Article Text

Abstract
Background and aims New options are needed for the management and prevention of colorectal cancer liver metastases. Interleukin 12 (IL-12) is an immunostimulatory cytokine with proven antitumour effect in animal models. Despite evidence indicating its biological effect in humans, neither the recombinant protein nor gene therapy vectors expressing IL-12 have shown a relevant benefit in patients with cancer.
Objective To develop a new approach to overcome the difficulties in obtaining a suitable expression pattern and the immunosuppressive milieu in the tumours which contribute to this poor performance.
Methods A high-capacity (‘gutless’) adenoviral vector carrying a liver-specific, mifepristone (Mif)-inducible system for the expression of IL-12 (HC-Ad/RUmIL-12) was used in combination with chemotherapy. Tumours were established in the liver of C57BL/6 mice by inoculation of MC38 colon cancer cells.
Results Intrahepatic injection of HC-Ad/RUmIL-12 and tailored induction regimens allowed the maintenance of safe and efficient levels of IL-12 in vivo. An individualised, stepwise increase in the dose of Mif (125–4000 μg/kg) was needed to compensate for the progressive but transient downregulation of the inducible system. Repeated cycles of Mif induction (every 24 h for 10 days) were needed for optimal tumour eradication. However, complete protection against tumour rechallenge was seen in <25% of the animals. The administration of oxaliplatin (5 mg/kg intraperitoneally) 3 days before starting the induction regimen achieved efficient elimination of liver metastases with a single cycle of IL-12 induction, and improved protection against tumour rechallenge. This was associated with a shift in the tumour microenvironment towards a more pro-immunogenic phenotype, with an increase in the CD8+/T regulatory cell ratio and a reduction in myeloid-derived suppressor cells. These effects were not seen with 5-fluorouracil, irinotecan or gemcitabine.
Conclusions Long-term controlled expression of IL-12 using an HC-Ad vector in combination with oxaliplatin is effective and clinically applicable against hepatic colon cancer metastases.
- Interleukin 12
- oxaliplatin
- colorectal cancer
- liver
- adenovirus
- adenoviral vectors
- chemotherapy
- colorectal metastases
- gene therapy
- immunotherapy
Statistics from Altmetric.com
- Interleukin 12
- oxaliplatin
- colorectal cancer
- liver
- adenovirus
- adenoviral vectors
- chemotherapy
- colorectal metastases
- gene therapy
- immunotherapy
Significance of this study
What is already know about this subject?
IL-12 is an immunostimulatory cytokine with antitumour effects in animal models of colorectal cancer.
Methods to control the location, intensity and duration of IL-12 production are needed to increase the safety and efficacy of genetic immunotherapy.
Tissue-specific, drug-inducible expression systems can be used to modulate the expression of transgenes in specific organs, but the biological effects of IL-12 can interfere with their functions.
High-capacity (“gutless”) adenoviral vectors are efficient in long-term gene transfer of the liver.
What are the new findings?
Using the HC-Ad/RUmIL-12 vector, individualised stepwise increases in the dose of inducer allowed the maintenance of therapeutic levels of IL-12 for >10 days, in cycles that can be repeated over a period of several months in mice.
Oxaliplatin cooperates with IL-12 in the stimulation of an efficient immune response against cancer cells, with complete eradication of pre-established colorectal cancer liver metastases and prevention of experimental relapses in a syngeneic model.
The mechanism of cooperation involves a reduction in the immunosuppressive microenvironment of tumours, with an increase in the ratio of CD8+/T regulatory cells and CD8+/myeloid-derived suppressor cells.
Cooperation with IL-12 is not seen with other chemotherapeutic drugs such as 5-flurouracil, irinotecan and gemcitabine.
How might they impact on clinical practice in the foreseeable future?
The protocol described here is compatible with conventional oxaliplatin-based chemotherapeutic regimens, and it can be applied in conjunction with surgical or ablative treatments to avoid recurrence of colorectal cancer liver metastases.
Introduction
Metastatic liver cancer is a life-threatening condition frequently observed in patients with colorectal cancer. Hepatic lesions are found in 10–25% of cases at the time of diagnosis, and 30% of them have no evidence of dissemination in any other organ. In addition, recurrence after surgical removal of the colorectal tumour occurs mainly in the liver, with a 20–25% rate of metachronous liver metastases.1 Overall, more than 50% of patients with colorectal cancer will have liver involvement during the course of their disease.2 Surgical resection is potentially curative only in the most favourable cases, with a general recurrence rate of 60–70%. Regional treatments achieve local control, but a significant increase in long-term survival is not guaranteed. Standard chemotherapy combinations consisting of 5-fluorouracil and leucovorin plus oxaliplatin or irinotecan (FOLFOX and FOLFIRI) achieve survival rates close to 10% in advanced colorectal cancer.3 The addition of biological agents such as cetuximab can increase the response rate4 and allows resection of liver metastases that were not candidates for surgery before treatment.5
However, recurrence rate exceeds 70% and it is clear that new therapeutic options are needed to improve the clinical management of hepatic metastases from colon cancer. Diverse strategies aimed to stimulate the immune responses against cancer cells offer the opportunity to block progression of disseminated tumour deposits and prevent relapses.6 However, unravelling this potential is still a challenge in the clinical setting. Tumours avoid the attack of the immune system by promoting the development and accumulation of leucocytes with an immunosuppressive phenotype. The importance of regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSC) in this process has been recently demonstrated (reviewed by Gabrilovich and Nagaraj; Nizar et al).7 8 The effect of cancer treatments on these cell populations is still poorly understood, but it can greatly influence their efficacy. Exploring the opportunities of cooperation with standard treatments is especially important, in light of the recent realisation that certain cytotoxic drugs can favour the immune response against tumours.9 These include some of the most active agents against colorectal cancer, such as oxaliplatin (OXP), 5-fluorouracil (5-FU) and gemcitabine.10–14
