Article Text

Abstract
Objective Although polymorphisms of the NOD2 gene predispose to the development of ileal Crohn's disease, the precise mechanisms of this increased susceptibility remain unclear. Previous work has shown that transcript expression of the Paneth cell (PC) antimicrobial peptides (AMPs) α-defensin 4 and α-defensin-related sequence 10 are selectively decreased in Nod2−/− mice. However, the specific mouse background used in this previous study is unclear. In light of recent evidence suggesting that mouse strain strongly influences PC antimicrobial activity, we sought to characterise PC AMP function in commercially available Nod2−/− mice on a C57BL/6 (B6) background. Specifically, we hypothesised that Nod2−/− B6 mice would display reduced AMP expression and activity.
Design Wild-type (WT) and Nod2−/− B6 ileal AMP expression was assessed via real-time PCR, acid urea polyacrylamide gel electrophoresis and mass spectrometry. PCs were enumerated using flow cytometry. Functionally, α-defensin bactericidal activity was evaluated using a gel-overlay antimicrobial assay. Faecal microbial composition was determined using 454-sequencing of the bacterial 16S gene in cohoused WT and Nod2−/− littermates.
Results WT and Nod2−/− B6 mice displayed similar PC AMP expression patterns, equivalent α-defensin profiles, and identical antimicrobial activity against commensal and pathogenic bacterial strains. Furthermore, minimal differences in gut microbial composition were detected between the two cohoused, littermate mouse groups.
Conclusions Our data reveal that Nod2 does not directly regulate PC antimicrobial activity in B6 mice. Moreover, we demonstrate that previously reported Nod2-dependent influences on gut microbial composition may be overcome by environmental factors, such as cohousing with WT littermates.
- INFLAMMATORY BOWEL DISEASE
- CROHN'S DISEASE
- BACTERIAL INTERACTIONS
- IBD - GENETICS
- INTESTINAL EPITHELIUM
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Polymorphisms of nucleotide-binding, oligomerisation domain 2 (NOD2) are the most frequently replicated disease alleles associated with Crohn's disease (CD).
-
NOD2 is highly expressed in Paneth cells (PC), which are small intestinal epithelial cells that secrete a multitude of antimicrobial molecules into the gut lumen. The most potent class of these antimicrobial peptides (AMP) is the α-defensins.
-
Previous work using Nod2-deficient mice suggests that Nod2 regulates PC α-defensin expression, which in turn modulates the composition of the gut microbiota.
What are the new findings?
-
When Nod2-deficient mice are examined on pure genetic background (C57BL/6), they show no differences in α-defensin levels relative to wild-type (WT) mice.
-
With the exception of the molecule cryptdin-related sequence 1C, expression of all major PC AMP classes are unaffected by Nod2 status in C57BL/6 mice.
-
The composition of the mouse faecal microbiota is independent of Nod2 status when WT and Nod2−/− littermates are housed in the same cages.
How might it impact on clinical practice in the foreseeable future?
-
This work guides future studies of CD pathogenesis by underscoring the importance of controlling for mouse genetic background and housing conditions when studying PCs in mouse models of CD risk alleles.
-
The finding that environmental influences can overcome the impact of host genetics on gut microbial composition suggests that microbial modulation strategies (such as probiotics) are feasible, even in the face of strong genetic influences on the intestinal microbiota.
Introduction
Crohn's disease (CD) is a chronic intestinal inflammatory disorder that results from the interaction of numerous genetic and environmental factors. Of the genetic loci associated with CD, single nucleotide polymorphisms (SNPs) of the NOD2 gene display the strongest correlation with disease development.1 Although NOD2 is found primarily in monocytes and dendritic cells, it is also constitutively expressed in small intestine epithelial cells known as Paneth cells (PC).2 PCs contribute to mucosal enteric immunity through the production of antimicrobial peptides (AMPs). The most potent class of PC-derived AMPs are the α-defensins (cryptdins).3 Genetic alterations that diminish PC α-defensin activity are associated with compositional changes in the intestinal microbiota.4 Because components of the gut microbiota are believed to drive intestinal inflammation in CD, dysregulation of α-defensin function may be an important factor in CD pathogenesis.5
Although the precise regulation of α-defensin activity remains unclear, various studies have suggested a role for NOD2 in modulating the function of these molecules. Previous reports have demonstrated that ileal CD patients possess attenuated expression of the two human PC α-defensins (HD-5 and HD-6),6 with the greatest reduction in patients with a specific NOD2 polymorphism.7 Subsequent work using a mouse model of Nod2-deficiency revealed that Nod2−/− mice also display reduced mRNA expression of particular α-defensins.8 Specifically, Nod2−/− mice exhibit ∼100-fold less transcript expression of the molecules α-defensin 4 (Defa4) and α-defensin-related sequence 10 (Defa-rs10) relative to wild-type (WT) mice.
