Article Text

Abstract
Objective Immune responses are important in dictating non-alcoholic steatohepatitis (NASH) outcome. We previously reported that upregulation of hedgehog (Hh) and osteopontin (OPN) occurs in NASH, that Hh-regulated accumulation of natural killer T (NKT) cells promotes hepatic stellate cell (HSC) activation, and that cirrhotic livers harbour large numbers of NKT cells.
Design The hypothesis that activated NKT cells drive fibrogenesis during NASH was evaluated by assessing if NKT depletion protects against NASH fibrosis; identifying the NKT-associated fibrogenic factors; and correlating plasma levels of the NKT cell-associated factor OPN with fibrosis severity in mice and humans.
Results When fed methionine-choline-deficient (MCD) diets for 8 weeks, wild type (WT) mice exhibited Hh pathway activation, enhanced OPN expression, and NASH-fibrosis. Ja18‒/‒ and CD1d‒/‒ mice which lack NKT cells had significantly attenuated Hh and OPN expression and dramatically less fibrosis. Liver mononuclear cells (LMNCs) from MCD diet fed WT mice contained activated NKT cells, generated Hh and OPN, and stimulated HSCs to become myofibroblasts; neutralising these factors abrogated the fibrogenic actions of WT LMNCs. LMNCs from NKT-cell-deficient mice were deficient in fibrogenic factors, failing to activate collagen gene expression in HSCs. Human NASH livers with advanced fibrosis contained more OPN and Hh protein than those with early fibrosis. Plasma levels of OPN mirrored hepatic OPN expression and correlated with fibrosis severity.
Conclusion Hepatic NKT cells drive production of OPN and Hh ligands that promote fibrogenesis during NASH. Associated increases in plasma levels of OPN may provide a biomarker of NASH fibrosis.
- Biomarker
- fibrosis
- natural killer T cells
- non-alcoholic steatohepatitis
- Spp1
- non-alcoholic steatohepatitis
- partial hepatectomy
- liver disease in pregnancy
- liver regeneration
- liver cirrhosis
- molecular genetics
- liver metabolism
- IBD
- statistics
- gene expression
- cancer genetics
- cell biology
- cirrhosis
- cancer
- hepatocellular carcinoma
- carcinogen metabolism
- fibrogenesis
- hepatic fibrosis
- molecular biology
- fatty liver
- obesity
- liver immunology
- liver
- basic sciences
Statistics from Altmetric.com
- Biomarker
- fibrosis
- natural killer T cells
- non-alcoholic steatohepatitis
- Spp1
- non-alcoholic steatohepatitis
- partial hepatectomy
- liver disease in pregnancy
- liver regeneration
- liver cirrhosis
- molecular genetics
- liver metabolism
- IBD
- statistics
- gene expression
- cancer genetics
- cell biology
- cirrhosis
- cancer
- hepatocellular carcinoma
- carcinogen metabolism
- fibrogenesis
- hepatic fibrosis
- molecular biology
- fatty liver
- obesity
- liver immunology
- liver
- basic sciences
Significance of this study
What is already known about this subject?
Immune responses are important in dictating liver disease outcomes.
Natural killer T (NKT) cells accumulate in fibrosing non-alcoholic steatohepatitis (NASH) of mice and humans.
Hedgehog (Hh) pathway activation occurs in non-alcoholic fatty liver disease progression.
Osteopontin (OPN) is a proximal effector of Hh and promotes hepatic stellate cell activation.
What are the new findings?
Livers from NKT-deficient (CD1d−/− and Jα18−/−) mice contained significantly less Hh and OPN, and had dramatically less fibrosis than wild type mice after diet-induced NASH.
Human NASH livers with advanced fibrosis contain more NKT cells, and more Hh and OPN protein than NASH livers with early fibrosis.
NKT cell activation is associated with increased expression of Hh ligands and OPN.
Plasma OPN mirrors hepatic OPN content.
Plasma OPN may be a useful biomarker of advanced NASH fibrosis.
How might it impact on clinical practice in the foreseeable future?
Modulating NKT cell function or numbers may be a useful strategy to inhibit fibrosis progression.
