Article Text

Abstract
Objective Mechanisms of action (MoA) of anti-tumour necrosis factor α (TNFα) therapies in Crohn's disease (CD) may critically involve induction of immune cell apoptosis via membrane-bound TNFα (mTNFα) binding. Certolizumab pegol (CZP), which is effective in induction and maintenance of remission in CD lacks the ability to induce apoptosis. The aim of this study was to analyse transcriptomal responses of reverse signalling induced by the TNFα binding agents infliximab (IFX) and CZP in myelomonocytic cells.
Design Induction of transcriptional patterns upon anti-TNFα stimulation was assessed using oligonucleotide microarrays. mRNA expression of GDF-1/ LASS1, which was identified as a shared target, was studied in inflammatory bowel disease by real-time PCR, while signalling pathways induced by growth and differentiation factor 1 (GDF-1) were investigated using western blots and ELISA.
Results IFX and CZP induced a common signature of 20 transcripts that could be categorised into control of cell cycle, transcription activation and pre-mRNA processing. We selected GDF-1/LASS1 for functional follow-up, which was found to be upregulated in inflamed CD tissues. We show that downregulation of GDF-1/LASS1 depends on autocrine release of transforming growth factor β after mTNFα ligation. We demonstrate that GDF-1 itself acts as a novel proinflammatory factor via induction of interleukin 6 and signal transducer and activator of transcription 3 and is downregulated after IFX treatment.
Conclusion Commonalities in the MoA of IFX and CZP comprise modulation of non-apoptotic pathways through downregulation of proinflammatory GDF-1. Further characterisation of the molecular role of GDF-1 in complex inflammatory processes in vivo is warranted to decide whether this proinflammatory molecule is a promising therapeutic target in patients with CD.
- TNFα blockade
- Crohn's disease
- GDF-1
- infliximab
- certolizumab pegol
- inflammatory bowel disease
- bacterial interactions
- TGFβ
- cell biology
- arthritis
- autoimmune disease
- carcinogenesis
- apoptosis
- hepatoma
- cancer
- cytokines
- immunohistochemistry
- IBD basic research
- IBD genetics
- antibacterial mucosal immunity
- infliximab
- genetics
- signal transduction
Statistics from Altmetric.com
- TNFα blockade
- Crohn's disease
- GDF-1
- infliximab
- certolizumab pegol
- inflammatory bowel disease
- bacterial interactions
- TGFβ
- cell biology
- arthritis
- autoimmune disease
- carcinogenesis
- apoptosis
- hepatoma
- cancer
- cytokines
- immunohistochemistry
- IBD basic research
- IBD genetics
- antibacterial mucosal immunity
- infliximab
- genetics
- signal transduction
Significance of this study
What is already known on this subject?
-
Infliximab (IFX) and certolizumab pegol (CZP) are effective in the therapy of Crohn's disease (CD).
-
IFX but not CZP induces apoptotic pathways.
-
Transforming growth factor β (TGFβ) has been shown to be induced by IFX.
What are the new findings?
-
Mechanisms of action of IFX and CZP comprise modulation of non-apoptotic pathways through downregulation of proinflammatory signals via growth and differentiation factor 1(GDF-1).
-
Downregulation of GDF-1 by IFX and CZP depends on autocrine release of TGFβ after membrane-bound tumour necrosis factor α ligation.
-
GDF-1 acts as a proinflammatory factor via induction of interleukin 6 and signal transducer and activator of transcription 3.
-
GDF-1 is upregulated in inflamed tissues from patients with CD.
-
GDF-1 is downregulated after IFX treatment in patients whose condition responds to IFX.
How might it impact on clinical practice in the foreseeable future?
-
Studying the exact physiological role of GDF-1 in complex inflammatory processes in vivo may answer the question of whether this proinflammatory molecule could be a novel promising target in the therapy of patients with CD.
Introduction
As many as 1.4 million people in the USA and 2.2 million in Europe have inflammatory bowel disease (IBD), a lifelong disease that can be differentiated into two major sub-phenotypes, Crohn's disease (CD) and ulcerative colitis (UC). The pathophysiology of IBD is characterised by a highly activated state of the mucosal immune system and excessive mucosal destruction. Although the aetiology is unknown, it is assumed that IBD is a multifactorial disease caused by the interplay of genetic, environmental and immunological factors. Despite advances in the understanding of the complex and diverse early events of disease precipitation, a pathophysiological hallmark of the inflammatory processes observed in CD is the preponderance of the proinflammatory cytokines, for example, tumour necrosis factor α (TNFα), interleukin 6 (IL-6) and IL-12, which play critical roles in the initiation and perpetuation of inflammation in CD. TNFα is an important mediator of inflammatory processes and is likely to be at the apex of the inflammatory cascade in CD,1 as it is increased in intestinal tissue and stools of patients with CD. Clinical evidence for efficacy of systemic inhibition of TNFα is given by the fact that a single infusion of a chimeric monoclonal anti-TNFα antibody (infliximab, IFX) has been shown to induce remission and significantly improve clinical symptoms in patients with CD in multiple studies.2 ,3
TNFα secretion represents a complex process by which membrane-bound TNFα (mTNFα) is expressed and then cleaved by TNFα-converting enzyme.4 It was reported that ligands of the TNF superfamily could act as receptors and are able to elicit bidirectional signals (‘reverse signalling’),5 whereas systematic knowledge about the impact of therapeutically administered TNFα binding proteins on reverse signalling via mTNFα is still missing.6 We have previously shown that infliximab transiently activates the p38 mitogen-activated protein kinase (MAPK) and p44/42 extracellular signal-regulated kinase (ERK1/2) in monocytic cells in vitro and in vivo. A differential phosphorylation of p38 MAPK was observed in patients with CD who respond and do not respond to infliximab therapy.7 Reverse signalling events via mTNFα led to an enhanced secretion of transforming growth factor β (TGFβ), which in turn was responsible for the activation of p38 and ERK1/2 and the induction of apoptosis via caspase 3.8 Further in vivo and in vitro studies also reported caspase-dependent and therefore pro-apoptotic effects of infliximab on peripheral blood mononuclear cells (PBMCs) and T cells. In contrast, the anti-TNFα compound etanercept, a recombinant TNFR2:Fc fusion protein, failed to induce apoptosis in peripheral and lamina propria lymphocytes9 and also did not induce clinical remission in patients with CD.10 Therefore, induction of apoptosis was thought to be a critical mechanism of action of anti-TNFα therapy in active CD.9 ,11 ,12 This theory has been questioned since certolizumab pegol (CZP), a pegylated Fab-fragment without IgG-Fc, has been shown to be clinically effective in CD but does not induce apoptosis.11 ,13 Taken together, the previous studies suggest a complex intracellular signalling cascade downstream of the engagement of mTNFα by anti-TNFα compounds.