Despite the evidence supporting the role of NOD2 in α-defensin regulation, these findings remain subject to a degree of uncertainty. First, a follow-up study in patients with ileal CD demonstrated that reduced α-defensin expression is associated with inflammation, as opposed to NOD2 status.9 Second, the precise background of the Nod2−/− mice used to implicate Nod2 as a regulator of α-defensin expression is unclear. This is critical, because recent work has demonstrated that mouse background strain plays a key role in defining α-defensin expression patterns.10 The Nod2−/− mice used in the original study were constructed using 129S1/Sv-derived W9.5 embryonic stem (ES) cells injected into C57BL/6 (B6) blastocysts, leaving open the possibility that these animals were on a mixed background at the time of analysis. Therefore, in light of the confounding variable of a mixed genetic background, the precise role of Nod2 in regulating mouse α-defensin expression remains unclear. Further characterisation of the impact of Nod2 on AMP expression is essential to define its precise function in PC biology, as well as its role in CD pathogenesis.
In the present study, we sought to determine the regulatory effects of Nod2 on mouse PC-derived AMP expression, controlling for background strain. This was accomplished by evaluating the PC AMP repertoire of Nod2−/− versus WT littermates on a pure B6 background. These animals are available commercially through Jackson Labs, and represent the original Nod2−/− strain8 that has since been backcrossed to completion on the B6 background. We hypothesized that these Nod2−/− B6 mice would display diminished AMP expression and attenuated PC function relative to their WT counterparts. Unexpectedly, we found that the majority of mouse PC-derived AMP classes showed equivalent expression in Nod2−/− and WT B6 mice. Furthermore, α-defensin antimicrobial activity and global faecal microbial composition were also strikingly similar in both Nod2−/− and WT groups. These findings suggest that PC antimicrobial function is independent of Nod2 status in this mouse strain.
Materials and methods
Mice
Nod2+/+ (WT) and Nod2−/− B6 mice were obtained from Jackson Laboratories (Bar Harbor, ME), and were housed in specific pathogen-free conditions consistent with guidelines established by the American Association for Laboratory Animal Care and Research. WT and Nod2−/− mice were crossed to generate Nod2+/− heterozygous animals, which were then bred to produce WT and Nod2−/− littermates that were housed together for comparison in our studies (see online supplementary figure S1). Importantly, Nod2+/− offspring were recrossed to generate a continuous pool of WT and Nod2−/− littermates, all derived ultimately from the same founders. This is in contrast with previous work, which used littermates from a single Nod2+/− heterozygous cross to start homozygous Nod2+/+ and Nod2−/− lines that were housed separately for the duration of the study.11 This distinction is critical, as the mice in our study were never housed separately based on Nod2 status alone, allowing for more consistent environmental exposure to both experimental groups. All mice were housed in the same animal room and sacrificed within 8–12-weeks of age. Nod2 genotyping was performed on genomic DNA extracted from mouse-tail clippings (see online supplementary methods). Identification of WT and Nod2-null mice was confirmed by PCR product sequencing (see online supplementary figures S2 and S3).
Quantitative reverse-transcriptase PCR
Quantitative RT-PCR was performed using TaqMan or SYBR Green assays (Applied Biosystems, Foster City, California, USA) per manufacturer's instructions. Details are described in the online supplementary methods. β-Actin was used as an internal control, and ΔΔCt values were calculated to obtain fold changes relative to the baseline group.
Acid urea polyacrylamide gel electrophoresis
Ileal tissue protein was extracted as previously described.10 Protein extracts were analysed by acid urea-polyacrylamide gel electrophoresis (AU-PAGE) followed by mass spectrometric analysis using matrix assisted laser desorption ionisation-time of flight tandem mass spectrometry (MALDI-TOF/TOF MS). Details are provided in the online supplementary methods.
Immunohistochemistry
Primary staining was accomplished using a rabbit polyclonal antilysozyme (Lyz) antibody (1:1500, Diagnostic BioSystems, Pleasanton, California, USA). Biotinylated antirabbit IgG was used as a secondary antibody (1:200, Vector Laboratories, Burlingame, California, USA). Details are provided in the online supplementary methods.
Flow cytometry
Ileal epithelial cells were isolated using EDTA/dispase digestion as previously described.12 Cells were fixed in 4% paraformaldehyde for 15 min and resuspended in saponin permeabilisation buffer (Invitrogen, Carlsbad, California, USA) with Lyz-FITC antibody (1:10, Dako, Carpenteria, California, USA) and CD45-A647 antibody (1:1000, BD Biosciences, San Jose, California, USA) for 30 min. Flow analysis was performed per established protocols.13
Bactericidal gel overlay assay
Bacterial strains were grown to mid-log phase in trypticase soy broth media and resuspended in warm 1% low-melt agarose as previously described.10 Ileal protein samples were prepared by electrophoresis on a small-scale AU-PAGE gel. Gel overlay plates were incubated overnight at 37°C and imaged for band-associated zones of bacterial clearance. Details are provided in the online supplementary methods.
Bacterial composition analyses
Total DNA was extracted from faecal samples as previously described.14 Bacterial composition of isolated DNA samples was characterised by PCR amplification of the V1-3 variable region of the 16S rRNA gene.15 Taxonomic and phylogenetic analyses of 16S rRNA sequence data are detailed in the online supplementary methods.16 ,17
Statistics
AMP expression and PC number comparisons were performed using GraphPad Prism 5 (GraphPad, San Diego, California, USA). All variables were found to have a normal distribution. Means were compared using Student's t test (2-tailed), and are expressed as mean±SEM.