Plasma OPN may be used as one of the biomarkers of advanced NASH fibrosis.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of hepatic dysfunction. Compared with individuals with simple hepatic steatosis, individuals with steatosis plus hepatocyte injury, cell death and inflammatory changes (dubbed non-alcoholic steatohepatitis, NASH) are more likely to develop progressive liver fibrosis, cirrhosis and cancer.1 ,2 Although the mechanisms which drive disease progression remain obscure, recent studies suggest that cells of the innate immune system, such as Kupffer cells, natural killer T (NKT) cells and NK cells may participate.3–6
The liver contains a large number of NKT cells, which provide intravascular immune surveillance.7 ,8 NKT cells respond to glycolipid antigens presented by major histocompatability class 1-like molecule, CD1d, and when activated, secrete both T helper 1 (Th1) (pro-inflammatory/anti-fibrotic) and Th2 (anti-inflammatory/pro-fibrotic) cytokines.8–11 In the hepatitis B virus-transgenic mouse,12 NKT-derived interleukin 4 (IL4) and IL13 were reported to be responsible for the fibrogenic outcomes. Conversely, CD1d knockout (CD1d−/−) mice, which lack NKT cells, were protected from xenobiotic-induced hepatic inflammation, injury and fibrosis.13 These findings complement human studies showing NKT accumulation in primary biliary cirrhosis14 ,15 and chronic viral hepatitis C.16
The role of NKT cells in NAFLD progression is only beginning to emerge. NKT cells are generally depleted in fatty livers,17–19 but appear to accumulate in livers with NASH-related fibrosis.19 ,20 Recently, we reported that hedgehog (Hh)-mediated accumulation of NKT cells contributes to development and progression of NASH in mice and humans.21 Mice with an overly active Hh pathway express higher levels of the NKT cell chemokine, CXCL16, vascular cell adhesion molecule 1 (VCAM-1) and IL15, a factor that promotes NKT viability.22 These mice accumulate more hepatic NKT cells and develop worse NASH fibrosis. Consistent with this, we found that individuals with NASH cirrhosis harboured fourfold more NKT cells than those with healthy livers.
In addition to secreting the classical cytokines, IL4 and IL13, NKT cells can also secrete osteopontin (OPN), a cytokine and matricellular protein that exacerbates concavalin A induced hepatitis,23 and promotes NAFLD progression.24 Recently, we showed that NKT cells also secrete the fetal morphogen, sonic Hh (Shh).25 Shh induces the transition of quiescent hepatic stellate cells (HSCs) into activated, collagen-producing myofibroblasts26 and amplifies the ‘repair-associated inflammatory response’.27 Intriguingly, inhibition of the Hh pathway in vivo leads to the amelioration of fibrosis in mice with cholestatic liver injury.28
In this study, we evaluated the hypothesis that fibrosis progression in NASH is NKT cell dependent. We fed mice that were genetically deficient in NKT cells (Jα18−/− and CD1d−/−) methionine-choline-deficient (MCD) diets to induce NASH, evaluated if NKT depletion protected against NASH fibrosis, and assessed if NKT-cell-mediated fibrogenesis required OPN and Hh. Findings in mice and cultured HSCs were then corroborated by evaluating similar endpoints in patients with NASH.
Methods
Animal experiments
Mice
Wild type (WT) (Jackson Laboratories, Bar Harbor, Maine, USA), CD1d-deficient (CD1d−/−) and Jα18-deficient (Jα18−/−) mice (RIKEN, Saitama, Japan) of similar backgrounds (B6) were fed MCD diet or control chow for 8 weeks. Animal care and procedures were approved by the Duke University Institutional Animal Care and Use Committee as set forth in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Analysis of liver architecture, morphometry and immunohistochemistry
Formalin-fixed paraffin embedded (FFPE) livers were analysed. Haematoxylin and eosin (H&E) and picrosirius red staining with quantification by morphometric analysis was performed as previously described.24 The primary antibodies used were OPN (R&D, Minneapolis, MN, USA, AF808; 1:40) and α smooth muscle actin (αSMA) (Abcam, Cambridge, MA, USA, Abcam 5694; 1:800). Polymer-HRP anti-rabbit (Dako, Carpinteria, CA, USA, DAKO4003) and anti-goat (Santa Cruz, California, USA, sc-2768; 1:250) were used as secondary antibodies. Antigens were demonstrated by diaminobenzidine (DAB) (Dako). Omitting primary antibodies eliminated staining, demonstrating specificity. For OPN and αSMA quantification, 15 randomly selected, 40× fields (excluding the major bile duct in each portal tract from consideration) were analysed with the MetaView software.
ALT determination
Serum alanine aminotransferase (ALT) was measured using kits commercially available from Biotron Diagnostics (66-D; Hemet, California, USA) according to the manufacturer's instructions.
RNA extraction and mRNA quantification
Total liver RNA extraction and mRNA quantification by real-time PCR (qRT-PCR) were performed as published.24 Details of primer sequences are given in online supplemental table 1.
Hydroxyproline assay
Hydroxylproline content in whole liver specimens was quantified colorimetrically as previously described.29
Effect of primary liver NKT cells on activation of primary HSC
Primary liver mononuclear cells (LMNCs) were isolated as previously described,25 and cultured in complete NKT media,30 with or without the NKT cell ligand α-galactosylceramide (αGC) (100 ng/ml; San Diego, California, USA; Cat. No 306027) for 24 h. αGC specifically activates invariant NKT (iNKT) cells.21 ,31 αGC-activated LMNC conditioned medium (LMNC-CM) was then added to primary murine HSCs in the presence or absence of the Hh neutralising antibody 5E1 (10 μg/ml; Iowa Hybridoma Bank, Iowa City, Iowa, USA), the Smoothened antagonist (inhibitor of Hh signalling) GDC (GDC-0449; Selleck, Houston, Texas, USA) or vehicle for 48 h and RNA was harvested for qRT-PCR. Experiments were performed in duplicate wells and repeated twice.