The cytokine interleukin 12 (IL-12) can serve as a link between the innate and adaptive immune responses because it can activate the proliferation of T lymphocytes and natural killer (NK) cells, the secretion of other mediators such as interferon γ (IFNγ) and the cytotoxic activity of these effector cells.15 The ability of IL-12 to increase the number and activity of tumour-specific lymphocytes has been verified in humans,16 and its antitumour effect has been observed.17 However, clinical responses tend to be poor, and intensification of the treatment is difficult owing to the toxicity associated with systemic exposure to IL-12. The use of gene therapy vectors encoding the p35 and p40 IL-12 subunits to obtain expression of the cytokine in specific locations is an attractive alternative.18–20 However, clinical experience indicates that refinement of these vectors is needed. Intratumoral administration of a replication-deficient adenovirus carrying the IL-12 gene was well tolerated in patients with hepatic metastases.21 Biological effect was demonstrated, but antitumour responses were very modest. The poor performance of the vector as indicated by intensity and duration of expression was recognised as a key limitation in this approach. Additional obstacles are the high heterogeneity in the infectivity of tumour nodules among different patients, and the rapid appearance of neutralising antibodies against adenovirus, which makes subsequent administrations ineffective.22
To circumvent these problems, high-capacity adenoviral vectors (HC-Ad) have been designed. The lack of viral genes reduces their toxicity and avoids the elimination of transduced hepatocytes by the immune system.23–26 The incorporation of a liver-specific, drug-inducible expression system in the GL-Ad/RUmIL-12 vector (called here HC-Ad/RUmIL-12) allowed intense, controlled and long-term expression of the transgene upon intravenous administration of the vector.27 The possibility of controlling the intensity of IL-12 expression by adjusting the dose of inducer makes this approach safer and more clinically applicable. However, the function of drug-inducible expression systems is influenced by the immunostimulatory properties of IL-12 acting at the transcriptional level.28 This causes a progressive but reversible inhibition of transgene expression that might limit the therapeutic effect of this approach, especially in the case of pre-established liver metastases.29 30 We hypothesised that optimisation of the induction regimen and combination with chemotherapy would produce a more efficient and clinically relevant protocol. We describe here an individualised induction scheme that prevents toxicity, compensates the downregulation of the system and allows several rounds of sustained IL-12 expression. More importantly, we found that the combination with systemic OXP favoured the immune response against tumours, alleviated the need for long-term IL-12 expression and increased protection from an experimental tumour rechallenge. This treatment achieved complete eradication of hepatic lesions and long-term, disease-free survival of animals in a syngeneic model.29
Materials and methods
Cell lines
The MC38Luc1 cell line has been previously described.29 31 Cell lines were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/ml streptomycin and 100 μg/ml penicillin (all from Gibco, Invitrogen, Paisley, UK), 2 mM glutamine (Cambrex, Wiesbaden, Germany) and 400 μg/ml G418 (Geneticin, Gibco, Invitrogen).
Mice and tumour models
C57BL/6 and athymic (nu/nu) female mice (5–8 weeks old) were purchased from Harlan and Charles River, respectively (Barcelona, Spain). Hepatic tumours were established by direct implantation of 5×105 MC38Luc1 cells, as previously described.29 For subcutaneous tumour formation (rechallenge experiments), a total of 106 cells were injected in the right hind flank. Tumour size was monitored at indicated time points by measuring two perpendicular tumour diameters using a precision calliper. Tumour volume was calculated using the following formula: V= length×width2×0.5. Survival was checked daily and mice were euthanised if their general status deteriorated or subcutaneous tumours exceeded 20 mm in diameter. All in vivo experiments were performed in accordance with the local animal commission.
Bioluminescence imaging
Anaesthetised animals received the substrate D-luciferin (Promega, Madison, WI, USA) by intraperitoneal injection at 150 mg/kg. Ten minutes later, light acquisition was performed in an IVIS CCD camera system (Xenogen, Alameda, CA, USA) and analysed with the Living Image 2.20 software package (Xenogen). Typically, a circular region of interest measuring 3 cm in diameter was defined in the abdomen of mice, and quantification of light emission was performed in photons/second.
Vector and treatment procedures
The high-capacity (gutless) adenoviral vector HC-Ad/RUmIL-12 (also named GL-Ad/RUmIL-12) has been previously described.27 In this vector, all viral genes have been deleted and substituted by human non-coding DNA (“stuffer”), plus a transgene region containing a mifepristone (Mif)-inducible system for the expression of p35 and p40 subunits of murine IL-12. The transactivator of this system is expressed under the control of a liver-specific promoter (transthyretin). Both IL-12 subunits are co-expressed owing to an internal ribosomal entry site. Amplification of the vector was carried out as previously described.32 The virus was administered by direct intrahepatic injection surrounding the tumours after laparotomy.30 Typically, 2.5×108 infectious units (IU) of virus were mixed in a total volume of 50 μl saline solution before injection. Activation of the inducible system was carried out by intraperitoneal injection of Mif (RU486, Sigma, St Louis, MO, USA) dissolved in sesame oil (Sigma). Inductions started 2 weeks after implantation of cells. Administration of chemotherapy drugs was carried out by intraperitoneal injection. OXP (Eloxatin) was from Sanofi-Aventis (Essex, UK); 5-FU was from Ferrer Farma (Barcelona, Spain); irinotecan was from Hospira (Warwickshire, UK); gemcitabine (Gemzar) was from Lilly (Madrid, Spain). Animals treated with 5-FU received 50 mg/kg folinic acid (from Genericos Espanoles Laboratorio (GES, Madrid, Spain)) 1 h before 5-FU, and another dose together with 5-FU.
Determination of IL-12, IFNγ and HMGB1
Serum concentration of murine IL-12 and IFNγ were determined by OptE1A mouse IL-12 (p70) and mouse IFNγ ELISA kits (BD Biosciences PharMingen, San Diego, California, USA). High mobility group box 1 protein (HMGB1) in serum or supernatant of cells was quantified by ELISA kit from IBL International GmbH (Hamburg, Germany).
Flow cytometry
Spleens, healthy livers and livers with tumours were harvested and treated with 400 U/ml collagenase D and 50 μg/ml DNase I (Roche Diagnostics, Indianapolis, IN, USA). After mechanical tissue dissociation, cells were passed through a 70 μm nylon mesh filter (BD Falcon, BD Bioscience, San Jose, California, USA) and washed. To enrich liver cell suspension in leucocytes, hepatocytes were removed with Percoll gradients. The single-cell suspension obtained was pretreated with anti-CD16/32 (clone 2.4G2; BD Biosciences Pharmingen) to reduce non-specific binding to Fc receptors. After this, cells were stained with the following fluorochrome-conjugated antibodies: CD8 (53-6.7), CD4 (clone RM4-5), CD25 (PC61), CD11b (M1/70), Ly6C (AL-21), F4/80 (BM8) and Ly6G (1A8) from BD Pharmingen. Intracellular staining for FoxP3 was performed using mouse regulatory T cell staining kit (BD Biosciences), according to the manufacturer's instructions. Cells were acquired on either a FACSCalibur or a FACSCanto flow cytometer (BD Biosciences). Flow cytometry data were analysed using FlowJo software (Tree Star, Ashland, OR, USA). CD8/Treg or CD8/MDSC ratios—that is, CD8 T-cell count divided by either Treg cell or MDSC count were calculated.