We systematically compared transcriptomal signatures elicited by the therapeutic TNFα-binding molecules CZP and IFX in myelomonocytic cells to characterise unique and shared molecular mechanisms that may help to explain the clinical efficacy of these agents in the therapy of CD. Whole genome expression screening in THP-1 cells, a human monocytic leukemia cell line, revealed 20 transcripts, which are significantly coregulated by IFX and CZP. Among these we characterised the growth and differentiation factor 1 (GDF-1), a member of the TGFβ superfamily, as a novel proinflammatory mediator, which is regulated by anti-TNFα agents.
Materials and methods
Study population
There were four different patient cohorts in this study. Samples were categorised by the following abbreviations: hospitalised normals (HN), patients with CD and no inflammation (CD_ni), patients with CD and active inflammation (CD_i), patients with UC and no inflammation (UC_ni), patients with UC and inflammation (UC_i) and disease control patients with unspecific intestinal inflammation (DC_i).
Patient cohort I includes 109 patients: 85 patients with IBD and 24 controls for real-time PCR (RT-PCR) quantification experiments with approximately half of the samples taken from active disease in the patient groups (for detailed characterisation of all patients, see online supplementary table 1). The normal control group (HN) included 24 subjects without abnormal endoscopy findings. Group II (20 patients: 5 HN, 5 CD_i, 5 UC_i and 5 DC_i) and group III (23 patients: 3 HN, 5 CD_I, 5 CD_ni, 5 UC_i and 5 UC_ni) had ELISA and western blot analyses.
Group IV included 13 patients with CD with biopsies taken directly before and 1 week after infliximab therapy. Patients who showed a clinical response to IFX were defined as patients whose CD activity index (CDAI) decreased at least 70 CDAI points 2 weeks after IFX infusion, similar to seminal clinical trials.3 ,14 Of this group, eight patients were classified as clinical responders and five were classified as clinical non-responders to IFX treatment.
Biopsies were taken from the sigmoid and active disease was defined by respective disease activity indices (CDAI >150, colitis activity index (CAI) >4). Medication included 5-aminosalicylic acid, azathioprine or glucocorticoids (<25 mg) according to the clinical requirements of the patients, but not an active biological therapeutic regime. The endoscopies were part of regular patient management. All patients agreed to participation by giving informed consent at least 24 h before the procedure and the study was granted prior approval by the local ethics committee.
Cell culture, transfection and reagents
All analysed cell lines were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). PBMCs were isolated from 100 ml of peripheral blood drawn from healthy volunteers and cultured as described previously.15
For stimulation experiments, the following main stimuli were used: a monoclonal mouse-human chimeric anti-TNFα antibody IFX (Remicade; Centocor, Horsham, Pennsylvania, USA), an irrelevant pegylated (Fab')2 fragment and a pegylated (Fab')2 fragment of a humanised monoclonal antibody CZP (CDP870; Cimzia; UCB, Belgium), recombinant human TGFβ (Biosource, Invitrogen, Carlsbad, California, USA) or recombinant human GDF-1 (H00002657-P01-10, Abnova, Heidelberg, Germany). An irrelevant human IgG1 antibody (Alpha Diagnostic International Inc, San Antonio, Texas, USA) served as the control antibody.
Microarray quantification of gene transcripts
THP-1 cells were plated at 1×106/2 ml, grown for 24 h and incubated for 6 and 24 h with IFX or CZP (both 10 μg/ml) or were left untreated. Colonic biopsies from anti-TNF naïve CD patients prior to or after treatment with IFX were collected and snap frozen as previously described.16 Total RNA was isolated from THP-1 cells as well as from colonic biopsies according to the manufacturer's instruction (QIAGEN Inc, Valencia, California, USA). RNA integrity was verified using an Agilent Bionanalyzer (Agilent, Böblingen, Germany) according to the manufacturer's guidelines. Microarrays (Affymetrix HG U 133 plus 2.0; Affymetrix, Santa Clara, California, USA) were prepared and processed as previously described.17 After acquiring the data using Affymetrix GeneChip Command Console (AGCC), data were normalised using the robust multiarray averaging (RMA) method (R, Bioconductor). For microarray analysis of THP-1 cell samples, differential expression was determined using three filter criteria: transcripts had to be present in all samples of at least one experimental group; as a relative measure for significance, we employed a rank-sum test, allowing a maximum of one outlier per measurement (for the two controls vs three treatment samples comparison presented here, a rank sum difference below seven corresponds to two or more outliers, a rank sum difference of seven corresponds to one outlier, while a rank sum difference of nine corresponds to no outliers); and the fold change, which was calculated based on the ratios of the medians, had to be either bigger than 1.5 or smaller than −1.5. Genes meeting these criteria were subjected to further analysis, which was carried out using TIBCO Spotfire (TIBCO, Palo Alto, California, USA).
The microarray data (raw and normalised) were processed according to minimal information about a microarray experiment (MIAME) guidelines and submitted to Gene Expression Omnibus18 and are accessible through GEO Series accession number GSE33585 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33585).
Expression data for GDF-1 was obtained from a previously published dataset in which data were normalised using the RMA method.16 Differential expression of GDF-1 within this dataset was determined using the Mann–Whitney U test.
mRNA isolation and reverse-transcription PCR
Total RNA was isolated from collected cell pellets using the RNeasy kit (QIAGEN Inc) following the manufacturer's instructions. Reverse transcription was performed by using Advantage-RT-for-PCR (Clontech, Palo Alto, California, USA) and expression of target genes and the reference transcript G3PDH was assayed by using standard PCR procedures15 and sequence-specific primers (online supplementary table 3). PCR reactions were amplified using a thermo cycler (Gene Amplification PCR System 9700, Perkin Elmer, Applied Biosystems, Foster City, California, USA). For each gene, the number of cycles was chosen directly above the detection threshold.