In separate experiments, LMNC-CM was added to primary murine HSCs with either sham aptamers (OPN-R3-2; 100 nmol/litre) or OPN aptamers (OPN-R3; 100 nmol/litre) (both synthesised by Dharmacon, Lafayette, Colorado, USA)24 for 48 h, and RNA harvested as above. This concentration of OPN aptamer has been shown to inhibit adhesion, migration and invasion in the MDA-MB-231 breast cancer cell line (which highly expresses OPN and is a standard tool for evaluating OPN actions).32
ELISA assays for plasma OPN
Peripheral blood was collected at the time of sacrifice, and plasma obtained and stored at −80°C till analysis. Plasma OPN was measured using the commercially available OPN ELISA kit (R&D, Minneapolis, MN, USA, DY441) and in accordance with the manufacturer's instructions. All samples were run in duplicate, and expressed as pg/ml.
Human subjects
Immunohistochemistry
FFPE liver sections of de-identified subjects with biopsy-proven NASH-related early and advanced fibrosis (n=10/group) from the Departments of Pathology at Duke University were studied in accordance with National Institutes of Health and Institutional guidelines for human subject research.
The primary antibodies used were OPN (R&D, AF1433, 1:40); Indian hedgehog, Ihh (Abcam, Ab39634, 1:1000), Shh (Epitomics, Burlingame, CA, USA, 1843-1; 1:7500) Gli2 (Genway, San Diego, CA, USA, 18-732; 1:4500) CD57 (NeoMarkers, Fremont, CA, USA, MS136). Polymer-HRP anti-mouse (Dako; K4001) was used as secondary antibodies. Antigens were demonstrated by diaminobenzidine (DAB) (Dako). Omitting primary antibodies eliminated staining, demonstrating specificity.
Plasma OPN measurements
Peripheral blood was taken from patients with early (n=25) or advanced NASH (n=25) at the time of liver biopsies, and plasma stored at −80°C until analysis. Plasma OPN was measured using the commercially available OPN ELISA kit (R&D; DY1433) and in accordance with the manufacturer's instructions. All samples were run in duplicate, and expressed as pg/ml.
Statistical analysis
Results are expressed as mean ± SEM. Statistical significance was determined using the Student t test. Significance was accepted at the 5% level.
Results
NKT-deficient mice develop less fibrosis than WT mice after MCD diets
Compared with WT mice that were fed normal chow (n=10), MCD-diet-treated mice (n=10) developed significant steatohepatitis and fibrosis after 8 weeks (figure 1A, online supplemental figure 1). The latter was demonstrated by increased Sirius red staining (figure 1B,C), and hepatic hydroxyproline quantification (figure 1D). Collagen deposition was accompanied by the accumulation of αSMA-immunoreactive cells (figure 2A,B), and induction of pro-fibrogenic genes, including α-SMA, collagen 1α1, and transforming growth factor β (figure 2C,D, online supplemental figure 2A).

Natural killer T (NKT)-deficient mice develop less fibrosing non-alcoholic steatohepatitis (NASH) after methionine-choline-deficient (MCD) diet. Wild type (WT), Jα18−/− and CD1d−/− mice were fed control chow (n=10 per strain) or MCD diet (n=10 per strain) for 8 weeks and then sacrificed. (A) Representative haematoxylin and eosin (H&E) and (B) Sirius red (SR) staining after MCD diet. (C) Sirius red quantification by morphometric analysis. The results are expressed as fold change relative to WT control chow-fed mice and presented as mean ± SEM. (D) Hepatic hydroxyproline content at the end of the treatment period. *p<0.05 versus control chow-fed mice.

Natural killer (NKT)-deficient mice exhibit attenuated fibrogenic response compared with wild type (WT) mice. Mice were fed control chow and methionine-choline-deficient (MCD) diets as described in figure 1. (A) α Smooth muscle actin (αSMA) immunoreactivity and (B) αSMA morphometry. Sections from four animals were used and 15 randomly selected, 40× fields chosen for analysis by the Metaview software. Small inserts in (A) show representative staining from chow-fed mice. Whole liver tissues were harvested for RNA analysis by quantitative real-time PCR. (C) αSMA mRNA, (D) collagen Iα1 mRNA, and (E) alanine aminotransferase (ALT) (IU/litre). The results are expressed as fold change relative to WT control chow-fed mice and presented as mean ± SEM; *p<0.05 versus control chow-fed mice.
Jα18−/− mice, which lack iNKT cells, developed significantly less hepatic fibrosis (figures 1B–D, 2A–D) and injury (figure 2E) than WT mice after MCD diet. This finding in mice that lack iNKT cells is consistent with our previous report that CD1d−/− mice (which lack all NKT subsets) exhibited reduced fibrogenic responses after diet-induced NASH21 (figures 1C,D, 2B–D, online supplemental figure 2A).
