it is possible to discover significant sites exclusively in one sex, otherwise undetectable when combined sex group analysis is performed (figure 7C).
Age and ISG expression
As observed during the COVID-19 pandemic, sex and age are crucial factors to consider in treating critically ill patients; therefore, we investigated age-dependent change in expressions of ISG genes, between sexes and in different contexts of IFNL4 genotypes. The mean expression of interferon module genes (m6) upregulated between non-CC versus CC (lfc >1, n=14) was used as metagene to assess correlation with age. A bivariate correlation analysis shows, particularly in females, a significant positive correlation of ISG expression and age in non-CC patients (online supplemental figure S8a). In CC patients overall we did not observe a strong correlation of the same ISGs with ageing; however, higher expression and correlation could still be noticed in CC females. The effect size analysis (including in analysis of variance model age, sex, cirrhosis status and interaction term for age and sex) confirmed for non-CC patients a significant contribution of age and sex to the expression of interferon module metagene (online supplemental figure S8b IFNL4, right plot).
Supplemental material
To investigate further postmenopausal effects, standard GSEA was performed in older patients (>55 years old) to test sex enrichments in CC and non-CC groups; the interferon module was found significantly enriched only in non-CC-females (FDR=0.23, phenotype permutation; FDR<<0.01, gene set permutation), while immunoglobulin and T-cell modules were found highly enriched in CC males (FDR<<0.01) (online supplemental figure S8c). Non-CC men still show an enrichment of T-cell module (FDR=0.08) confirming the tendency in males to an increased cellular response, even in older age. Postmenopausal CC women might not maintain a higher expression of immunoglobulin genes as observed in preranked GSEA analysis (adjusting for age; figure 6).
Impact of host gene networks in response to therapy in the BOSON study
In the study of the impact of antiviral therapies in the BOSON trial (sofosbuvir+ribavirin for 16 or 24 weeks or in combination with pegylated interferon alpha), a clear impact of both IL28B/IFNL4 status and sex was seen, with CC patients and females at least at risk of relapse with the partially effective directly acting antiviral therapy.12 The data in this study reveal the marked underpinning transcriptional differences which result from both genotype and sex prior to therapy. We have previously shown a recovery of interferon signalling in CC patients on antiviral therapy34: in that study, at the end of therapy (after 16 weeks) a recovery of interferon response was measured using the average of expression of a selection of ISGs (n=27) and it was observed exclusively in patients with CC. In this cohort, the same subset of ISGs appears to be upregulated in females (adjusting for all covariates). This suggests that a specific subset of IFN genes represent the first ISGs to be promptly activated in contexts of more functional immune responses (CC/females) and provides an explanation for the impact of sex on the treatment response seen (online supplemental figure S8d).
Comparability of interferon module in other conditions
To address the generalisability of these findings outside HCV and the liver, we analysed enrichment of network modules detected in our dataset in a range of publically available gene expression datasets; these were from a SARS murine model35 (GSE59185), human lung tissue from postmortem patients with 36 (GSE171524) and sorted CD4 cells from HIV post-treatment patients.37 The m6 module was found highly enriched (FDR<<0.01) in mice infected with the wild-type strain of SARS-CoV-1 when compared with a mutated/attenuated one, associated with milder disease (online supplemental figure S9a). This first result provides evidence of a cross-tissue and cross-species conservation of this HCV-derived module, and also the impact of viral variants. To address the influence of sex on this module, we assessed a study of SARS-CoV-2 postmortem lung. Here an enrichment of the m6 module in females (FDR=0.003) was still observed analysing lung pseudo-bulk transcriptomics38 (online supplemental figure S9b, preranked GSEA controlling for age and postmortem interval). Finally to further assess the relevance of m6 to therapy, we analysed an HIV cohort undergoing treatment withdrawal. A highly significant enrichment (FDR<<0.01) of the interferon module was also observed in CD4 transcriptomics of ‘post-treatment controllers’37: patients with HIV with a late viral load rebound after treatment interruption (online supplemental figure S9c). This result supports the involvement of m6 gene modules in other conditions, an interaction with sex, and a possible beneficial role in therapy as observed in BOSON.