Statistical analysis
A two-tailed unpaired t-test was used to compare two groups of values when n>10. For smaller groups, the Mann–Whitney non-parametric test was used. Comparisons of more than two groups were performed using analysis of variance with Bonferroni correction. Survival curves were compared by log rank test (GraphPad Prism software).
Results
Individualised and progressive adjustment of Mif compensates transduction variability and downregulation of the inducible system
Therapeutic use of IL-12 requires efficient methods to control the plasmatic levels of this potent immunostimulatory cytokine in order to avoid toxicity. We had previously determined in our MC38 syngeneic tumour model that seric IL-12 <20 ng/ml has no antitumour effect, and levels >700 ng/ml are associated with toxicity.27 The two main obstacles to keeping to this therapeutic range are the variability in transduction among individuals and the progressive silencing of the drug-inducible system mediated by the immune system.28 We have focused on the favourable dose–response effect of the Mif-inducible system (figure 1A) to design a new induction protocol. Once the liver has been transduced with the vector (typically 2.5×108 IU), a suboptimal amount of Mif (125 μg/kg) is administered for the first 2 days in order to prevent toxicity. The concentration of IL-12 is measured in serum 10 h after the first induction, and based on this information, a stepwise increase in Mif is scheduled according to table 1. Figure 1B shows the kinetics of IL-12 production obtained with the adjusted protocol. In contrast with a fixed amount of inducer (constant protocol), doubling the dose of Mif every 3 days contributes to the stabilisation of IL-12 levels for at least 10 days. Since downregulation of the system is due to a reversible inhibition of the transthyretin promoter,28 the induction protocols can be repeated after a resting period of at least 2 weeks in order to obtain several cycles of IL-12 expression (figure 2B). We have verified that the system is functional for more than 5 months, with a slow decrease in the intensity of expression in each cycle owing to the non-integrative nature of adenoviral vectors.
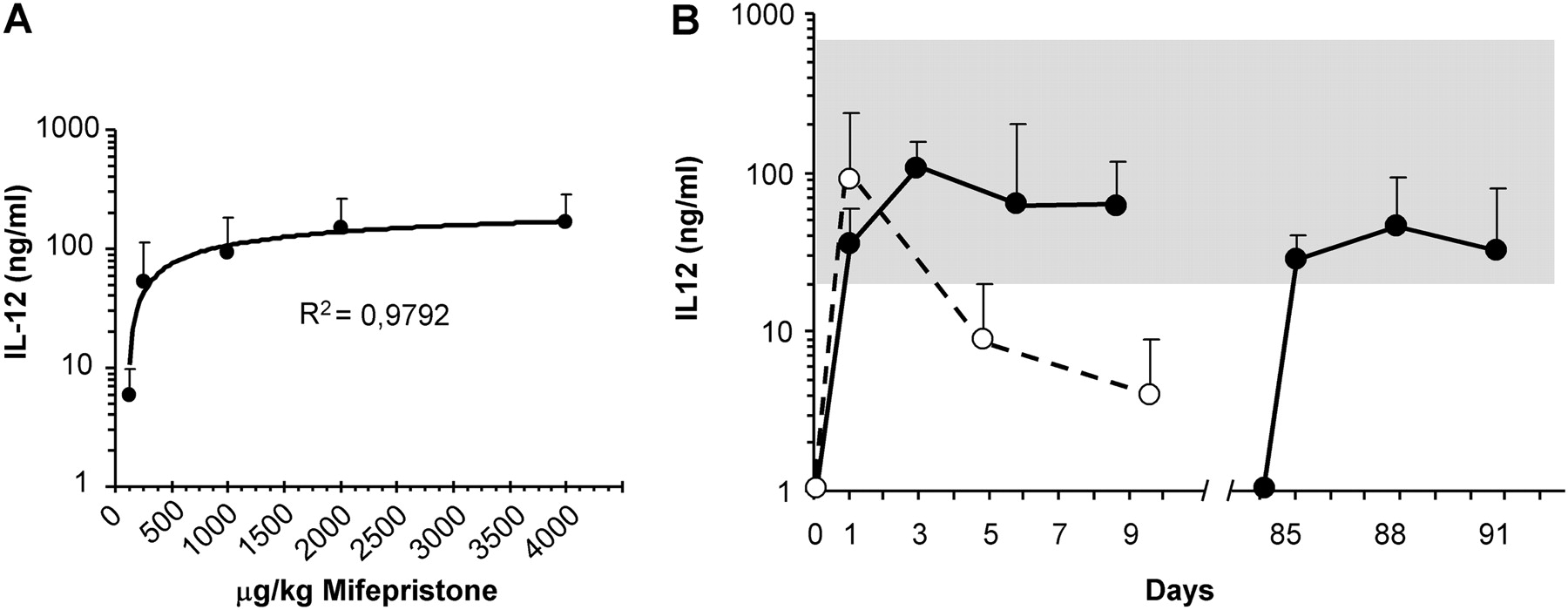
Progressive increase in the dose of mifepristone (Mif) allows several cycles of sustained interleukin 12 (IL-12) expression in mice treated with the HC-Ad/RUmIL-12 vector. The vector was administered at 2.5×108 IU/mouse in C57BL/6 mice by intrahepatic injection. (A) Two weeks later, a single dose of Mif (125; 250; 1.000; 2.000 or 4.000 μg/kg) was administered intraperitoneally to different groups of animals (n=5). The concentration of IL-12 was measured in serum 10 h later. (B) Mice received daily injections of 250 μg/kg Mif for 10 consecutive days (constant protocol, white circles, n=10). A different set of animals received an adjusted protocol (black circles, n=8) consisting of 125 μg/kg Mif days 1–2; 250 μg/kg days 3–5; 500 μg/kg days 5–7 and 1000 μg/kg days 9–11. The concentration of IL-12 in serum was determined 10 h after induction at the indicated days. More than 2 months after the last Mif administration, a second cycle of inductions was repeated in the same group of animals using an adjusted protocol that started at 250 μg/kg Mif. The grey area represents the range of IL-12 concentrations considered therapeutic in the MC38 tumour model. Error bars represent SD.