TaqMan real-time PCR
GDF-1/LASS1 mRNA transcript levels were measured using quantitative RT-PCR. cDNA was arrayed on 384-well plates using the expression assay Hs00242151_m1 (Applied Biosystems), which detects exon boundary 3–4 in the open reading frame of GDF-1/LASS1 on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems) according to the manufacturer's protocols. Relative transcript levels were determined using the standard curve quantisation method and β actin as the endogenous control gene.
Determination of viable cell mass
The CellTiter 96 non-radioactive cell proliferation assay based on the reduction of MTS (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) by living cells was performed with THP-1 and PBMCs (20 000 cells/well/100 μl in 96-well microtitre plate) according to the manufacturer's instruction (Promega Corp, Madison, Wisconsin, USA).
Caspase-Glo 3/7 assay
THP-1 cells and PBMCs were cultivated and stimulated with IFX or CZP in a microtitre plate (96 wells) and analysed using the Caspase-Glo 3/7 assay according to the manufacturer's instructions (Promega Corp).
SDS-PAGE and immunoblotting
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting experiments with whole protein lysates isolated from cell lines or colonic biopsy samples were performed as previously described (for antibodies see online supplementary table 4).15
ELISA
Supernatants of cell cultures were collected for measurement of secretion of IL-1β and IL-6 by specific ELISA (BioSource Europe S.A., Nibelles, Belgium) according to the manufacturer's protocol.
For human GDF-1 and TGFβ ELISA, colonic biopsy samples were collected from patients with CD and UC (n=20), and from hospitalised patients (n=3) and lysed according to the manufacturer's instructions using native lysis buffer (Cell Signaling Technology, Inc, Danvers, Massachusetts, USA). ELISA experiments were performed according to the manufacturer's instructions (GDF-1 ELISA, USCN Life Science Inc, Wuhan, China; TGFβ ELISA, Invitrogen).
siRNA-mediated knockdown of ACVR1C
HEK293T cells were seeded at a density of 1×105/well in six-well plates. Next day, transfection of cells with 25 nM ACVR1C-specific small interfering RNA (siRNA) or 25 nM of negative control siRNA for 24 h was carried out using Lipofectamine2000 (Invitrogen) according to the manufacturer's manual. PBMCs were transfected with 300 nM siRNA for 24 h by electroporation using 1×107 cells/sample and the Amaxa Human T cell Nucleofector Kit (Lonza, Walkersville, Maryland, USA) according to the manufacturer's instructions. Synthetic siRNA targeting ACVR1C was purchased from Applied Biosystems/Ambion (Foster City, California, USA). Target sequences were as follows: ACVR1C siRNA (ID s43499) sense 5′-GGUCCUUAUAUGACUAUUtt-3′ and antisense 5′-AAUAGUCAUAUAAGGAGCCct-3′. As control, unspecific siRNA Negative Control #1 (Applied Biosystems/Ambion) was used.
Representation of data and statistical analysis
Statistical significance was determined by the Mann–Whitney U test or the Wilcoxon matched pairs test (only figure 5) using GraphPad Prism 5.0 software. Results were displayed as means ± SD. A p-value≤0.05 was considered statistically significant (*) and p≤0.01 was considered highly significant (**). Experiments and measurements were replicated at least three times.
Results
Whole genome expression analysis
To systematically analyse cellular responses initiated by IFX and CZP in THP-1 cells a systematic expression analysis using oligonucleotide microarrays was performed.
Using rank-sum-based statistics, a total of 1058 transcripts were regulated in which 1684 regulatory events were observed (ie, some transcripts were regulated in more than one condition). In detail, 979 transcripts were regulated in response to IFX (99 upregulated, 880 downregulated), 99 transcripts were regulated in response to CZP (17 upregulated, 82 downregulated), 959 transcripts were regulated only by IFX (98 upregulated, 861 downregulated), 79 transcripts were regulated by CZP only (16 upregulated, 63 downregulated) and 20 transcripts were regulated by CZP and FIX (1 upregulated, 19 downregulated). A more detailed list of the regulated transcripts is presented in online supplementary table 2, while the 20 transcripts regulated by both CXP and IFX are presented in online supplementary table 5. The top 50 regulated genes for IFX only, IFX and CZP and for CZP only are presented in figure 1. Only among the IFX-regulated transcripts was a cluster of apoptosis-associated transcripts detected, which confirms the pro-apoptotic action of IFX detected in previous studies. Analysis of this cluster revealed a downregulation of apoptosis-inhibitory genes (eg, CIAPIN1, API5)19 ,20 and an upregulation of apoptosis-promoting genes (eg, PDCD4, PDCD6, BTG1, BIRC4BP, BNIP3L)21–25 (table 1). Differential induction of pro-apoptotic and anti-apoptotic signalling events by IFX and CZP in THP-1 was validated in PBMCs by RT-PCR, MTS and Caspase Glo assays. As shown in online supplementary figure 1, API5 was upregulated by CZP (6 and 24 h) but not by IFX treatment (online supplementary figure 1A). Furthermore, MTS assays revealed that IFX led to a marked reduction of cell viability in THP-1 and PBMCs. Using this method, CZP also elicited a slight but significant reduction of cell viability in PBMCs, while no effect on THP-1 cells could be observed (online supplementary figure 1B–D). Using a luminometric assay for caspase activation, IFX, but not CZP, activated caspases 3/7 in both cell types (online supplementary figure 1E,F).

Whole genome expression analysis. THP-1 cells, a human monocytic cell line, were stimulated with infliximab or certolizumab pegol (both 10 μg/ml) for 6 and 24 h or were left untreated (control). Three different comparisons are presented: transcripts, regulated when comparing infliximab with control (A), certolizumab pegol with control (B), and transcripts regulated by both stimuli (C). For (A) and (B) the top 50 regulated transcripts are presented (selected by fold change and significance). Columns represent samples while each row represents a transcript. Where no gene symbol was available, the public reference was listed in brackets. Transcripts are ordered according to their expression similarities (correlation). The heatmap displays relative expression intensities (z-score normalised). Arrows to the right of each heatmap group the transcripts into regulation categories.