Supplemental material
Discussion
The rationale for this study was to identify the key intrahepatic host gene networks responding to HCV infection, addressing in particular the influence of sex and dissecting the impact of host IL28B/IFNL4 genotype. We identified a number of immunologically meaningful modules where the effect of these factors can be identified. It is evident that HCV-liver transcriptomics show clear sex divergence in expression of genes involved in immune responses. Overall, we observed in females an enhanced expression of interferon signalling genes while in male patients a higher expression of T-cell-associated genes is visible (figures 5 and 6). These distinct responses to the same infection are likely linked to the divergent clinical outcomes in this disease.
The gene expression analysis between sexes in separate IFNL4 genotypes shows a clear overexpression of interferon genes in non-CC females—revealing an important interaction between host genotype and host sex in an antiviral response. Other interactions were seen which were more complex. For example, looking at the B-cell module, in the CC subset we noted a significant upregulation of relevant immunoglobulin genes in females, but interestingly in non-CC samples this gene set was upregulated in males. We also observed an impact of viral variants on immune relevant genes able to perturb the direction of the sex response. In particular, the immunoglobulin response seems to be very sensitive to a combinatorial effect of host genotype, virus mutations and sex. The impact of sex on the differential gene expression driven by host genetic variation was confirmed by eQTL analyses. Assuming distinct sex responses in an infectious disease is quite a general effect, sex-specific eQTL analysis could be an important approach elsewhere. As yet undiscovered genotypes not visible in studies of mixed populations but visible in studies of separated sexes could help explain the pathogenesis of sex-biased pathologies.
Broadly each module could be considered to have a specific clinical/pathological significance. The clearest of these are m6, as discussed, associated with interferon responses and inversely with viral load. Module m4 was associated with T-cell responses—this was correlated to ALT, indicating a pathological role for cellular responses (interestingly this was positively correlated with viral load). The content of m7 was more closely linked to B-cell responses and again associated with immune pathology. The other modules, m1 (signalling), m2 (matrix organisation), m3 (metabolic and synthetic pathways) and m5 (transforming growth factor-β signalling/catabolism) were all most closely associated with cirrhosis—with a strong positive association for m2 and a negative association for m3. IFNL4 genotype and sex had wide impacts on the modules, but were strongly linked to the innate response (m6) and negatively to adaptive responses (m4 and m7). Although we did not extensively validate these modules in other clinical settings, we believe they could have broad value. In particular, m6 was found across species and organs, and a sex-associated impact was found in COVID-19.
We also examined the recovery of interferon responsiveness in therapy. Importantly, this analysis could be performed in blood as we previously observed in sequential blood sampling a clear ‘bounce’ in gene expression seen over the course of the BOSON trial12 in those undergoing successful cure. This is relevant as it indicates that some of the key liver modules can also be observed in blood, broadening the application. The overlap between genes upregulated in blood and those enriched in women in this study may help explain the clear impact of sex seen in the BOSON study. Women appear to efficiently induce the expression of a coordinated set of ISGs providing an effective antiviral response. Thus, potentially the quality or dynamics rather than just the amplitude of antiviral gene expression could be critical. While therapies for HCV are now highly effective in all groups, in other clinical settings the role of sex-driven transcriptional and immune networks needs further exploration as it could impact on patient stratification or guide the use of specific interventions.