Adaptation of the induction regimen to the initial concentration of interleukin 12 (IL-12) in serum. Mifepristone (Mif) is administered daily by intraperitoneal injection. The first 2 days, a suboptimal dose of 125 μg/kg is given to all mice. The concentration of IL-12 is determined in the serum 10 h after the first Mif injection (first column on the left). Based on this information, the dose of Mif is established for days 3–5, and subsequently duplicated every 3 days

Oxaliplatin (OXP) increases the antitumour effect of interleukin 12 (IL-12) in a syngeneic model of colorectal cancer liver metastases. (A) Schematic representation of the experiment. MC38Luc1 cells (5×105 cells/mouse) were inoculated in the liver of C57BL/6 mice at day 0. Control animals (Cont) were left untreated (intrahepatic injection of saline solution), or treated with OXP alone (5 mg/kg intraperitoneal, group OXP). Other groups received the HC-Ad/RUmIL-12 vector by intrahepatic injection surrounding the tumours at day 5. Daily inductions with mifepristone (Mif) started at day 12 using an adjusted protocol (group IL-12). One subset of these animals received OXP (5 mg/kg intraperitoneal) 3 days before the first Mif administration (group IL-12+OXP). (B) Bioluminescence quantification performed at day 35. At this point, most animals in the control and OXP groups were dead. (C) Volume of hepatic tumours determined by direct measurement through laparotomy at day 40, or after necropsy at earlier time points in animals dead or killed for ethical reasons. (D) Survival of animals. Statistical significance: ***p<0.001; *p<0.05.
Combination with OXP increases the antitumour effect of IL-12 on hepatic colon cancer metastases
Once the induction protocol was established, we investigated whether conventional OXP-based chemotherapy might enhance the antitumour effect of sustained IL-12 expression. Tumours were obtained by direct inoculation of MC38Luc1 cells in the liver of syngeneic mice.29 Bioluminescence detection ensured engraftment of cancer cells and homogeneous tumour load in all experimental groups (not shown). The HC-Ad/RUmIL-12 vector was injected intrahepatically in the proximity of the tumour nodules, as described previously.30 One week later, daily inductions were started following the adjusted protocol described above. One subset of these animals received a single dose of OXP (5 mg/kg intraperitoneally) 3 days before the first Mif administration. Other mice were inoculated intrahepatically with saline solution instead of vector, with no additional treatment (control group), or received a single dose of OXP. A schematic representation of the experiments is given in figure 2A. Two weeks after completing one cycle of inductions (day 35 after cell implantation), most animals from the control or OXP-only groups were dead as a consequence of tumour progression (see figure 2D). At this point, bioluminescence quantification predicted the presence of hepatic tumours in approximately half of the animals treated with intrahepatic IL-12 alone, whereas the addition of OXP achieved a stronger and more homogeneous inhibition of the luciferase signal (figure 2B). To confirm this observation, all surviving animals underwent laparotomy 5 days later. Figure 2C shows the volume of tumours determined by direct measurement at this stage, or after necropsy in animals with shorter survival. The result clearly demonstrates that the combination of IL-12 and OXP achieves the best therapeutic effect, as measured by antitumour effect and long-term survival (figure 2D). Treatment with OXP alone caused a small, not statistically significant reduction in the final tumour volume, but no survival advantage was observed. The lack of efficacy of OXP in aggressive models based on MC38 cells has been described by other groups.33
OXP enhances the establishment of a protective immune response against a tumour rechallenge
Protection from tumour relapse or from the appearance of new liver metastases is crucial to improve the prognosis of patients with colorectal cancer. Therefore, we studied the immunological protection provided by our treatments against a tumour rechallenge. Animals that remained free from their hepatic tumours for more than 1 month after treatment with HC-Ad/RUmIL-12 (with or without OXP) received a new inoculation of MC38Luc1 cells in a distant location (subcutaneous). Tumour growth was monitored, showing that, on average, tumour progression was delayed in mice from both treatment groups in comparison with naïve animals (figure 3). Interestingly, the inhibition was more intense if the treatment regimen had included OXP, and the proportion of animals that was completely protected from the appearance of new tumours increased from 23% to 55%.

Immunological protection against cancer cells in animals treated with interleukin 12 (IL-12) plus oxaliplatin (OXP). MC38Luc1 cells (106 cells/mouse) were inoculated subcutaneously in C57BL/6 mice whose hepatic tumours were cured after being treated with the HC-Ad/RUmIL-12 vector in the presence (group IL-12+OXP) or absence (group IL-12) of OXP. The same amount of cells was inoculated in naïve mice as a control. The average tumour volume for each group is represented. The number of mice remaining tumour-free for the entire observation period is indicated in parenthesis. Day 0 indicates the moment of inoculation of subcutaneous cells, which corresponds to at least 1 month after completion of the previous treatments. *p<0.05.