Overview of apoptosis-associated transcripts regulated by infliximab (IFX)
Importantly, a signature of 20 shared transcripts was identified, which were significantly regulated by IFX and CZP (figure 1C). Among these, two were found to encode unknown proteins (LOC284702, LOC440944) and two other putative transcripts were not covered by spliced ESTs (expressed sequence tags) corresponding to the reported oligonucleotide probe. Twelve of the remaining 16 transcripts could be categorised into the following main cellular processes: cell cycle regulation (PRDX6, NACA, UHMK1, C13orf25, GDF-1/LASS1), mRNA processing (RNPS1, SFRS7, HNRPA1, TncRNA), transcription activation and nuclear transport (KPNA3, HNRPA1, ANKRD12, ARHGEF19), whereas the last four transcripts are part of the ribosomal 60S subunit (RPL17, RPL37A) or belong to the solute carrier family (SLC16A10 and SLC16A3) (online supplementary table 5).
To verify these results, cDNA was prepared from THP-1 and PBMCs stimulated with IFX or CZP. The mRNA data for THP-1 cells (GDF-1/LASS1, ARHGEF19, NACA, UHMK1 and PRDX6) and for PBMCs (c13orf25, UHMK1, ARHGEF19, KPNA3, NACA, PRDX6, RNPS1) in RT-PCR experiments also reflected data received from microarray expression study (online supplementary figure 2A,C). To exclude the effects of IgG1 or pegylated structures, THP-1 cells were stimulated with irrelevant IgG1 or pegylated Fab fragment for 24 h. cDNA was analysed by RT-PCR with regard to GDF-1/LASS1 expression. No regulation could be observed (online supplementary figure 2B).
GDF-1 is expressed ubiquitously in different tissues
GDF-1/LASS1 was selected for follow-up, as the protein GDF-1 represents the only soluble factor in the list of commonly regulated transcripts. This renders the protein generally accessible to future neutralisation studies, for example, by specific antibodies. Moreover, GDF-1 belongs to the TGFβ superfamily, members of which have been broadly implicated both in the etiopathogenesis of IBD and as important mediators of the mechanisms of action (MoA) of anti-TNF compounds.8 ,26
GDF-1 is transcribed into a bicistronic mRNA together with LASS1, the human orthologue of longevity assurance factor 1 from Saccharomyces cerevisiae. In addition to the bicistronic transcript variant, a second monocistronic variant encoding only LASS1 has been identified (online supplementary figure 2C). Data for the Probe-ID 229448_x_at that only detects the monocistronic variant of LASS1 mRNA were compared with data received from the Probe-ID 206397_x_at that detects the bicistronic mRNA variant GDF-1/LASS1. It could be shown that only the bicistronic mRNA variant of GDF-1/LASS1 was regulated by IFX/CZP, whereas no alteration of LASS1 mRNA expression could be detected. Comparison of received data sets revealed a very low detection signal for Probe-ID 229448_x_at (online supplementary figure 2D).
cDNA from a human tissue panel was used for mRNA expression analysis of GDF-1/LASS1 in different tissues. GDF-1/LASS1 transcript was ubiquitously expressed (online supplementary figure 3A) with highest expression in small intestine, brain, testis and skeletal muscles.
As GDF-1/LASS1 are translated from a single bicistronic mRNA, we investigated protein levels of GDF-1 and LASS1 in relevant cell lines to determine translation efficacy of the two putative ribosomal entry sites. GDF-1, which is initially translated as a proprotein and processed by proteolytic cleavage to yield the mature protein (figure 2A), was expressed in all examined cell lines with the highest expression in HEK293, HepG2 and Caco-2 (online supplementary figure 3B). Moderate protein expression was found in HeLa, SW480, HT-29 and THP-1. While an unclear protein band, corresponding to the predicted molecular weight of LASS1 (39 kDa), was found in cell lines, virtually no protein band was detectable in lysates from intestinal biopsies of HN, patients with IBD and DC (online supplementary figure 3B,C).

Growth and differentiation factor 1 (GDF-1) expression analysis. (A) Human GDF-1 proprotein and GDF-1 mature protein. The presented models illustrate processing of GDF-1 precursor to yield mature protein. Homology models were generated using GDF-1 amino acid sequence (GenBank AAB94786.1) and SWISS-Model programme software.27 (B) GDF-1/LASS1 upregulation in colonic biopsies from patients with Crohn's disease (CD). Real-time PCR was performed with cDNA derived from sigmoidal biopsies (n=150). Medians of data for subgroups (HN n=30, CD_ni n=31, CD_i n=29, UC_ni=29, UC_i n=31) are presented as black bars. (C) Western blot analysis of GDF-1 protein upregulation in colonic biopsies from patients with CD. Densitometric analysis of the blot was performed with ImageJ. (D) GDF-1-specific ELISA was performed with colonic biopsies from inflamed and non-inflamed tissue from patients with CD and ulcerative colitis (UC) (each n=5). CD_i, Crohn disease inflamed; CD_ni, Crohn's disease non-inflamed; UC_i, ulcerative colitis inflamed; DC_i, diseased controls inflamed; HN, hospitalised normals; *p≤0.05; **p≤0.01.
GDF-1 expression is upregulated in inflamed biopsies from patients with CD
We next investigated by RT-PCR whether GDF-1/LASS1 is differentially regulated in patients with CD (n=35) or UC (n=50) compared with HN (n=24). Normalisation of GDF-1/LASS1 data against β-actin data and a statistical analysis indicated a significant upregulation in CD_inflamed samples compared with tissues from HN and patients with UC (figure 2B).
To verify the obtained results at the protein level, protein extracts isolated from colonic biopsies (n=20) were applied to SDS-PAGE and immunoblotting. GDF-1 protein was upregulated in CD_inflamed samples when compared with UC_inflamed (p≤0.01) and DC_inflamed (p≤0.05) samples (figure 2C). In an additional set of colonic biopsies taken from patients with CD and UC (each n=10) using ELISA we show a similar pattern of upregulation in inflamed tissue from patients with CD compared with CD non-inflamed and UC tissue (irrespective of inflammation state; figure 2D), although the findings do not reach formal significance levels.
GDF-1 expression is regulated by TGFβ-mediated signalling
As previous findings demonstrate that mTNFα ligation by anti-TNFα agents induces elevation of TGFβ secretion, a putative influence of TGFβ-mediated signalling transduction on GDF-1 expression was analysed.8
PBMCs were stimulated with IFX/CZP for 6 or 24 h or were left untreated and mRNA levels of TGFβ and GDF-1/LASS1 were determined by RT-PCR. A reciprocal time-dependent upregulation of TGFβ mRNA and downregulation for GDF-1/LASS1 mRNA was observed (figure 3A). In contrast, stimulation of THP-1 monocytes and PBMCs with TGFβ for 12 h led to downregulation of GDF-1/LASS1 mRNA expression in a concentration-dependent manner. TGFβ mRNA expression was not altered by TGFβ stimulation (figure 3B). These findings were also reflected at the protein level for precursor and mature GDF-1 in THP-1 (figure 3C).