Results
Mouse PC AMP expression is predominantly independent of Nod2
To determine the effects of Nod2 deletion on PC AMP gene expression, qRT-PCR was used to quantitate ileal AMP levels in WT and Nod2−/− B6 mice. The major PC AMP classes expressed in B6 mice include: α-defensins (cryptdins), cryptdin-related sequence peptides (CRS1C and CRS4C), lysozyme-P (Lyz), angiogenin-4 (Ang4) and regenerating islet-derived protein 3 γ (Reg3γ).18 Consistent with earlier reports in CD patients6 and Nod2−/− mice,19 we observed no differences in Lyz or Reg3γ expression between Nod2−/− and WT mice (figure 1A,B). Furthermore, Nod2-deficiency did not alter transcription of the Ang4 gene (figure 1C). Interestingly, Nod2 deletion also did not affect global levels of α-defensin transcripts (figure 1D), using primers that detect all known PC α-defensin genes.20
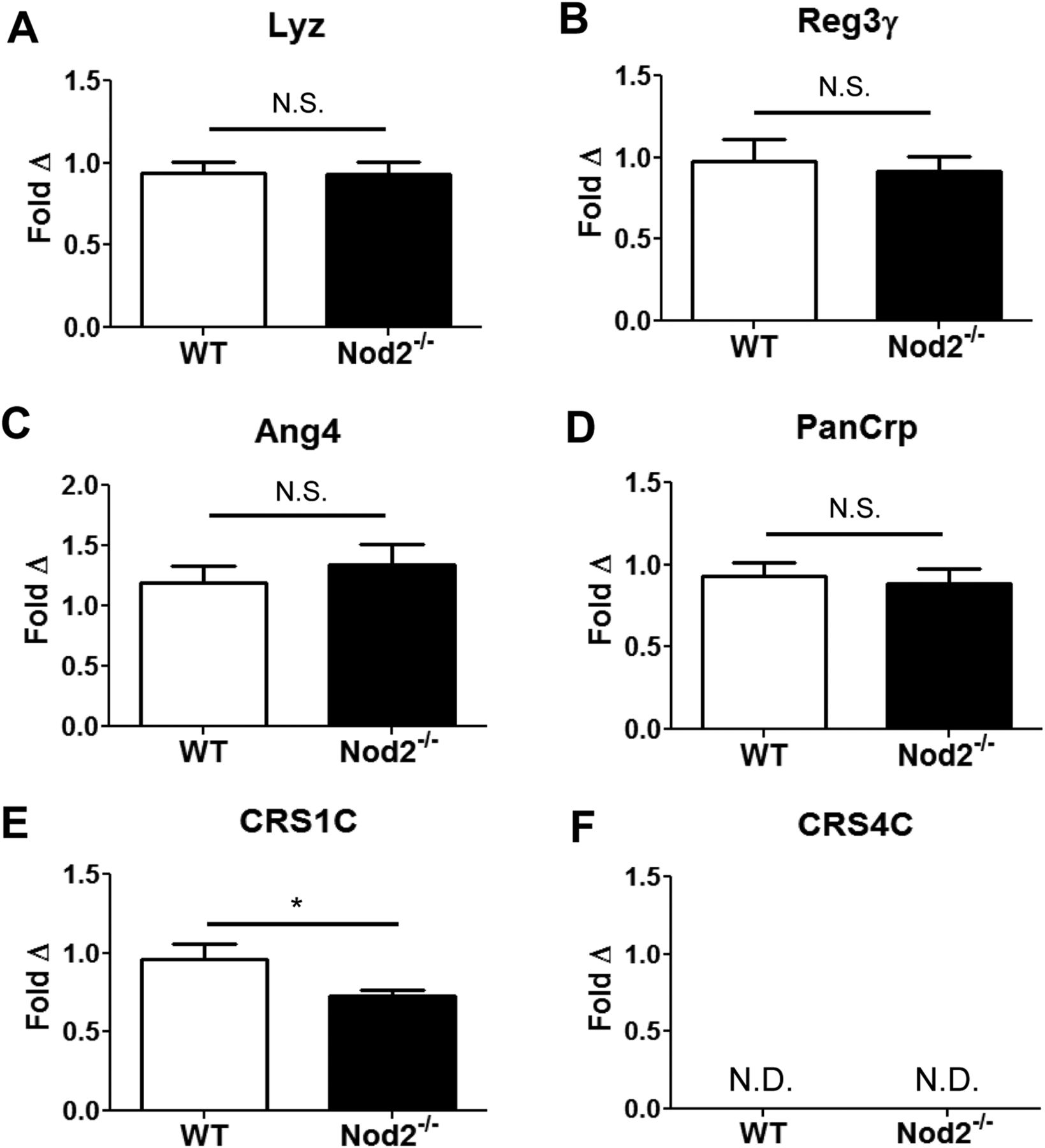
Paneth cell (PC) antimicrobial peptide transcript expression is primarily unaffected by Nod2. Ileal transcript levels of (A) lysozyme-P (Lyz); (B) regenerating islet-derived protein 3 gamma (Reg3γ); (C) angiogenin 4 (Ang4); (D) total PC α-defensins (PanCrp); (E) cryptdin-related sequence (CRS)1C and (F) CRS4C are shown for wild-type (WT) and Nod2−/− mice (n=8–10 mice/group). CRS4C was not detected (ND) in any animal. Copy number is normalised to β-actin and expressed as a fold Δ relative to the WT group. Data are shown as means with SEM. *p<0.05; N.S.—not significant (p>0.6), based on Student's t test.