We reported that Hh pathway activation induces the transition of quiescent HSC to collagen-producing myofibroblasts,26 and promotes fibrogenic repair in NASH.29 Consistent with this, Indian Hh (a Hh ligand), and Gli2 (a Hh-regulated transcription factor) were significantly induced during diet-induced NASH (online supplemental figure 2B,C). Conversely, NKT-deficient mice, which developed less fibrosis, exhibited attenuated Hh signalling (online supplemental figure 2B,C).
NKT-deficient mice express less OPN during MCD-diet-induced NASH than WT mice
We recently showed that OPN acts in paracrine and autocrine fashions to promote HSC activation and fibrogenesis.24 After MCD diet, mice genetically deficient in OPN exhibited reduced fibrosis, while mice that overexpressed OPN developed worse fibrosis. Here, we show that livers from MCD-fed WT mice upregulate OPN protein expression by nearly fivefold (figure 3A,C). In contrast, livers of both NKT-cell-deficient strains that developed less fibrosis than WT mice after diet-induced NASH (figures 1B–D, 2A–D, online supplemental figure 2) had significantly reduced levels of OPN (figure 3A–C, online supplemental figure 3). These findings raise the possibility that NKT cell-associated factors modulate OPN expression and resultant hepatic fibrogenesis.

Natural killer T (NKT)-deficient mice harbour less liver and plasma osteopontin (OPN) than wild type (WT) mice after diet-induced non-alcoholic steatohepatitis (NASH). Mice were fed control chow or methionine-choline-deficient (MCD) diet for 8 weeks and then sacrificed as described in figure 1. Matched plasma was collected for OPN ELISA. (A–C) Representative OPN immunostaining after control chow or MCD diets in WT and Jα18−/− mice (final magnification 200×), and quantification by morphometry. Sections from 10 animals were used per group and 10 randomly selected, 200× fields chosen for analysis by the Metaview software. The results are expressed as fold change (% positive staining) relative to WT control mice and presented as mean ± SEM. (D) Plasma OPN. Plasma from 10 animals per group was used for OPN ELISA, and displayed as pg/ml. Each sample was run in duplicate; *p<0.05 versus WT control mice.
Previously, others reported that plasma OPN levels correlated with severity of liver fibrosis in subjects with chronic hepatitis B and C.33 ,34 In this study, plasma from WT mice with fibrosing NASH contained nearly twofold more OPN by ELISA than chow-fed controls (figure 3D). Consistent with reduced hepatic OPN expression, plasma OPN levels were twofold lower in NKT-cell-deficient mice after diet-induced NASH. The aggregate data, therefore, suggest that elevated plasma OPN levels may be a biomarker of advanced NASH fibrosis.
Primary mouse liver NKT-cell-associated OPN promotes myofibroblastic activation of primary HSC
In addition to HSCs and cholangiocytes,24 OPN is expressed by activated immune cells23 (figure 3). Therefore, we examined if activated liver NKT cells generate OPN that acts in a paracrine fashion to promote HSC fibrogenesis. LMNCs were isolated from healthy mice and incubated with αGC (which specifically activates iNKT cells) or vehicle overnight. LMNC-conditioned medium (CM) was then collected for OPN ELISA. αGC-activated LMNC-CM contained fourfold more OPN protein than vehicle-treated LMNC-CM, and similar levels of OPN as CM from cholangiocytes, which are known to be a source of OPN in liver (figure 4A). To ascertain if activated LMNCs in vivo also express higher amounts of OPN, we harvested primary LMNCs from chow or MCD-fed WT mice (n=4/group), and OPN expression was analysed by qRT-PCR. Primary LMNCs from MCD-fed mice harboured 12-fold more OPN mRNA than LMNC from chow-fed mice (figure 4B).