Detection of influence of transforming growth factor β (TGFβ)-induced signal transduction on GDF-1/LASS1 expression. (A) Inverse regulation of GDF-1/LASS1 and TGFβ by infliximab (IFX) and certolizumab pegol (CZP). Peripheral blood mononuclear cells (PBMCs) were stimulated with IFX or CZP (both 10 μg/ml) for 6 or 24 h. Real-time PCR (RT-PCR) experiments were performed with cDNA and sequence-specific primers. (B) Downregulation of GDF-1/LASS1 mRNA by recombinant human TGFβ (rhTGFβ) signalling transduction. Stimulation of cells with rhTGF-β was performed for 18 h. mRNA expression was analysed by RT-PCR. (C) Western blot analysis of GDF-1 protein regulation by rhTGFβ signaling. THP-1 was stimulated with rhTGFβ (1 ng/ml, 10 ng/ml) for 1 and 4 h. (D) Reversion of IFX/CZP-induced effects by TGFβ blockade. THP-1 cells were stimulated with IFX and CZP (both 10 μg/ml) for 6 and 24 h in the presence or absence of anti-TGFβ antibody (1 μg/ml). mRNA expression was analysed by RT-PCR. (E) TGFβ-specific ELISA was performed with colonic biopsies from inflamed and non-inflamed tissue from patients with Crohn's disease (CD) and ulcerative colitis (UC) (each n=5) as well as from hospitalised normals (n=3). CD_i, Crohn's disease inflamed; CD_ni, Crohn's disease non-inflamed; DC_i, diseased controls inflamed; UC_i, ulcerative colitis inflamed; GDF-1, growth and differentiation factor 1; HN, hospitalised normals; **p≤0.01.
To further study the role of TGFβ-mediated signalling transduction as a potential autocrine regulator of GDF-1 after mTNFα ligation, we analysed GDF-1/LASS1 mRNA levels in THP-1 after stimulation with IFX and CZP for 6 or 24 h in the presence or absence of anti-TGFβ antibody. Pre-incubation of THP-1 with anti-pan TGFβ antibody (1 μg/ml) for 30 min prior to IFX stimulation enhanced GDF-1/LASS1 mRNA expression in a time-dependent manner. The presence of anti-pan TGFβ antibody abolished the repressive effects of both anti-TNFα compounds on GDF-1/LASS1 mRNA expression levels (figure 3D).
TGFβ expression was analysed in inflamed and non-inflamed colonic biopsy samples from patients with CD and UC and from healthy normals by TGFβ-specific ELISA experiments. Significant upregulated TGFβ expression was found in inflamed UC tissue compared with non-inflamed UC tissue. No regulation could be detected for CD tissue in comparison to UC or HN samples (figure 3E).
GDF-1 acts as a proinflammatory cytokine by regulating IL-6
Finally, we studied the influence of recombinant human GDF-1 (rhGDF-1) protein on inflammatory responses in PBMCs.
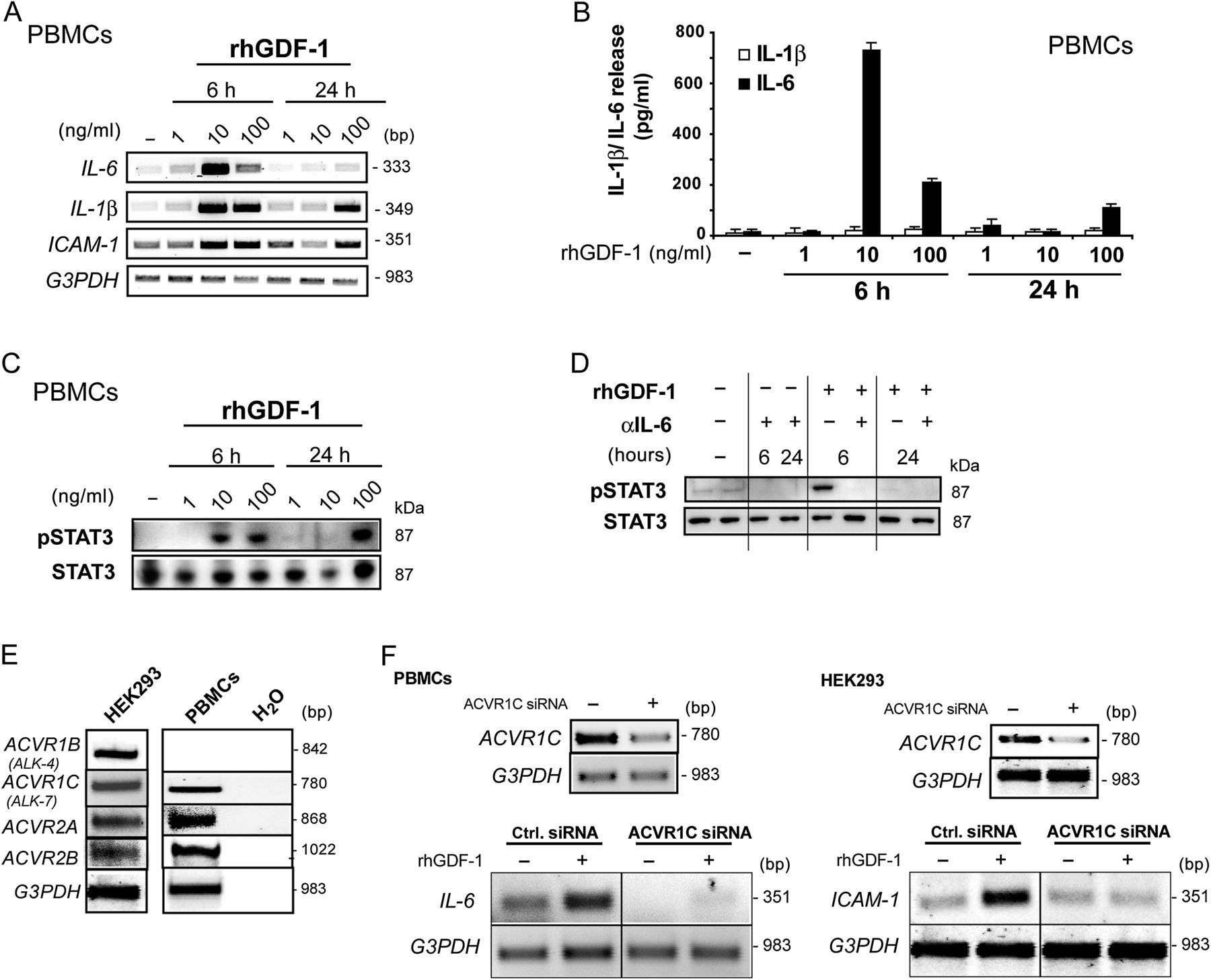
Cells were stimulated for 6 and 24 h with various concentrations of rhGDF-1. mRNA expression of the proinflammatory cytokines IL-6, IL-1β and the inter-cellular adhesion molecule 1 (ICAM-1) was upregulated in a time-dependent manner with peaking after 6 h of stimulation (figure 4A). Upregulation of IL-6 transcript levels was accompanied by an increased IL-6 secretion as assessed by ELISA, whereas no secretion of mature IL-1β was observed (figure 4B).