By contrast with the AMPs described above, mRNA levels of the CRS peptides analysed in this study did display a unique expression pattern in Nod2−/− mice. Specifically, transcript levels of the CRS1C subgroup were significantly decreased in Nod2−/− mice relative to WT controls (∼25% reduction, p<0.05) (figure 1E). Furthermore, we were unable to detect transcripts of the second CRS peptide subgroup, CRS4C, in either WT or Nod2−/− B6 mice (figure 1F), though this subgroup was prominently expressed in the ilea of 129/SvEv mice (see online supplementary figure S4A). This is particularly important because a member of this subgroup, Defa-rs10, has been reported to be reduced in Nod2−/− mice relative to WT controls.8 Despite multiple attempts, we were unable to detect Defa-rs10 transcripts in either WT or Nod2−/− B6 mice, though this transcript was also highly expressed in 129/SvEv animals (see online supplementary figure S4A).
In summary, with the exception of the CRS1C group, we failed to reject the null hypothesis that the major mouse AMP classes had equal mRNA expression. Moreover, the 95% CIs of the mean AMP levels for the WT and Nod2−/− groups were highly overlapping (see online supplementary table S1), suggesting that if there is a real difference in AMP expression between WT and Nod2−/− mice, it is small in magnitude.
Mouse PC α-defensin expression is independent of Nod2
Because previous work has demonstrated reduced expression of the α-defensin Defa4 in Nod2−/− mice,8 it was surprising to find equivalent global α-defensin levels in Nod2−/− and WT mice. To determine if a compensatory induction of specific α-defensin isoforms could explain these findings, we next measured mRNA levels of selected α-defensin AMPs from 3 different phylogenetic groups (Defa4, Defa3 and Defa5).21 As shown in figure 2A, Defa4 transcripts were undetected in both WT and Nod2−/− mice, though were present in 129/SvEv animals (see online supplementary figure S4B). Because Defa4 is expressed exclusively in non-B6 mouse strains,10 ,21 we also quantified transcript expression of Defa20, which is the B6 homologue of Defa4. Transcripts levels of Defa20 were equivalent in WT and Nod2−/− mice (figure 2B). Finally, Defa3 and Defa5 mRNA levels also showed no statistical differences between experimental groups (figures 2C,D).

Paneth cell α-defensin expression is independent of Nod2. mRNA expression of α-defensin isoforms (A) Defa4; (B) Defa20; (C) Defa3; and (D) Defa5 in the ileum of wild-type (WT) and Nod2−/− mice (n=8–10 mice/group). Copy number is normalised to β-actin and expressed as a fold Δ relative to the WT group. Data are shown as means with SEM. ND— not detected; NS— not significant (p>0.1), based on Student's t test. (E) acid urea-polyacrylamide gel electrophoresis demonstrates peptide expression patterns of α-defensin isoforms in the ileum of WT and Nod2−/− mice. First lane is recombinant Defa4 control; each additional lane represents an individual mouse. Individual bands (based on calculated mass determined via mass spectrometry): 1-Defa4 (recombinant); 2,7-Defa5; 3,8-Defa24; 4,9-Defa20/Defa21; 5,10-Defa2; and 6,11-Defa22.
At the protein level, PC α-defensins were assessed using AU-PAGE, which localises α-defensins to a series of bands at the cathodal end of the gel.22 Figure 2E shows that α-defensin banding patterns were identical between WT and Nod2−/− mice. Subsequent MALDI-TOF/TOF MS analysis demonstrated that the identities of the α-defensin bands in WT and Nod2−/− mice were also indistinguishable (see online supplementary table S2). This is further highlighted in online supplementary figure S4C, which reveals that pooled samples of WT B6 and Nod2−/− B6 ileal protein extracts have a distinct banding pattern from those of WT 129 mice. This supports the premise that mouse strain profoundly influences the ileal α-defensin profile, while Nod2-deficiency does not result in measurable changes in the expression of these molecules.