OXP counteracts the immunosuppressive microenvironment of tumours
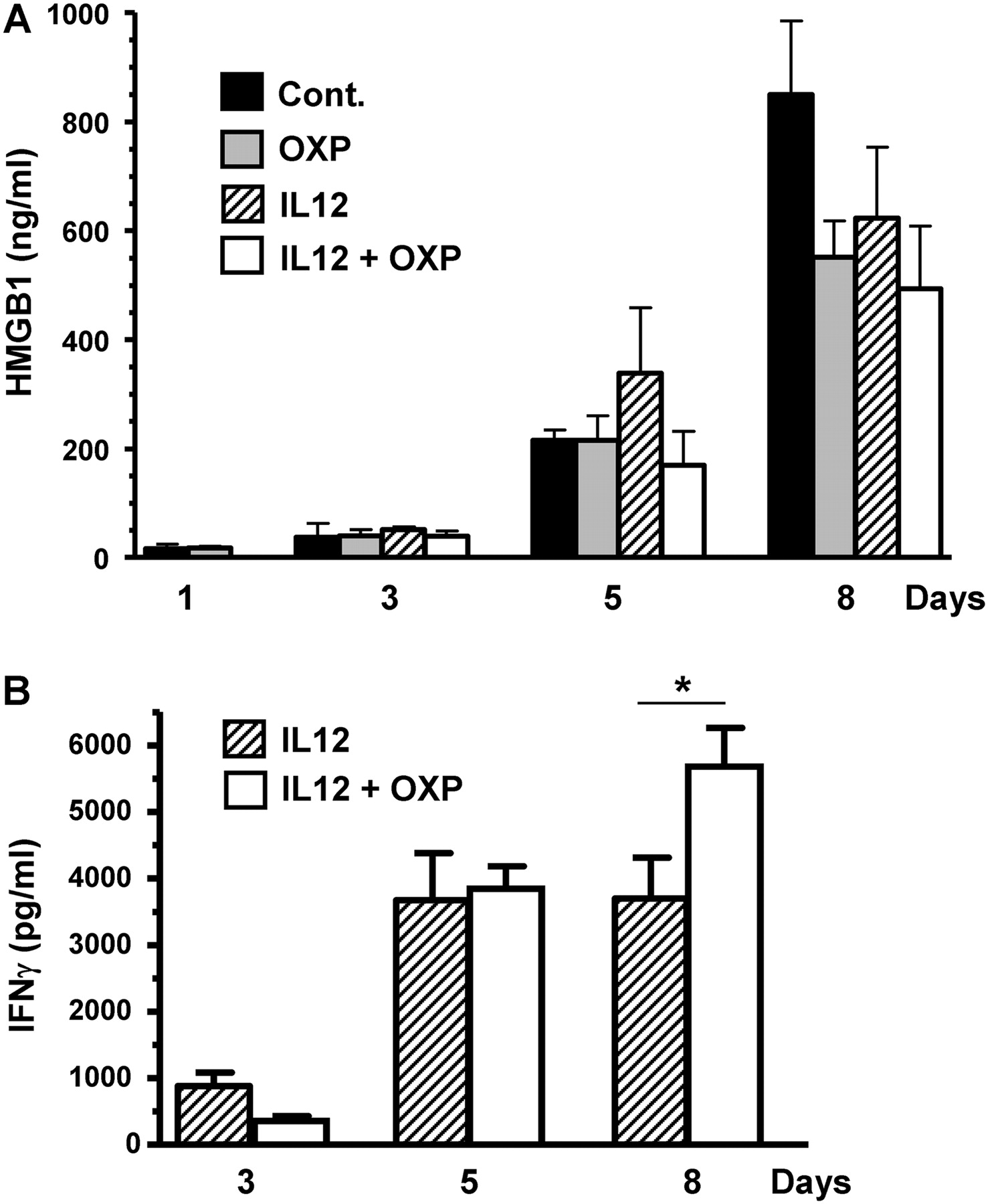
We investigated the mechanisms involved in the cooperation between IL-12 and OXP. First, we verified that IL-12 does not increase the cytotoxic activity of OXP in MC38Luc1 cells in vitro (online supplementary figure 1). Although the possible influence of direct cytotoxicity cannot be completely ruled out in vivo, experiments performed in athymic (nu-/nu-) mice showed a dramatic loss of therapeutic effect (supplementary figure 2), demonstrating a critical role for T-cell-mediated immune responses. Exposure of calreticulin (CRT) on the cell surface and release of HMGB1 have been proposed as key elements in the immunostimulatory properties of oxaliplatin.14 Although we did not detect a significant increase in the exposure of CRT upon treatment of MC38Luc1 cells with OXP (data not shown), the release of HMGB1 was evident (supplementary figure 1A). However, the biological significance of this finding in our tumour model is uncertain, because tumour-bearing animals treated with HC-Ad/RUmIL-12 plus OXP did not show an increase in the serum concentration of HMGB1 in comparison with untreated mice (figure 4A). This is probably due to the fact that MC38Luc1 tumours spontaneously release increasing amounts of HMBG1 as they progress in the liver of the animals, in correlation with the situation observed in patients with advanced colorectal cancer.34 This is in contrast with other tumour models such as glioblastoma multiforme in which HMGB1 levels are low in untreated animals, but show an acute increase in response to suicide gene therapy.35

Oxaliplatin (OXP) enhances the immunostimulatory action of interleukin 12 (IL-12). Mice bearing intrahepatic MC38 tumours were treated with the HC-Ad/RUmIL-12 vector, and IL-12 expression was induced with mifepristone (Mif) following an adjusted protocol (IL-12 group). A subset of animals received 5 mg/kg OXP intraperitoneally 3 days before the first induction (IL-12+OXP group). Other groups were left untreated (cont), or treated with OXP only. Blood was collected 24 h after the administration of OXP (considered as day 1), and 10 h after the first, third and sixth Mif inductions (days 3, 5 and 8, respectively). The concentration of high-mobility group box 1 protein (HMGB1) (A) and interferon γ (IFNγ) (B) was determined by ELISA (n=10). *p<0.05.
Next, we verified that OXP does not cause a significant increase in the production of IL-12 mediated by HC-Ad/RUmIL-12 (data not shown). However, the amount of IFNγ produced in response to IL-12 was higher in the group treated with OXP (figure 4B), suggesting a stronger activation of the immune system. This effect was seen 6 days after the initiation of IL-12 expression, and coincides with the peak of IFNγ stimulation. Using major histocompatibility complex tetramer staining specific for an MC38 epitope we detected a moderate increase in the frequency of CD8+ T lymphocytes in peripheral blood in response to IL-12, but the addition of OXP did not significantly increase this population (data not shown).
Therefore, we studied the tumour microenvironment to evaluate whether the combined treatment had an effect on the local immunosuppressive cell populations. A favourable balance between T-effector and T-suppressor cells has been associated with improved immune responses against tumours.36 37 Tumour, liver and spleen from control and treated animals were collected 3 days after the initiation of IL-12 expression, and leucocyte populations were analysed by flow cytometry. We observed no change in the NK cells in any of these tissues (data not shown). However, the ratio between CD8+ T lymphocytes and Tregs (CD25+FoxP3+ population) was significantly increased in the tumours of mice treated with IL-12 in combination with OXP, in comparison with IL-12 alone. Interestingly, this effect was not seen either in the surrounding liver, or in the spleen of these animals (figure 5A,B). Similar results were obtained in samples obtained on the sixth day of IL-12 induction (not shown). In addition, we found that the combined treatment caused a decrease in the monocytic myeloid-derived suppressor cells (MDSC), defined as the CD11b+Ly6C+Ly6G- population.38 The ratio CD8+/MDSC was slightly elevated in the tumours of mice treated with IL-12, but it was significantly enhanced if OXP was included in the therapeutic regimen (figure 5A,C). In this case, the ratio was also increased in the spleen, but not in the liver. Since monocytic MDSC have the potential to differentiate into tumour-associated macrophages,7 38 we determined the treatment-induced changes in the expression of the macrophage differentiation marker F4/80. We found that the vast majority of myeloid-derived cells in our tumour model remained in an undifferentiated state, and no changes were seen with any of the treatment conditions (supplementary figure 3).