Regulation of interleukin 6 (IL-6) via recombinant human growth and differentiation factor 1 (rhGDF-1). Peripheral blood mononuclear cells (PBMCs) were stimulated with rhGDF-1 (1–100 ng/ml) for 6 or 24 h or left untreated before being analysed by (A) real-time PCR (RT-PCR), (B) IL-6 and IL-1β-specific ELISA or (C) western blot. (D) Western blot analysis of signal transducer and activator of transcription 3 (STAT3) activation by rhGDF-1 depended on IL-6 signalling. PBMCs were stimulated with rhGDF-1 for 6 or 24 h in the presence or absence of anti-IL-6 antibody (1 μg/ml). (E) Expression analyses of ACVR1B, ACVR1C, ACVR2A or ACVR2B in PBMCs and HEK293T cells were performed by RT-PCR. All four receptors could be detected in both cell lines except for ACVR1B in PBMCs. (F) RNA interference-induced knockdown of ACVR1C. PBMCs (left panel) or HEK293T cells (right panel) were transfected with an ACVR1C-specific small interfering RNA (siRNA) or a negative control siRNA. Subsequently, cells were stimulated with rhGDF-1 (10 ng/ml) for 6 h or were left untreated. RT-PCR was performed using transcript-specific primers. ICAM-1, inter-cellular adhesion molecule 1.
To characterise signalling events underlying the proinflammatory effect of GDF-1 we analysed the activation of several transcription factors. While activation of transcription factor Smad2/3 was only induced after 6 h of stimulation of PBMCs with rhGDF-1 (online supplementary figure 4), we detected a strong activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) as assessed by phospho-specific western blot (figure 4C).
As IL-6 activates STAT3 phosphorylation via binding to IL-6R and gp130, it was analysed whether the observed STAT3 phosphorylation depends on autocrine IL-6 secretion induced by rhGDF-1. PBMCs were stimulated with rhGDF-1 in the presence or absence of an anti-IL-6 antibody for 6 and 24 h. Stimulation with rhGDF-1 for 6 h induced activation of STAT3 that was completely prevented by pre-incubation of cells with anti-IL-6 antibody for 30 min prior to rhGDF-1 stimulation (figure 4D). To unravel which receptor is engaged by GDF-1 signalling, expression analyses of known GDF-1-specific receptors ACVR1B and ACVR1C as well as of the unspecific receptors ACVR2A and ACVR2B were performed by RT-PCR using PBMCs and HEK293 cells serving as an additional model system. As demonstrated in figure 4E, all four receptors were expressed in HEK293 cells, while in PBMCs expression of ACVR1C, ACVR2A and ACVR2B but not of ACVR1C could be detected. Therefore, knockdown of ACVR1C in PBMCs and HEK293T cells was induced by an ACVR1C-specific siRNA before stimulation of cells for 6 h with 10 ng/ml rhGDF-1. Finally, mRNA expression of ACVR1C, IL-6 or ICAM-1 was analysed by RT-PCR. In both PBMCs and HEK293 cells, GDF-1 induced upregulation of IL-6 or ICAM-1 mRNA expression was prevented by RNA interference (RNAi)-mediated knockdown of ACVR1C (figure 4F).
Downregulation of GDF1/LASS1 expression by IFX therapy is restricted to IFX responders
To underline the role of GDF-1 as an important mediator of MoA of IFX and CZP in vivo, we investigated GDF-1 levels using a microarray dataset from colonic biopsy samples of anti-TNF naïve patients with CD prior to and 1 week after IFX therapy. Data were categorised into responders and non-responders. Clinical responders to IFX were defined to be patients whose CDAI decreased at least 70 CDAI points after 1 week of IFX treatment. As presented in figure 5A, GDF-1/LASS1 was significantly downregulated after IFX therapy in IFX responders but not in IFX non-responders, whereas the proinflammatory marker transcript ICAM-1 only showed a moderate trend towards lower transcript levels (see figure 5B).

Regulation of GDF-1/ LASS1 by infliximab (IFX) therapy of patients with Crohn’s disease (CD). cDNA from CD colonic biopsy samples collected prior and after IFX treatment were analysed by microarray analysis. (A) GDF-1/LASS1 expression (Probe Set ID 229448_at) was significantly downregulated after IFX treatment only in responders (n=8) but not in non-responders (n=5). (B) Inter-cellular adhesion molecule 1 (ICAM-1) (Probe Set ID 202637_s_at) expression was also found to be downregulated in responders (n=8) but not in non-responders (n=5) after IFX treatment but failed to reach statistical significance. GDF-1, growth and differentiation factor 1.
Discussion
TNFα antagonists are able to bind to mTNFα and activate a physiological reverse signalling cascade. We investigated transcriptome regulation by two TNFα-binding molecules that are therapeutically active in CD. By determining the communality in regulatory mechanisms elicited by IFX and CZP we aimed to uncover essential molecular principles of MoA triggered by these two agents.