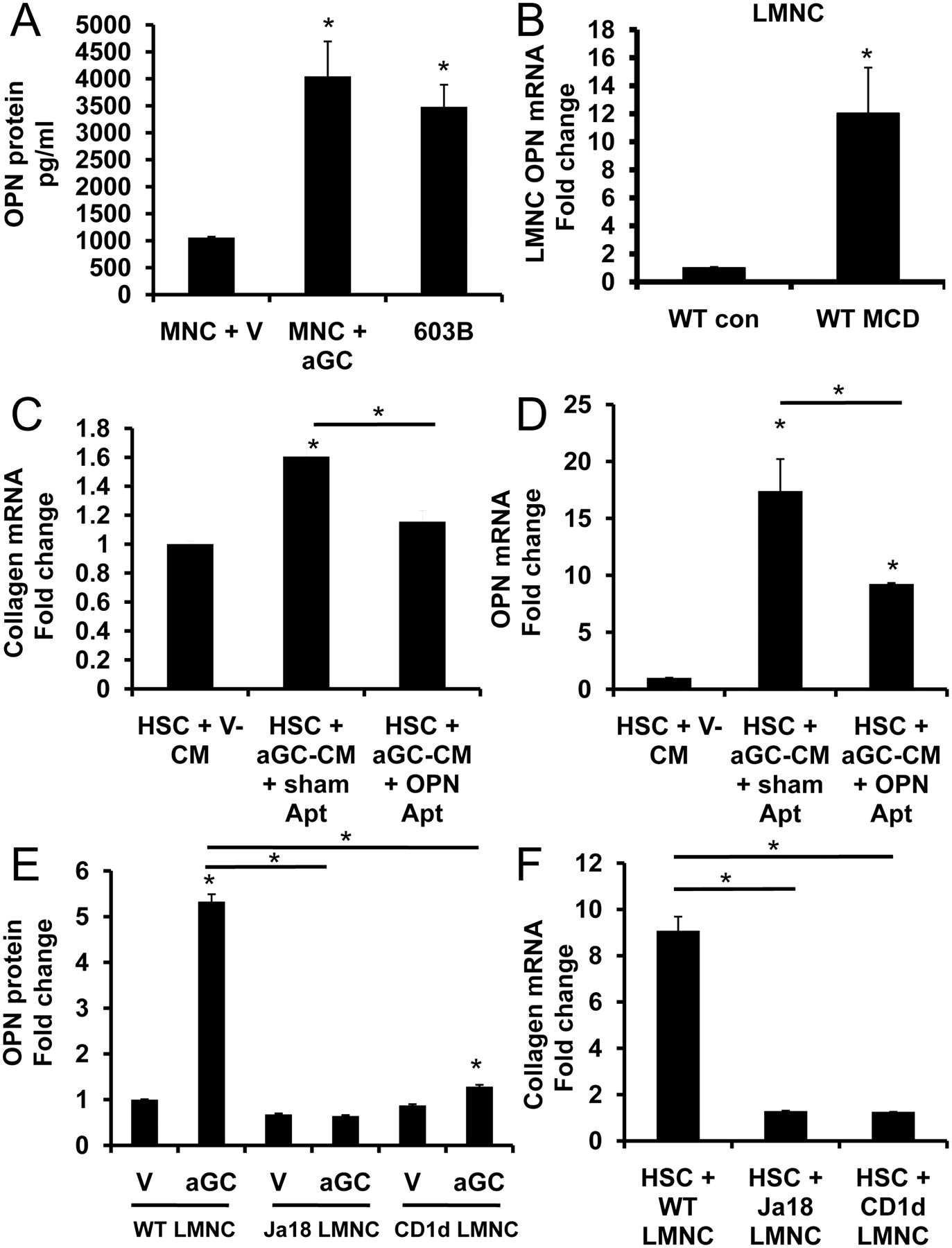
α Galactosylceramide (αGC)-activated liver natural killer T (NKT)-cell-associated osteopontin (OPN) promotes primary hepatic stellate cell (HSC) activation. Primary liver mononuclear cells (LMNCs) (1×105) from wild type (WT) mice were cultured in complete NKT media, and treated with vehicle (V) or αGC (to activate NKT cells) for 24 h. Conditioned medium (CM) was collected for OPN ELISA; 603B mouse cholangiocyte-CM was used as a positive control. (A) OPN protein was quantified by ELISA. Each LMNC-CM sample was run in duplicate and expressed as pg/ml. (B) LMNCs were isolated from WT mice that were fed control chow (n=4) or methionine-choline-deficient (MCD) diet (n=4) for 8 weeks, and OPN mRNA expression was analysed by quantitative real-time PCR (qRT-PCR). Results are expressed as fold change relative to control-chow-derived LMNCs. (C, D) Mouse primary HSCs were cultured with CM from αGC-activated WT LMNCs (αGC-CM) plus OPN aptamers or sham aptamers for 48 h. HSC expression of collagen I α1 (C) and OPN (D) mRNA were assessed by qRT-PCR. Results are expressed as fold change relative to HSCs that were treated with CM from vehicle-treated WT LMNCs (V-CM). (E) OPN was quantified by ELISA in CM from LMNCs that were harvested from WT, Jα18−/− or CD1d−/− mice and treated with either vehicle (V) or αGC to activate NKT cells. Results are expressed as fold change relative to vehicle (V)-treated WT LMNCs. Mouse primary HSCs were cultured for 48 h in CM from LMNCs that were harvested from WT, Jα18−/− and CD1d−/− mice and treated with vehicle or αGC to activate NKT cells. HSC expression of collagen I α1 mRNA was assessed by qRT-PCR (F). The results are expressed as fold change relative to HSCs that were cultured with CM from respective vehicle-activated LMNCs. Mean ± SEM of duplicate experiments are presented; *p<0.05.
αGC-activated LMNC-CM was then added to primary cultures of mouse HSC with either OPN aptamers or sham aptamers for 48 h. The addition of OPN aptamers to neutralize OPN activity in LMNC-CM resulted in a significant repression of HSC fibrogenic genes (OPN, collagen) (figure 4C,D).