Nod2 does not regulate PC numbers
Our findings thus far demonstrate minimal differences in PC AMP expression in ileal tissue from WT and Nod2−/− mice. However, if Nod2-deficiency results in decreased AMP expression at a cellular level, it is possible that a compensatory increase in PC numbers could result in similar total AMP levels between WT and Nod2−/− groups. To ensure that alterations in PC number were not masking some degree of Nod2-mediated AMP regulation, we enumerated PCs in WT and Nod2−/− mice. H&E staining of ileal sections from both experimental groups showed that WT and Nod2−/− mice have normal tissue architecture, and display no signs of inflammation (figure 3A). Quantification of intestinal crypts revealed no differences between the two groups of mice, and therefore, no effect of Nod2 on crypt development (figure 3B). Using anti-Lyz staining as a PC marker, immunohistochemistry displayed neither ectopic placement of PCs, nor hyperplastic growth of these cells (figure 3C). Furthermore, there was no significant difference in the number of Lyz+ cells per crypt between WT and Nod2−/− mice (figure 3D). Finally, to minimise the bias of counting PCs from selected high-power fields, flow cytometry of epithelial cell preparations from the entire ileum of WT and Nod2−/− mice was performed. Using CD45 as a haematopoietic cell maker, PCs were identified as the Lyz+CD45− cell fraction (figure 3E). Quantification from three independent experiments confirmed that WT and Nod2−/− ilea contained an equivalent percentage of PCs within their epithelia (figure 3F). Total numbers of epithelial cells from each group were not significantly different. In summary, this analysis demonstrates that Nod2 does not regulate α-defensin expression on a per-cell basis.

Nod2 does not influence Paneth cell (PC) development. (A) H&E staining of ileal tissue shown at 400 × magnification from wild-type (WT) and Nod2−/− mice was used to quantify intestinal crypts. Panel (B) represents average data from 18–20 high power fields/group (3–4 mice/group). Panel (C) shows immunohistochemical staining of ileal tissue for lysozyme (Lyz), allowing for the quantification of PCs per crypt. Panel (D) represents average data from 40–45 crypts/group (5–6 mice/group). (E,F) Flow cytometry was used to measure the percentage of Lyz+ CD45− PCs from the ileal epithelium of WT and Nod2−/− mice. Data are representative of three independent experiments (NS— not significant, p>0.1, based on Student's t test).
PC α-defensin antimicrobial activity is unaffected by Nod2
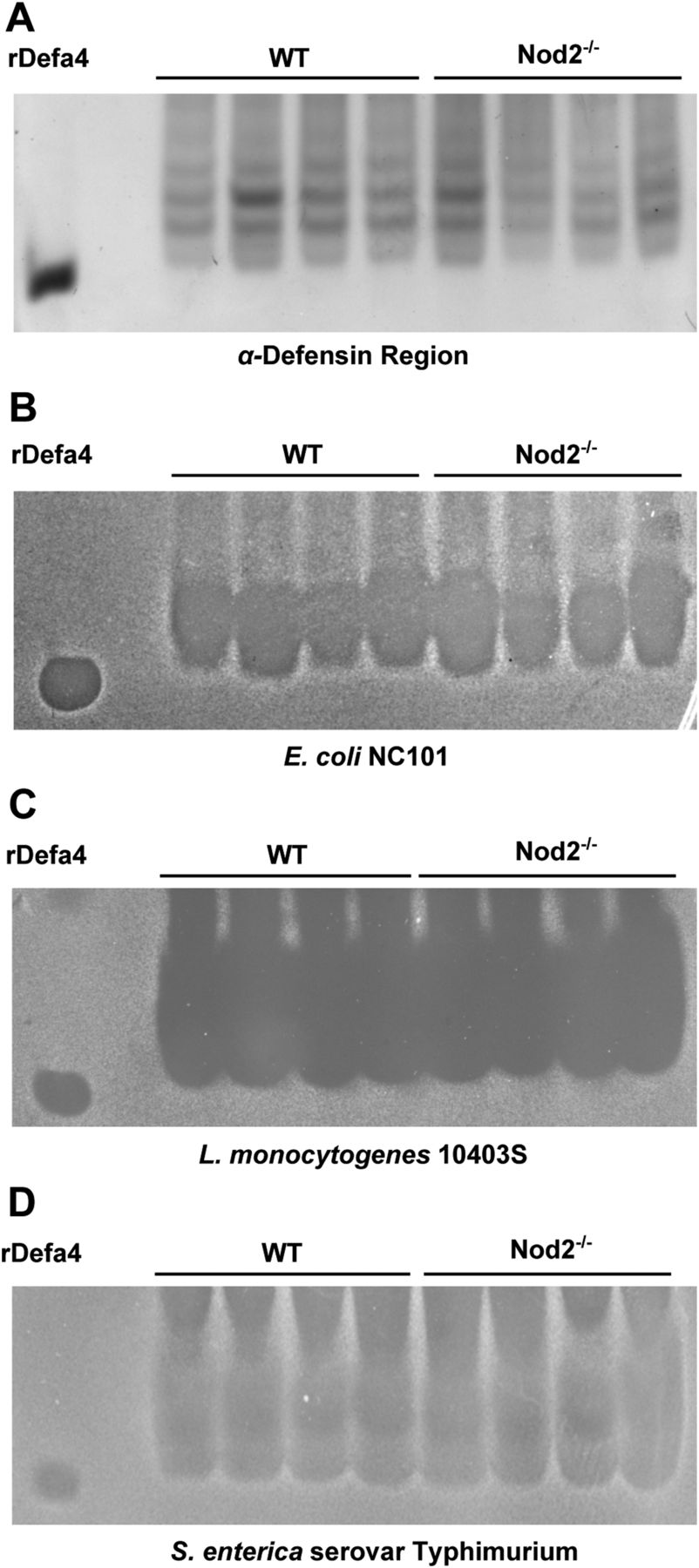
The similarity in the AU-PAGE banding pattern of the α-defensin region between WT and Nod2−/− mice indicates that Nod2 does not regulate post-translational processing or conformational folding of PC α-defensins. To confirm that Nod2-deficiency does not affect the functional microbicidal activity of PC α-defensins, we tested the ability of ileal protein extracts from WT and Nod2−/− mice to inhibit the growth of commensal and pathogenic bacterial strains. Specifically, we assessed α-defensin antimicrobial activity against the commensal bacterium Escherichia coli strain NC101 (which has been shown to induce intestinal inflammation in genetically engineered mice),23 as well as the intestinal pathogens Listeria monocytogenes and Salmonella enterica. To test α-defensin antimicrobial activity against these organisms, we excised the α-defensin zone of an AU-PAGE minigel (figure 4A), overlaid this onto agarose plates of confluent bacteria, and observed for zones of bacterial inhibition. Figure 4B–D demonstrates comparable zones of α-defensin-mediated bacterial inhibition for all tested bacteria, regardless of host Nod2 status. Therefore, Nod2 does not influence the bacteriostatic activity of PC α-defensins against relevant proinflammatory commensal and pathogenic bacterial strains. It is interesting to note that Nod2−/− mice are reported to have increased susceptibility to oral L monocytogenes infection.8 The present data suggest this is unlikely due to defective α-defensin antimicrobial function.