A systematic microarray-based expression analysis in THP-1 demonstrated that signalling of IFX and CZP, respectively, induces distinctly different transcriptomal responses. Commonalities include downregulation of transcripts involved in cell cycle progression. This finding corresponds to previous studies in which IFX treatment was connected to inhibition of proliferative processes and the induction of cell cycle arrest in different subsets of human cells.26 ,28 Both molecules have also been found to interfere with the nuclear factor κB (NF-κB) pathway or other transcriptional activators to diminish a sustained transcription activity as detected in active CD.29 Suppression of sustained NF-κB activation in intestinal biopsies of patients with CD has been previously shown after medical treatment with IFX that induced upregulation of the NF-κB inhibitor proteins IκBα and IκBγ.2 ,30 The data further suggest a link of IFX and CZP to the control of pre-mRNA processing and transport. Currently, little is known about inflammatory conditions and pre-mRNA splicing events. However, alterations in the morphology of nuclear speckles and therefore pre-mRNA splicing has been detected during inflammatory stimulation of colonic epithelial cell lines31 and a first report has shown that splicing patterns may be systematically altered in IBD.32 In conclusion, IFX and CZP target fundamental processes (cell cycle, transcription activation, pre-mRNA processing) most likely through mTNFα reverse signalling that are pivotally involved in perpetuation of chronic inflammatory responses in CD.
Among the shared transcripts that were regulated by both anti-TNFα compounds, we selected the downregulated bicistronic transcript GDF-1/LASS1 for a detailed functional follow-up.
In the present study, we detected moderate expression levels for GDF-1/LASS1 in the adult colon and high constitutive expression in the adult small intestine. We demonstrate a highly significant upregulation of mRNA levels as well as a tendency for upregulation of protein levels of the TGFβ superfamily member GDF-1 in the inflamed colonic mucosa of patients with CD. While the reduced dynamics of the observed GDF-1 regulation on the protein level may in part be explained by the fact that secreted proteins are sometimes harder to detect in complex intestinal tissue samples, the findings still suggest a potential role of GDF-1 in inflammatory processes in CD.
Furthermore, our data point to a negative correlation of expression levels of GDF-1 and the main family member TGFβ in monocytic cells (ie, THP-1 and PBMCs). It is generally accepted that inflammatory signalling events induced by proinflammatory cytokines like TNFα and IL-6 are counteracted by the anti-inflammatory cytokine TGFβ. By binding to its receptors (TGFβ RII and activin receptor-like kinases 1/2/5), TGFβ activates main downstream targets, for example Smad2/3 and MAPKs, to induce antiproliferative processes.33 However, TGFβ has been linked to some cancer types and autoimmune diseases.33 ,34 This study is the first to report in monocytes that ACVR1C (ALK-7)-mediated signalling transduction triggered by GDF-1 leads to a significant induction of proinflammatory cytokines and indirect activation of the transcription factor STAT3. Absence of IL-6 secretion after 24 h of stimulation with GDF-1 might point to a complex network of cellular signalling cascades, such as initiation of negative feedback loops, that controls excessive activation of signalling pathways and in that way counteracts with GDF-1 signalling cascade. It must be further noted that it could not be excluded that the second GDF-1-specific receptor ACVR1B might play an important role in GDF-1 signalling in other cellular contexts. We identified the proinflammatory cytokine IL-6 to be a major autocrine amplifier of GDF-1 signalling, as STAT3 activation downstream of GDF-1 could be prevented by a blocking IL-6 antibody.
IL-6 is a pleiotropic cytokine that induces inflammatory and proliferative processes.35 Sustained IL-6 production and therewith gp130-mediated activation of STAT3 have been linked to proliferative and survival effects in normal and premalignant intestinal epithelial cells and therefore are involved in tumourigenesis.36 Several publications also discussed the function of IL-6 trans-signalling via gp130 and STAT3-induced resistance to apoptosis in IBD,37 while blocking of membrane-bound and sIL-6R has been found to diminish disease activity in patients with CD.38
In intestinal epithelial cells, TGFβ has been shown to inhibit the IL-6/STAT3 signalling pathway via Smad2 activation.39 Activation of the TGFβ downstream target Smad2/3 is blocked by the upregulation of its inhibitor Smad7.40 Vice versa, in murine models of colitis, treatment with Smad7 antisense oligonucleotides initiates restoration of TGFβ signalling and therefore diminishes inflammation.41 Hyperactivation of STAT3 in murine models is accompanied by the activation of Smad742 and thus prevents anti-inflammatory signal transduction by TGFβ.