To ascertain if differences in the fibrogenic outcomes observed in figure 1 could be attributed to fewer NKT cells, and hence, reduced NKT-associated OPN, we isolated primary LMNCs from WT, Jα18−/− and CD1d−/− mice, and incubated them with αGC or vehicle overnight (as above), and LMNC-CM was collected for OPN ELISA or added to primary cultures of HSCs. We found that WT-LMNC mice secreted fivefold more OPN protein (figure 4E) and induced greater collagen expression in HSC than NKT-deficient LMNCs (figure 4F). These findings, in concert, support the hypothesis that activated NKT-cell-associated OPN promotes HSC activation in a paracrine fashion.
NKT-cell-associated Hh promotes myofibroblastic activation of primary HSCs
Previously, we reported that progressive NASH is associated with Hh pathway activation,29 that activated hepatic NKT cells are capable of secreting Hh ligands,25 and that Hh ligands could directly induce HSC activation and fibrogenesis.26 Because OPN expression in HSCs is regulated, at least in part, by Hh signalling,24 we examined whether NKT-derived Hh ligands could directly promote OPN expression in HSCs and mediate fibrogenic responses. Primary WT LMNCs were incubated with αGC or vehicle (as above). LMNC-CM was then added to primary cultures of mouse HSCs, with or without Hh neutralising antibody, 5E1, or GDC, a pharmacological inhibitor of the Hh pathway. Forty-eight hours later, HSC RNA was harvested for PCR analysis. We confirm by fluorescence activated cell sorting analysis that primary hepatic NKT cells express the Hh ligand (online supplemental figure 4A,B). CM from αGC-LMNCs (that contained activated NKT cells) upregulated HSC expression of collagen and OPN (figure 5A,B). The addition of 5E1 or GDC (inhibition of Hh signalling) led to a significant abrogation of these fibrogenic effects (figure 5A,B). The aggregate data suggest that in addition to generating OPN (figure 4A,E), activated hepatic NKT cells secrete Hh ligands to promote liver fibrogenesis.

α-Galactosylceramide (αGC)-activated liver natural killer T (NKT)-cell-associated hedgehog (Hh) ligand contributes to hepatic stellate cell (HSC) activation. Primary cultures of mouse HSCs were cultured for 48 h with conditioned media (CM) from vehicle-treated (V-CM) or αGC-activated (αGC-CM) mouse primary liver mononuclear cells (LMNCs) (1×105) plus Hh-neutralising antibody (5E1), a Hh signalling antagonist (GDC) or control vehicle (DMSO, dimethyl sulfoxide). Quantitative real-time PCR analysis was used to quantify the effects on expression of (A) collagen I α1 and (B) OPN mRNA. The results are expressed as fold change relative to HSCs that were cultured in CM from vehicle-treated LMNCs (V-CM). Mean ± SEM of duplicate experiments are presented; *p<0.05.
NASH progression in humans is accompanied by increased liver and plasma OPN
NAFLD progression in mice is associated with activation of the Hh pathway, induction of Hh-regulated fibrogenic factors, such as OPN,24 and the accumulation of intrahepatic NKT cells.19 ,21 We demonstrate that fibrogenesis in mice with diet-induced NASH depends on hepatic NKT cells, and show the mechanism involves NKT-cell-mediated production of Hh ligands and OPN (figures 1–5). NKT cells comprise a much greater proportion of the resident hepatic immune cells in mice than humans.4 ,35 Therefore, we examined livers from patients with NASH to assess the relationship between hepatic NKT cell accumulation, Hh pathway activation, OPN expression and fibrosis in humans.