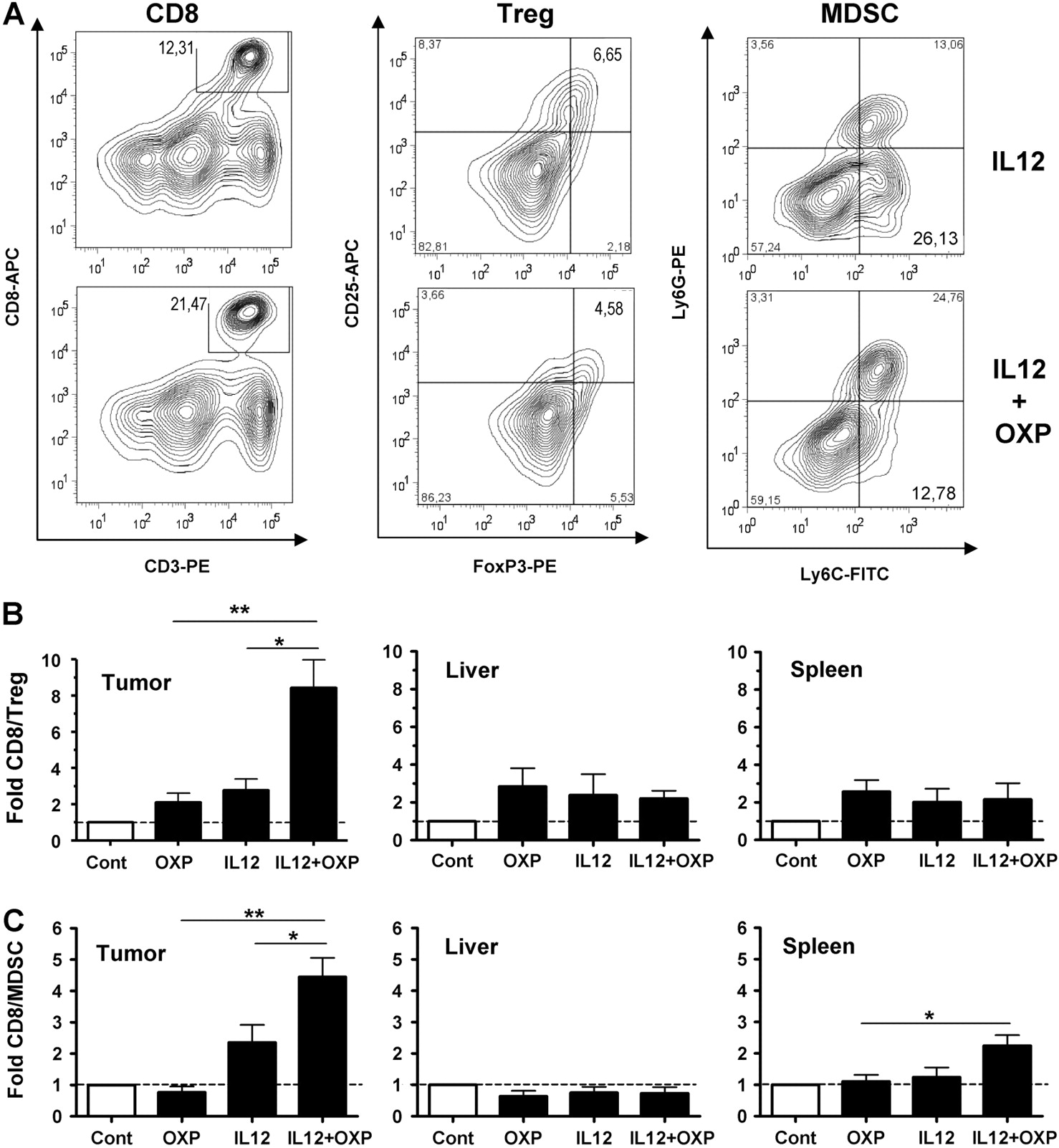
Oxaliplatin (OXP) counteracts the immunosuppressive microenvironment of tumours. C57BL/6 mice bearing intrahepatic MC38 tumours were treated with the HC-Ad/RUmIL-12 vector in the absence or presence of OXP, as indicated (n=10). Interleukin 12 (IL-12) expression was obtained using an adjusted induction protocol with mifepristone (Mif), and animals were killed after the third day of induction. Control groups include untreated animals (Cont) and mice treated with OXP alone (n=10). Leucocytes were isolated from liver, spleen and tumour samples. CD8+ T cells, regulatory T cells (Treg; CD25+FoxP3+) and monocytic myeloid-derived suppressor cells (MDSC; CD11b+Ly6C+Ly6G-) were identified by flow cytometry. (A) Countour plots showing the percentage of CD8 T cells (CD3+CD8+), Tregs (CD4+CD25+FoxP3+) and MDSC (CD11b+ly6C+Ly6G-) in representative mice treated with the HC-Ad/RUmIL-12 vector in the absence or presence of OXP (IL-12 and IL-12+OXP groups, respectively). Countour plots are gated on alive cells (CD8 T cells), CD4 cells (Tregs) or CD11b+ cells (MDSC). (B and C) The fold increase in the ratio of CD8/Treg (B) and CD8/MDSC (C) are represented, considering the untreated group as a reference.