Antimicrobial activity of Paneth cell α-defensins is unaffected by Nod2. (A) wild-type (WT) and Nod2−/− mouse-derived ileal protein extracts were resolved by acid urea-polyacrylamide gel electrophoresis. An excised gel strip containing the α-defensins was placed onto bacteria-laden agarose. Bacterial clearance zones are shown for (B) Escherichia coli NC101, (C) Listeria monocytogenes 10403S, and (D) Salmonella enterica serovar Typhimurium. Similarly sized zones of bacterial growth inhibition are seen for all bacterial strains, regardless of Nod2 status.
Nod2 has minimal effects on global faecal microbial composition
Given the similarities in PC AMP function between WT and Nod2−/− mice, we next sought to determine if there were differences in the global microbial composition of our experimental groups. Previous studies have demonstrated that Nod2−/− mice have alterations of their intestinal microbiota relative to WT animals.11 ,24 However, as described in our methods, the breeding strategy used in the present study allows for more rigorous control of environmental influences through cohousing of Nod2−/− and WT littermates. By principal coordinates analysis of samples based on deep sequencing of the 16S gene, it appeared that the cage the mice were housed in had a stronger effect on microbial community composition than did the WT or Nod2−/−genotype (figure 5). Indeed, the null hypothesis that the cage has no effect on gut microbial structure was rejected for five of the first eight coordinates at a 10% false discovery rate (table 1). By contrast, we failed to reject the null hypothesis that genotype has no effect on microbial composition for any of the first 15 coordinates at a p<0.05 threshold (table 2). This was also true at the individual taxa level, where we failed to reject the null hypothesis that individual taxa were not significantly different between WT and Nod2−/− at a 10% false discovery rate (see online supplementary table S3). These data suggest that the cage environment plays a stronger role in structuring the microbial community than the absence of Nod2 expression.
Effects of mouse cage on gut bacterial composition
Effects of Nod2 genotype on gut bacterial composition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Local cage environment overrides Nod2-mediated influences on the intestinal microbiota. Principal coordinates analysis (PcoA) using the first two coordinates of a PcoA based on Bray-Curtis dissimilarity of 454 sequences of 16S rRNA from stool samples. Nod2−/− mice (KO) are indicated by circles, while wild-type (WT) mice are shown with squares. The numbers by each symbol indicate the cage of the animal, and each cage is depicted in a unique colour. PC, principal coordinate.
Discussion
Polymorphisms of the NOD2 gene remain the most replicated risk alleles for the development of CD.25 Despite this high reproducibility, the precise mechanisms by which NOD2 dysfunction increases CD susceptibility are unclear. Previous work using Nod2−/− mice suggests a key role for Nod2 in the transcriptional regulation of specific PC α-defensins.8 However, in the present study, we demonstrate that there are no profound effects of Nod2-deficiency on PC α-defensin mRNA expression, protein levels, or antimicrobial activity when mouse background is strictly controlled. Moreover, using cohoused WT and Nod2−/− littermates, we reveal that local cage conditions outweigh Nod2-mediated effects on faecal microbial composition.