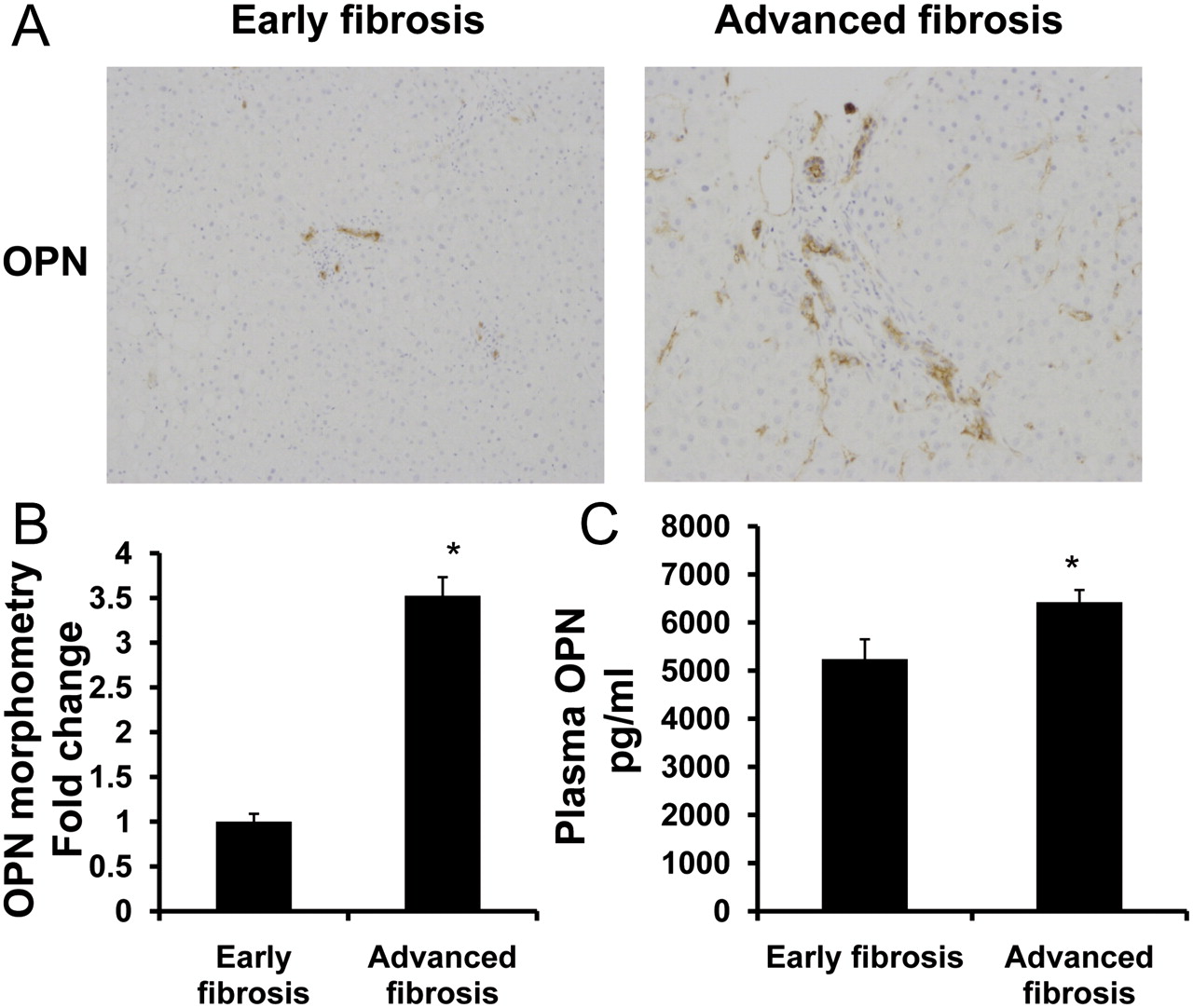
Human livers with advanced NASH (stage 3–4 fibrosis) contained threefold more of the Hh-regulated cytokine, OPN, particularly in the fibrous septa, than those with early NASH (no fibrosis or stage 1–2 fibrosis) (figure 6A–B). Livers from individuals with advanced NASH also contained more cells that expressed the Hh ligands, Shh and Ihh (online supplemental figure 5A–C), and the Hh-regulated transcription factor, Gli2 (online supplemental figure 6A). Previously, we reported that the hepatic content of cells that co-express CD57 and CD3 or CD56 and CD3 (ie, NKT cells) is increased significantly in patients with NASH-related cirrhosis.21 Here we show that NKT cells that accumulate during fibrosing NASH express Ihh and OPN protein (online supplemental figure 6B–E). Moreover, we found that increased hepatic OPN expression was mirrored by higher plasma OPN levels in NASH patients with advanced fibrosis than those with mild fibrosis (figure 6C). Overall plasma OPN levels in NASH were approximately twofold higher than levels detected in healthy volunteers (mean OPN level, normal: 3000 pg/ml).33 The findings in humans with NASH, therefore, are similar to those in mice with NASH (figure 3) and together suggest that OPN may be a useful biomarker of advanced NASH fibrosis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Non-alcoholic steatohepatitis (NASH) progression in humans is accompanied by upregulation of liver and plasma osteopontin (OPN). Coded liver sections from 10 patients with early and advanced NASH fibrosis were stained for OPN, and OPN expression was quantified by computer-assisted morphometry. (A) Representative photomicrographs from patients with early and advanced NASH fibrosis. (B) Quantitative analysis of liver OPN-positive cells in all patients. Numbers of OPN-positive cells are expressed as percentage of stained cells per high-powered field. (C) Plasma was collected from individuals with early or advanced NASH (n=25/group) and plasma OPN measured by ELISA. Plasma samples were run in duplicate and the results are presented as mean ± SEM; *p<0.001 versus early fibrosis.
Discussion
This study shows that hepatic NKT cells are critical drivers of fibrogenesis in NASH. It also demonstrates that the mechanism involves NKT-cell-mediated stimulation of hepatic Hh pathway activity and resultant increases in the liver content of OPN, a Hh-regulated, pro-fibrogenic factor. Importantly, plasma levels of OPN reflect the activity of this process in liver, and thus, may prove to be a useful non-invasive marker of advanced NASH.