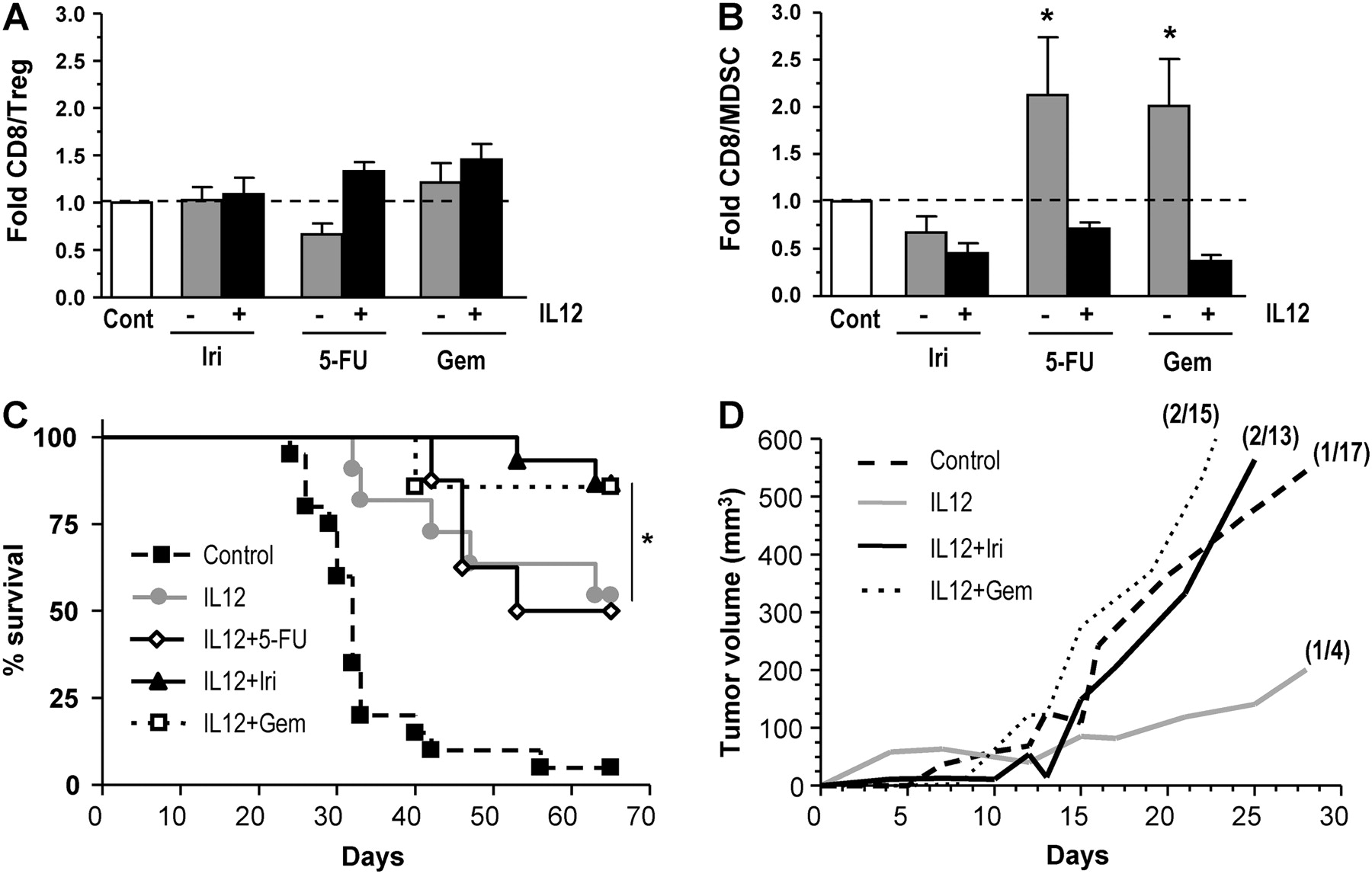
Together, these data indicate that OXP collaborates with IL-12 to reduce the immunosuppressive microenvironment of tumours. This effect is not shared by other agents frequently used against colorectal cancer. For instance, treatment with irinotecan alone or in combination with HC-Ad/RUmIL-12 did not increase the CD8/Treg and CD8/MDSC ratios (figure 6A and B). Interestingly, we found that 5-FU and gemcitabine caused an increase in the CD8/MDSC ratio, in accordance with other reports.39 40 However, this effect was reversed when the drugs were combined with IL-12. Although irinotecan and gemcitabine cooperated with IL-12 in the eradication of hepatic lesions and increased the survival of animals (figure 6C), they did not improve the immunological protection against tumour rechallenge (figure 6D), suggesting that alternative mechanisms are taking place.

Combination of interleukin 12 (IL-12) and other chemotherapeutic drugs. Mice bearing intrahepatic MC38 tumours were treated with the HC-Ad/RUmIL-12 vector, and IL-12 expression was induced with mifepristone (Mif) following an adjusted protocol (IL-12 group). Subsets of animals received additional treatment with one of the following chemotherapeutic drugs: 25 mg/kg 5-fluorouracil (5-FU) plus 100 mg/kg leucovorin on 2 consecutive days, starting 3 days before the first Mif induction (IL-12+5-FU); a single dose of 150 mg/kg irinotecan (Iri), 3 days before induction (IL-12+Iri); two doses of 60 mg/kg gemcitabine (Gem), 3 days and at the time of induction (IL-12+Gem). Other groups were left untreated (cont), or treated with the chemotherapeutic drugs only. Five animals from each group were killed after the third day of induction and tumours were processed for analysis of CD8+ T cells, regulatory T cells (Treg; CD25+FoxP3+) and monocytic myeloid-derived suppressor cells (MDSC; CD11b+Ly6C+Ly6G-) by flow cytometry. The fold increase in the ratio of CD8/Treg (A) and CD8/MDSC (B) are represented, considering the untreated group as a reference. The remaining animals completed the full induction protocol and survival was monitored (C). Death of animals was due to tumour progression, except for three mice in the group IL-12+Gem that died before the completion of the treatment owing to toxicity and were excluded from the survival curve. Animals treated with the chemotherapy drugs without IL-12 had no survival benefit compared with the untreated group, and are not represented in the graphic. Cured animals from the IL-12, IL-12+Iri and IL-12+Gem groups received a subcutaneous rechallenge with MC38Luc1 cells (106 cells/mouse). The same amount of cells was inoculated in naïve mice as a control. The average tumour volume for each group is represented (D). The number of mice that remained tumour-free for the entire observation period is indicated in parenthesis. *p<0.05.