The differing α-defensin expression results demonstrated by our analysis versus those previously reported8 likely stem from the variability of mouse background used in the former study. Numerous investigations demonstrate that B6 and 129 mouse strains possess distinct α-defensin profiles and antimicrobial properties.10 ,21 ,26 Such findings underscore the importance of clearly defining mouse background strain when performing α-defensin studies. The original Nod2-null mice used to demonstrate Nod2-dependent α-defensin regulation were constructed by injecting genetically manipulated 129S1/Sv-derived W9.5 ES cells into B6 blastocysts. However, no mention of backcrossing to a pure background strain was indicated, and hence, the precise background of mice used in the final analysis remains unclear. Indeed, the detection of Defa4 and Defa-rs10 transcripts in their experimental groups suggests that the mice previously studied possessed at least a component of the original 129S1/Sv genetic background, as these genes are not found in B6 strains.10 ,21 The background ambiguity of these mice leaves open the possibility that formerly observed differences in α-defensin expression may be due to variations in mouse strain between experimental groups, as opposed to a true effect of Nod2 deficiency.
In the present study, Nod2−/− mice were obtained commercially through Jackson Labs, and represent the original strain generated by Kobayashi et al that has since been backcrossed to completion on the B6 background. Using B6 Nod2−/− and WT littermates, we found no differences in α-defensin expression between the two groups. The lack of Defa4 and Defa-rs10 expression in both experimental groups is consistent with the absence of these genes in the B6 mouse strain.10 ,21 The lack of Nod2-mediated α-defensin regulation was also true at the protein level, and could not be explained by differences in PC number between WT and Nod2−/− mice. Similar to the α-defensins, there was no evidence of Nod2-driven transcriptional regulation of other AMP classes, including Lyz, Reg3γ and Ang4, consistent with previous reports.19
Interestingly, the CRS AMP subclasses did display a unique expression pattern in our experimental groups. CRS peptides comprise a prominent family of AMPs, which were first described as PC-specific transcripts that are highly similar to the α-defensins.27 These molecules are divided into two primary groups, CRS1C and CRS4C, based on their C-terminal peptide sequence.28 CRS4C peptides form homodimers and heterodimers that expand the diversity of AMPs within PC secretions.29 They include Defa-rs10, which was previously reported to be decreased in Nod2−/− mice relative to WT controls.8 However, our evaluation of the NIH B6 mouse genome assembly demonstrated no evidence of the Defa-rs10 gene in B6 animals.10 This again implies that the background of mice used previously possessed a non-B6 component. Indeed, we found no transcript expression of the CRS4C class in either WT or Nod2−/− mice on a B6 background. Similarly, no mRNA expression of Defa-rs10 was detected in our experimental groups. This was confirmed using TaqMan quantitative RT-PCR primers, as well as the SYBR green primer sequences published originally by Kobayashi et al.8
By contrast with CRS4C, mRNAs for CRS1C have been detected in the B6 mouse small intestine.30 Accordingly, we found a modest ∼25% reduction of CRS1C expression in Nod2−/− mice relative to WT controls. Although previous work has suggested a role of Wnt/Tcf-4 signalling in the regulation of CRS1C expression,31 there have been no reports of Nod2 influencing transcript levels of these molecules. However, despite finding slightly reduced CRS1C expression in Nod2−/− mice, our examination of the promoter region of the CRS peptide genes revealed no canonical NF-κB binding sites (data not shown). Therefore, the direct effect of Nod2 on CRS1C mRNA expression remains unclear. Moreover, to our knowledge, CRS1C peptides have not been characterised or detected at the protein level. Therefore, it is unclear what effect this reduction in CRS1C gene expression may have on the bactericidal activity of PC secretions.
It is important to underscore that the present study focuses on Nod2-mediated regulation of PC AMP production, and does not examine the effects of Nod2-deficiency on PC secretion. It has been established that the ligand for Nod2, muramyl dipeptide (MDP), is a PC secretagogue.3 Subsequent work has supported this premise, demonstrating decreased bactericidal activity of Nod2-deficient intestinal crypt secretions stimulated by MDP.32 Furthermore, Nod2-deficiency also interfered with carbamyl-choline (CCh)-mediated PC secretion, suggesting that Nod2 may affect a common inductive signalling pathway for PC secretion. A presumptive link between Nod2 and global PC secretion has been made via the molecule KCNN4, which is an established risk allele for CD.33 Specifically, the mouse homologue of KCNN4 (Kcnn4) is a K+ channel that regulates Ca2+ flux within PCs.34 Patients with NOD2 risk polymorphisms display reduced levels of KCNN4 mRNA expression, leading to the hypothesis that Nod2-dysfunction may depress PC secretion by downregulating the expression of this potassium channel.33 Interestingly, we found no differences in Kcnn4 transcript levels in WT and Nod2−/− mice (data not shown). Nevertheless, a regulatory effect of Nod2 on PC secretion cannot be ruled out by the present study.