We further detected in this study that both IFX and CZP suppress the expression of GDF-1 via a TGFβ-dependent autocrine signalling loop in vitro. However, TGFβ-dependent paracrine signalling cannot be excluded. Previous studies also linked the anti-inflammatory action of TNFα antagonists, in particular IFX, to the secretion of TGFβ8 ,26 and interestingly to the inactivation of STAT3 in activated intestinal CD4+ T cells.43 Hence, it might be speculated that anti-TNFα treatment in active CD may reverse the imbalance between Smad2/3 and Smad7 by increasing TGFβ release, therefore allowing TGFβ to completely unfold its anti-inflammatory action (figure 6). It must be noted that a complex regulation of mucosal TGFβ levels has been described in IBD in different cell types40 On the whole biopsy level, some studies demonstrated excessive production of TGFβ in inflamed IBD tissues, while others suggested lesser TGFβ secretion by lamina propria mononuclear cells (LMPCs) isolated from patients with CD when compared with UC samples.40 ,44 Interestingly, whereas we detected a significant upregulation of GDF-1 levels in mucosal biopsies in IFX responders, we observed no significant regulation in TGFβ expression after IFX treatment in the same patients (data not shown). Taken together with the fact that TGFβ levels may not reflect the biological activity of the cytokine in the tissue (eg, as it has to be activated by proteolytic cleavage or due to Smad7 dysregulation) a more detailed investigation of cell-type specific regulatory events of this pathway after anti-TNF therapy is still warranted. In this study we have based the responder/non-responder categorisation on changes in a validated clinical activity index (CDAI) that has been used in previous anti-TNF clinical trials.3 ,14 Given the role of TGFβ in regeneration and tissue repair, it will be interesting to study the detailed role of GDF-1 and TGFβ in mucosal healing in this setting. Our functional data suggest that GDF-1 may serve as an additional layer of complexity that has to be taken into account.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
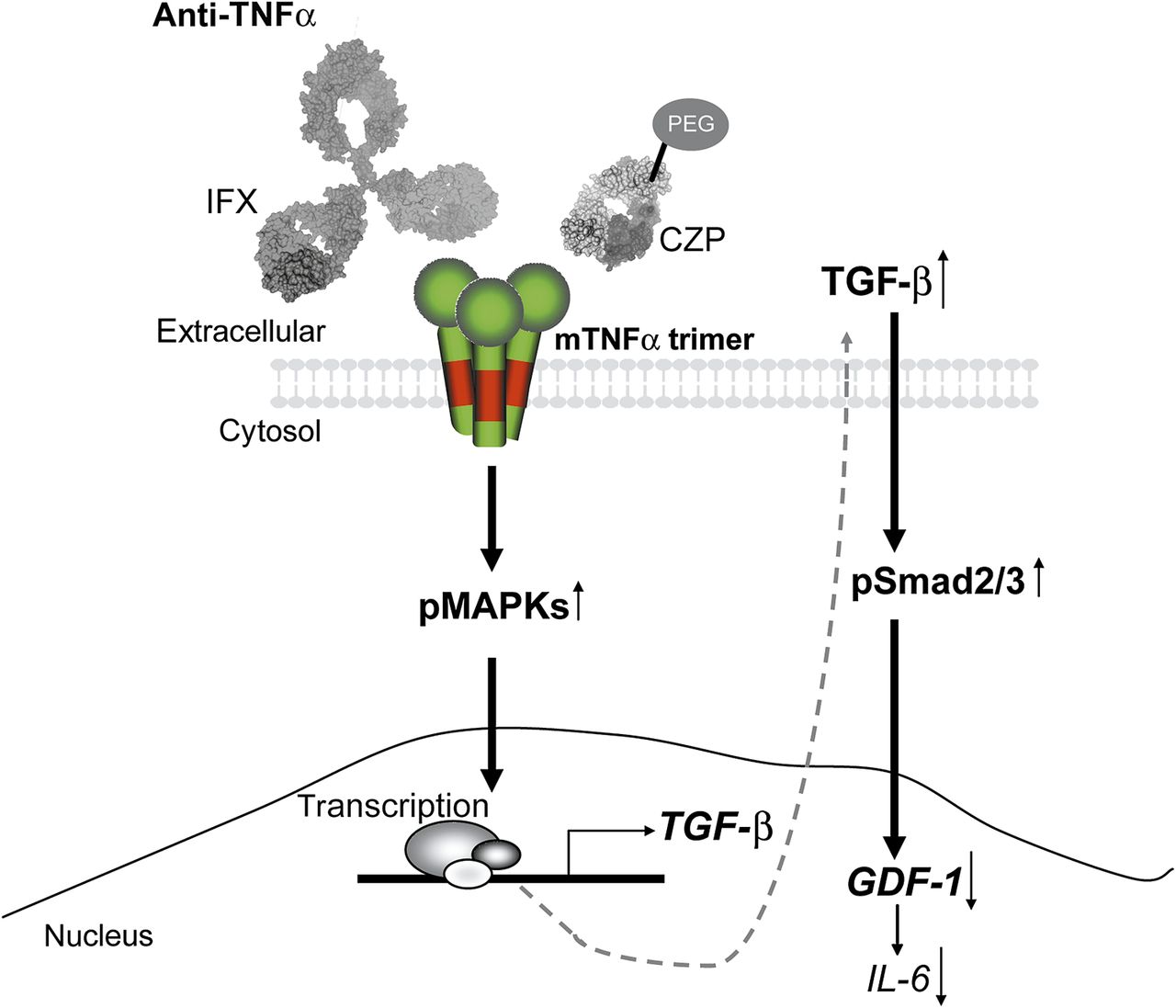
Model of membrane-bound tumour necrosis factor α (mTNFα) reverse signalling via infliximab (IFX)/certolizumab pegol (CZP). TNFα antagonists induce mTNFα reverse signalling leading to mitogen-activated protein kinase (MAPK) activation and enhanced transforming growth factor β (TGFβ) secretion. TGFβ in turn exerts anti-inflammatory action through triggering Smad2/3-dependent downregulation of proinflammatory mediators like growth and differentiation factor 1 (GDF-1) and interleukin 6 (IL-6). PEG, pegylated; pMAPKs, phosphorylated MAPKs; pSmad2/3, phosphorylated Smad2/3.
In conclusion, this study for the first time presents converging alleys of signalling networks triggered by the TNFα antagonists IFX and CZP. One of their main targeted cellular processes seems to be the regulation of cell cycle and proliferation. Here, we reveal a completely new role of the TGFβ superfamily member GDF-1 as a proinflammatory cytokine involved in inflammatory processes. It might be hypothesised that, in vivo, GDF-1 induces chronic inflammation as found in CD via sustained activation of the IL-6/STAT3 pathway that may further suppresses the anti-inflammatory effects of TGFβ and hence promotes ongoing proliferation of cells.36 ,42 Furthermore, GDF-1 is downregulated in responders, but not in non-responders after IFX therapy. Accordingly, the mechanism of action of IFX and CZP in CD might in part be explained by interfering with GDF-1 and hence IL-6 production via a TGFβ-dependent autocrine signalling loop initiated by mTNFα reverse signalling. Studying the exact physiological role of GDF-1 in complex inflammatory processes in vivo may answer the question of whether this proinflammatory molecule could be a novel promising target in the therapy of patients with CD.
Acknowledgments
The authors thank the patients without whom this study would not have been possible. The endoscopy staff and the physicians Tanja Kaacksteen, Dorina Oelsner, Yasmin Brodtmann and Melanie Schlapkohl are gratefully acknowledged for their expert technical assistance. We thank Nancy Mah and the other authors of the IFX response study for providing GDF-1 mRNA expression data. Andrew Nesbitt (UCB) is acknowledged for his kind gift of pegylated Fab.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Gut's education editor Mairi McLean discusses the paper with Philip Rosenstiel and Stefan Schreiber in the Gut podcast.
Footnotes
PCR and SS share scientific responsibility and senior authorship.
Funding This work was supported by an unrestricted grant of UCB S.A. (Brussels, Belgium) and the clusters of excellence Inflammation at Interfaces and The Future Ocean and the SFB877 (to SR-J, PCR and SS).
Competing interests Individual conflicts of interest are stated in the ICJME forms for SS, SN, TK, SRJ and PR.
Ethics approval Ethics approval was provided by the local ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The authors agree to share the data.