Repeated cycles of IL-12 plus OXP are efficient in the long-term management of colorectal liver metastases
Potential clinical application of immunochemotherapy will surely require repeated cycles of treatment to control the initial lesions and to prevent, or eventually cope with, relapses and the appearance of new metastases. In order to mimic this situation, we initiated a series of experiments in which mice bearing hepatic tumours were treated with two cycles of IL-12 induction after vector administration. Then, they were subjected to a subcutaneous tumour rechallenge and, finally, a third cycle of induction was completed. In total, mice were evaluated for 5 months from the establishment of the initial hepatic tumours (see schematic representation in figure 7A). Three days before each of these cycles, one group of animals received a single dose of OXP (5 mg/kg). Interestingly, we observed that two cycles of IL-12 induction separated by 2 weeks efficiently controlled the hepatic tumours and extended the survival time of mice (figure 7B, left part of the graphic). In this setting, the cooperation of OXP was not apparent because most animals were cured.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Long-term management of colorectal cancer. Cooperation of oxaliplatin (OXP) and interleukin 12 (IL-12) for the control of experimental relapses in distant locations. (A) Schematic representation of experiments. C57BL/6 mice bearing hepatic tumours were treated with the HC-Ad/RUmIL-12 vector and received two cycles of mifepristone (Mif) induction preceded or not by OXP (5 mg/kg, intraperitoneally). Animals cured from their hepatic tumours were subjected to a subcutaneous challenge with the same tumour cells (MC38Luc1), and received a third cycle of induction during the indicated days. (B) Overall survival of mice. The grey areas indicate the duration of each induction cycle. Most animals in the OXP and OXP+IL-12 groups were cured from their hepatic tumours (not shown). The day of subcutaneous tumour rechallenge is marked with a vertical dotted line. ‘Control rechallenge’ refers to naïve animals that were inoculated with tumour cells and received no treatment. Mice were killed when the tumour burden reached the ethical end point. Survival after rechallenge was significantly higher in all treated groups than in control (p<0.01). Survival of animals in the IL-12+OXP group was significantly higher than in the IL-12 group (p<0.04). The progression of individual subcutaneous tumours in the rechallenge experiment is represented in panels C (group IL-12) and D (group IL-12+OXP). The duration of the induction protocol (Mif) and the day of administration of OXP are indicated. All naïve animals inoculated with tumour cells developed tumours that progressed in the absence of treatment (not shown).
One month after the completion of the second cycle of induction, the subcutaneous tumour challenge was performed in mice that had rejected the hepatic lesions. Compared with the results obtained with a single cycle of induction, no increase in the immunological protection was seen with any of the treatments. Therefore, we had the opportunity to evaluate whether a third cycle of induction might control the progression of the subcutaneous tumours, as if they were distant metastases. As shown in figure 7C, IL-12 alone was unable to inhibit the growth of the tumours that had escaped the initial immunological protection. In contrast, the combination of IL-12 and OXP obtained a response in 60% of the tumours (figure 7C). Of note, these tumours had progressed for more than 3 weeks and some of them exceeded 1000 mm3 in volume before the third induction cycle started. As a result, the overall survival of animals, including deaths from the tumour rechallenge, was improved by the incorporation of OXP in the treatment (figure 7B, right part of the graphic).
Discussion
The development of a long-term expression vector based on adenovirus enables an efficient delivery of therapeutic genes into the liver.25 In this approach, the target cells for transduction are the “normal” hepatocytes, not the cancer cells. This reduces the variability of infectivity and makes the gene transfer more predictable. The possibility of regulating the intensity of IL-12 expression by adjusting the dose of an inducer is important for the clinical application of immunogene therapy. It allows the use of relatively high doses of vector, to ensure that the liver of all patients is efficiently transduced. Then, careful monitoring of the individual response to the inducer will guide the intensification of the regimen, a concept that is common to many oncological treatments.
There is abundant information about the safety of Mif at doses higher than those used here,41 42 suggesting that the potency of the system could be increased further. Predicting the duration of treatment that would be needed in patients based on our preclinical results is too speculative. However, we provide evidence indicating that sustained expression of IL-12 is beneficial. One cycle of Mif induction is suboptimal in our tumour model. Repetition of short cycles does not improve the efficacy (data not shown), thus both the number of cycles and their duration are important—that is, the total period covered by therapeutic levels of IL-12. This requirement will probably depend on the severity of the disease. Although suppressive mechanisms used by tumours to evade the immune attack may be activated by IL-12,43 44 the net balance on tumour control is still positive. Our data suggest that OXP may enhance the immune response against tumours by tipping the balance between effector and regulatory/suppressor cells in favour of effector cells, as has been previously described for other chemotherapeutic drugs.37 Stimulation of Treg development by MDSCs has been described in some tumour models,45 indicating that mutual influence of these suppressive populations is possible. Recently, it has been shown that the inhibition of MDSC accumulation by sunitinib increases the antitumour effect of IL-12,46 supporting the relevance of this mechanism. The need for a complementary mechanism that initiates the damage to cancer cells may be particularly important in the protocol proposed here, since expression of IL-12 takes place in the liver parenchyma surrounding the tumour. Although we did not detect a consistent increase in the exposure of CRT in MC38 cells exposed to OXP, we cannot rule out the possibility that small amounts may contribute to immunogenicity of the cells, as recently described for other murine colon cancer cells.14 Our results point to a previously unrecognised mechanism taking place when OXP is used in combination with IL-12, but further studies are needed to gain a more detailed understanding of the immunostimulatory properties of OXP.
The vector and the experimental conditions described here can be easily adapted to evaluate other immunochemotherapy combinations of new combined protocols. Careful testing of specific combinations is required, because the effect of drugs on the immune system may be different when used as monotherapy or together with IL-12 stimulation, as suggested by the results obtained with 5-FU and gemcitabine. This phenomenon is currently under investigation.
In summary, the finding that controlled expression of IL-12 in the liver complements OXP-based chemotherapy regimens opens new possibilities for the management of patients with advanced colorectal cancer.
Acknowledgments
We thank Maria Bunuales and Elixabeth Bolanos for technical assistance
References
Supplementary materials
Online only appendix
Footnotes
Funding This work was funded in part by Fundacion Ramon Areces; Fundacion MMA; grant SAF2009-11324 from the Spanish Department of Science; Fundacion Pedro Barrie de la Maza; Condesa de Fenosa; INMUNONET-SOE1/P1/E014; Instituto de Salud Carlos III and the UTE project CIMA. JM-E was supported by a fellowship from Spanish FIS. PB was supported by a Juan de la Cierva contract from Spanish MEC and a Miguel Servet contract from Spanish Instituto de Salud Carlos III. RHA was supported by a Ramon y Cajal contract from Spanish MEC. JC was in receipt of a grant from Gobierno de Navarra.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Digest