Even if we stipulate that Nod2-deficiency may result in some degree of defective PC secretion, the present study raises questions as to the biological relevance of such impairments. Previous work using Mmp7−/− mice that possess reduced α-defensin activity demonstrated profound shifts in the gut microbiota of these animals.4 Similar alterations of the intestinal microbiota would be expected in Nod2−/− mice, if these mice indeed possess impaired PC function. Previous studies have supported this concept, demonstrating increased levels of Bacteroides and Firmicutes bacteria in the ilea of Nod2−/− mice relative to WT controls,32 as well as global alterations of Nod2−/− gut microbial communities analysed by high-throughput pyrosequencing.11 ,24 While these studies did control for background strain by using pure B6 mice, the precise breeding and husbandry conditions were often unclear. When such conditions were clearly described (as in the case for Rehman et al),11 Nod2−/− and WT colonies were housed independently through the course of the study. While this approach can offer some insight into the effects of Nod2 on gut microbial composition, it is limited in its ability to detect confounding cage or maternally transmitted effects on the structure of gut bacterial communities. This is elegantly demonstrated in a recent study by Ubeda et al,35 which examined gut microbial composition in MyD88- and TLR-deficient mice. Importantly, this study analysed the intestinal microbiota of knockout (KO) mouse colonies that had been housed and bred in isolation, but also evaluated WT and KO littermates which were housed together prior to necropsy (similar to the strategy used in the present study). Their analysis revealed marked differences in the microbiota of mice from the independent mouse colonies, but minimal alterations in cohoused littermates. This supports the results of our study, which shows no apparent differences in gut microbial composition between cohoused Nod2−/− and WT littermates.
The lack of observed Nod2-dependent intestinal microbial alterations in the present study may be explained by two possible causes. First, local cage environment (including coprophagia) may be a strong influence on faecal microbial composition, able to suppress differences in the microbial community caused by genotype. Second, there may be a strong maternal influence on the faecal microbiota. This would lead to similar microbial profiles within individual mice of the same litter, regardless of genotype. Such effects have also been extensively described in the literature.35 ,36 Because our study design involved housing mice of the same litter within the same cage, we were unable to distinguish cage and litter effects in this study. In either case, there appears be a strong cage and/or litter effect that supersedes the influences of Nod2 on faecal microbial composition.
The findings described in the present study have important clinical implications. First, numerous human CD risk alleles have been associated with PC abnormalities,6 ,37–39 suggesting an important role for these cells in CD pathogenesis.40 However, to translate such findings to clinical practice, the mechanisms of these gene-mediated PC impairments must be elucidated. Mouse models provide an opportunity to study such mechanisms.41 In this study, we have established a paradigm for studying PCs in mouse models of CD risk allele dysfunction, highlighting the importance of controlling for mouse background and housing conditions in such investigations. Second, our data raise concerns with the commonly accepted hypothesis that NOD2 dysfunction leads to attenuated PC microbicidal activity, which in turn, alters the composition of the gut microbiota, thereby predisposing to the development of CD. This highlights the importance of identifying alternative roles of NOD2 in the control of intestinal inflammation, both within PCs as well as other components of the innate immune system. An elegant approach to characterising such functions will be to examine PC function in knock-in mice carrying the human NOD2 SNP13 polymorphism, which have been described previously.42 Finally, we demonstrate that environmental factors may overcome genetic influences on gut microbial structure. Ultimately, if we hope to modulate intestinal microbial communities as a treatment strategy for CD, it will be imperative to override the impact of host genetics on the gut microbiota. The homogenisation of the intestinal microbiota of Nod2−/− mice and their WT littermates suggests that strong environmental pressures may supersede gene-based influences on gut microbial composition. Future studies should focus on identifying specific environmental factors that can overcome the microbial effects of distinct CD risk alleles. This may lead to novel, patient-specific treatment strategies for this disease.
Acknowledgments
The authors wish to thank Dr Andre J Ouellette (University of Southern California) for his critical review of this manuscript, and for kindly providing the recombinant α-defensin 4 used in this study. We also acknowledge the services provided by the Histology and Microbiome Cores at the UNC Center for Gastrointestinal Biology and Disease.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online figure 1
- Data supplement 4 - Online figure 2
- Data supplement 5 - Online figure 3
- Data supplement 6 - Online figure 4
- Data supplement 7 - Online table 1
- Data supplement 8 - Online table 2
- Data supplement 9 - Online table 3
Footnotes
-
Contributors MTS, study concept and design; data acquisition, analysis, and interpretation; drafting of manuscript. IMC, data acquisition, analysis, and interpretation; drafting of manuscript. EG, data acquisition, analysis, and interpretation. AW, data acquisition, analysis, and interpretation. RJF, data acquisition, analysis, and interpretation. RB, data analysis and interpretation. AAF, data analysis and interpretation; critical revision of manuscript for intellectual content. SJH, study concept and design; critical revision of manuscript for intellectual content; obtained funding. RBS, study concept and design; critical revision of manuscript for intellectual content; obtained funding. ASG, study concept and design; data acquisition, analysis, and interpretation; drafting of manuscript; obtained funding.
-
Funding This work was funded by a Career Development Award from the Children's Digestive Health and Nutrition Foundation/Crohn's and Colitis Foundation of America (ASG), a Young Investigator Award from the Global Probiotics Council (ASG), grants National Institutes of Health KL2 RR025746 and TR000084 (Marshall S Runge, PI), NIH R01 DK53347 (RBS), NIH P40 RR018603 (RBS), NIH U01 DK085547 (SJH), and NIH P30 DK34987 (Robert S Sandler, PI).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.