Although the study was comparatively large, the use of bulk RNA-Seq does limit some of the interpretations, which could be further explored in future studies using single-cell RNA-Seq and spatial transcriptomic approaches. Since RNA-Seq is a bulk approach, the source of the interferon-driven networks in m6 could be further explored to identify the (sex-dependent) role of parenchymal and immune cell subsets. The status and localisation of T cells in the liver—including conventional and unconventional subsets, and their function and regulation require further analysis. The ability of such newer methods to explore metabolic pathways in individual cell types would also be of relevance to the strong (and opposing) impacts seen in modules m2 and m3. We would suggest on the basis of our data here that such future projects should all be designed to take into account the impact of sex as well as host genetics on such pathways.
Materials and methods
RNA extraction, library prep, sequencing and mapping for the BOSON cohort
The protocol followed as previously described by Ansari and coworkers.39 Liver biopsy samples were available for 198 patients. Liver biopsy samples were mechanically disrupted in the presence of lysis buffer and homogenised using a QIAshredder (Qiagen, 79654). Total RNA was extracted from patient liver biopsies at baseline (pretreatment) using RNeasy mini kits (Qiagen, 74104) following manufacturers’ instructions.
RNA yield was quantified using a NanoDrop spectrophotometer. Selected samples were also run on an Agilent TapeStation system to assess RNA quality and purity. Library preparation from purified RNA samples was performed using the Smart-Seq2 protocol,40 used along with previously described indexing primers during amplification.41
High- throughput RNA sequencing of prepared libraries was performed on the Illumina HiSeq 4000 platform to 75 bp PE at the Wellcome Center for Human Genetics (Oxford, UK). Reads were trimmed for Nextera, Smart-seq2 and Illumina adapter sequences using skewer-v0.1.125.42
RNA-Seq reads were aligned to GRCh38 human reference genome using HISAT43 sequence aligners. Rsubread44 R package was used for assignments and quantification of mapped reads to genomic features. Clustering analysis was performed using R packages,45 46 and assessment of variability drivers was executed with variancePartition13 method. Our own R code and limma47 package were used for preprocessing, normalisation and differential gene expression analysis. Standard GSEA and preranked GSEA were performed using software tool from Broad Institute website48 and fgsea49 R package. WGCNA23 was employed to infer genes in regulatory networks, and clusterProfiler,50 ReactomePA25 and fgsea49 were used to assess enrichment of Reactome pathways, GO categories and Hallmarks28 gene sets. CibertsortX32 algorithm was used to impute expressed genes in inferred immune cell population proportions. Raw GWAS data were generated and processed with PLINK,51 preprocessing and statistical analysis for eQTL on gene expression selections was performed using classes and methods in snpStats52 53 package.
Host genotyping
Host genome-wide genotyping was performed on 195 patients from the BOSON cohort as described previously.30
Expression data accession numbers
Preprocessed expression data of the BOSON cohort study have been deposited in the Gene Expression Omnibus Database (GEO, GSE149601).
Data availability statement
Data are available in a public, open access repository. Data are publicly available and deposited on online repository Gene Expression Omnibus Database (GEO, GSE149601)
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors EM developed the idea, performed the bioinformatics analysis and wrote the article. NR contributed to the interpretation and design of the bioinformatics analysis and reviewed the article. MAA and EB reviewed the article and contributed to the design and generation of the original transcriptional datasets. CH contributed to data analysis and the writing of the article. STOP-HCV provided patient information for effective experimental design and data analysis. PK contributed to the model, reviewed and wrote the article and gave final approval. PK and EM are guarantors for the work and the conduct of the study.
Funding EM, NR, CH, PK are funded by a Wellcome Trust grant (WT109965MA) and the NIHR Biomedical Research Centre, Oxford. EB is funded by the Oxford NIHR Biomedical Research Centre and is an NIHR Senior Investigator. The views expressed in this article are those of the author and not necessarily those of the NHS, the NIHR or the Department of Health. MAA is supported by a Sir Henry Dale Fellowship jointly funded by the Royal Society and the Wellcome Trust (220171/Z/20/Z). This research was funded in whole, or in part, by the Wellcome Trust (grant no. 220171/Z/20/Z). For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.