Previously, we reported that Hh pathway activity correlates with severity of NAFLD-related liver injury,29 and showed that Hh signalling enhances hepatic accumulation of immune cells, including CD1d-restricted NKT cells.21 Here, we prove that fibrogenesis in mice with diet-induced NASH directly depends upon NKT cells. Compared with WT mice, two strains of NKT-cell-deficient mice (Jα18−/− and CD1d−/−) exhibited dramatically attenuated fibrosis after 8 weeks of MCD diets. NKT-cell-deficient mice also demonstrated blunted Hh pathway activity (less expression of Hh ligand and the Hh-regulated transcription factor, Gli2), and reduced expression of OPN, a Hh-regulated cytokine which acts in both paracrine and autocrine manners to promote HSC activation and fibrogenesis.24
In the present study, LMNCs from WT mice with diet-induced NASH expressed 12-fold higher levels of OPN mRNA than chow-fed controls. CM from WT-mice-derived LMNCs that were treated with αGC to activate resident NKT cells stimulated cultured HSCs to express collagen mRNA, whereas similarly treated LMNCs from two different strains of NKT-cell-deficient mice did not. These results indicate that activated liver NKT cells promote the production of factors that drive fibrogenic responses in HSCs, and suggest that one of these pro-fibrogenic factors might be OPN. Indeed, ELISA demonstrated that CM from αGC-activated WT LMNCs contained four times more OPN than CM from similarly treated LMNC populations from NKT-cell-deficient mice. Moreover, neutralising OPN activity with OPN-specific RNA aptamers significantly abrogated the HSC fibrogenic responses to WT LMNC-derived factors, reducing HSC expression of collagen mRNA to basal levels. The aggregate findings, therefore, complement and extend earlier evidence that liver NKT cells are capable of producing OPN by showing that important fibrogenic actions of NKT cells are directly mediated by OPN.23 ,25 ,36
OPN expression is induced by activating Hh signalling in Hh-responsive cells.24 We previously reported that NKT cells produce and respond to Hh ligands.25 ,36 In the present study we show that adding the pan-Hh-neutralising antibody (5E1) to LMNC-CM completely blocked its ability to upregulate expression of collagen mRNA in HSCs. CM from LMNCs of NKT-cell-deficient mice is also completely unable to induce collagen gene expression in cultured HSCs. Thus, NKT cells appear to be a major source of the Hh ligands that drive fibrogenic responses in HSCs. The latter likely involves both NKT cell-derived OPN and HSC-derived OPN because treating HSCs that were grown in LMNC-CM with a drug that blocks intracellular Hh signalling (GDC) reduced HSC expression of OPN mRNA. Thus, fibrogenesis in NASH seems to result from the combined actions of several autocrine/paracrine signalling loops that re-enforce Hh pathway activation and OPN production in different types of hepatic non-parenchymal cells, particularly NKT cells and HSCs.
Our studies in human subjects support the pathophysiological relevance of excessive OPN production and Hh pathway activation during NASH-related fibrogenesis. Notably, we provide novel evidence that the upregulation of liver OPN production in advanced NASH is mirrored by increased plasma OPN levels. Recent reports suggest that high plasma OPN levels may also be predictive of cirrhosis in patients with chronic hepatitis B and C.33 ,34 In addition, elevated plasma OPN levels are associated with cancer development, recurrence and bad prognosis.37 ,38 Given these observations, it is conceivable that plasma OPN levels might be used to identify patients with NASH who are at high risk of bad outcomes. Further studies, in a larger cohort of individuals with NAFLD, will be necessary to confirm the clinical utility of this novel biomarker.
Acknowledgments
The authors would like to thank Patrice McDermott (Human Vaccine Institute Flow Cytometry Core Facility, Duke University) for her help with primary mononuclear cell sort, and Dr G J Gores (Mayo Clinic, Rochester, Minnesota, USA) and Yoshiyuki Ueno (Tohoku University, Sendai, Japan) for providing the murine immature ductular cell line (603B). CD1d-tetramers were provided by NIH, Atlanta, Georgia, USA. CD1d−/− mice were kindly provided by Dr Zhiping Li (Gastroenterology, John Hopkins University, Baltimore, Maryland, USA), while Jα18−/− mice were provided by Drs Hiroshi Watarai and Masaru Taniguchi (RIKEN Research Center for Allergy and Immunology, Yokohama City, Japan). In addition, the authors thank Mr Carl Stone for administrative support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding This work was supported by grants from the National Institute of Health, RO1 DK053792 (AMD) and RO1 DK077794 (AMD), and by the EASL fellowship (WKS) and BRET grant (WKS).
Competing interests None.
Patient consent The study involved the use of archived liver sections from which local ethics and informed consent had been obtained previously for the sampling of liver samples. For the isolation and FACS analysis of liver lymphocytes, informed consent was obtained from the patient undergoing liver resection (for the use of the resected livers for research).
Ethics approval Ethics approval was provided by Duke University and the South Birmingham Research Ethics Committee, UK (Birmingham local ethical approval 04/Q2708/41 and REC 2003/242).
Provenance and peer review Not commissioned; externally peer reviewed.