Article Text

Statistics from Altmetric.com
Introduction
Objective
The aim was to create updated guidelines for the management of adult coeliac disease (CD), but non-coeliac gluten sensitivity (NCGS) was not considered.
Recommendations
▸ Diagnosis of CD requires duodenal biopsy when the patient is on a gluten-containing diet and for the vast majority of adult patients also positive serology. (Grade B)
▸ Biopsy remains essential for the diagnosis of adult CD and cannot be replaced by serology. Follow-up should aim at strict adherence to a gluten-free diet. (Grade B)
Development of the guidelines
The British Society of Gastroenterology (BSG) guidelines on the management of adult CD were originally published in 1996. Recently the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) published updated guidelines for paediatric CD,1 but international guidelines for adult CD are scarce2–5 since the NIH consensus6 on CD in 2005 (despite more than 4000 PubMed publications about CD in the last 8 years). As a result, the Clinical Services and Standards Committee of the BSG commissioned these guidelines, subject to rigorous peer review and based on a comprehensive review of the recent literature, including data from any available randomised controlled trials, systematic reviews, meta-analyses, cohort studies, prospective and retrospective studies.
A multidisciplinary panel of 18 physicians from eight countries (Sweden, UK, Argentina, Australia, Italy, Finland, Norway and the USA), a dietitian and a representative and a patient advocate from Coeliac UK reviewed the literature on the management of CD. These individuals were involved in the original stakeholder meetings and with revision of the manuscript.
Intent and levels of evidence
All aspects of the contemporary diagnosis and management of patients with adult CD were considered. PubMed literature was searched from 1900 to 2012 to obtain evidence for these guidelines. Also there was input from all authors who have considerable expertise and experience in diagnosis and management of CD. The panel of international experts previously collaborated in the publication of definitions of CD7 and were invited by the BSG through coauthor DSS. Our task force contained representatives from the clinical disciplines gastroenterology, paediatrics, histopathology, neurology, dermatology, genetics and immunology.
The current literature of review papers was examined, focusing on 10 reviews8–17 to explore gaps in current reviews on CD. Nine working subgroups were then formed that examined the following areas of CD management: classification of CD: FB, MH, DSS, CC; genetics and immunology: KEAL, DaVH, PJC; diagnostic criteria, serology and endoscopy in the investigation of CD: MMW, JAM, FB, PHRG, JFL, KEAL; follow-up: DAL, PHRG, JCB, JFL; gluten-free diet (GFD): PJC, KK, CC, GLS; refractory CD (RCD) and complications: FZ, FB, DAL, PHRG; quality of life (QoL): GLS, JCB, TRC, FZ; novel therapy: JCB, KEAL; screening for CD: TRC, KK, JAM, JFL. The working groups wrote the sections, which were subsequently internally reviewed. Each final fully written and referenced section was then released to all group members for review by teleconference and email correspondence. Thereafter JFL created the first draft of the guidelines by amalgamating all documents. All authors then helped revise this draft until final document consensus was reached.
Between January 2012 and February 2013, six web surveys were performed using the web site ‘survey console’ (http://www.surveyconsole.com) to explore issues including coeliac topics of controversy; the role of endoscopy; the role of histopathology and serology in the diagnosis of CD; and follow-up of patients, including the use of follow-up biopsy. The web surveys were for the coauthors/Guidelines Development Group (GDG) members. Survey results were then discussed at teleconference and used to inform the direction of recommendations and outline areas where the GDG were not concordant. Disagreements were solved through discussion.
Studies used as a basis for these guidelines are graded according to the quality of evidence using the Oxford Centre for Evidence-based Medicine levels of evidence.18
Strength of recommendations
-
Directly based on category I evidence, for example, from systematic reviews and randomised controlled trials. This is the strongest recommendation of the four grades listed.
-
Directly based on category II or III evidence or extrapolated recommendation from category I evidence. This includes evidence from controlled non-randomised studies or time series; or indirect evidence from systematic reviews or randomised controlled trials.
-
Directly based on category IV evidence or extrapolated recommendation from category II or III evidence. This also includes evidence from non-experimental studies such as cohort studies or case–control studies.
-
Directly based on category V evidence or inconsistent or inconclusive studies of any level. This includes evidence from expert committees and respected authorities.
Background
CD is an immune-mediated small intestinal enteropathy that is triggered by exposure to dietary gluten in genetically predisposed individuals.7 Samuel Gee is credited with the first clinical description of CD in 1887, although Aretaeus of Cappadocia may have described the disease in the first century AD. In the 1930s and 1940s, Dicke demonstrated (then published in 195319) that a wheat-free diet was the key to management. The discovery of antigliadin antibodies in 1961 was the first non-invasive serological marker for CD.20
Until the 1980s, CD was considered a rare disease affecting mainly children (exceptions occurred21), but subsequently has been shown to occur at any age.22 Although the prevalence seems to vary considerably (from <0.25%23 to >1%24), a large-scale screening study in subjects from Finland, Italy, the UK and Germany found a prevalence of CD of around 1%,25–27 with a recent US study showing a prevalence of 0.71%.28 CD is more frequently diagnosed in women than in men with a ratio between 1.5 and 2,29 but this gender imbalance may vanish with age and has been absent in some screening studies.30
Traditionally patients with CD presented with malabsorption dominated by diarrhoea, steatorrhoea, weight loss or failure to thrive (‘classical CD’),7 but over time the proportion of newly diagnosed patients with malabsorptive symptoms has decreased,31 and ‘non-classical CD’7 and even asymptomatic CD have gained prominence. Newly diagnosed patients with CD can present with a wide range of symptoms and signs, including anaemia,32 vague abdominal symptoms (often similar to irritable bowel syndrome (IBS)33), neuropathy,34 ,35 ataxia,36 depression,37 short stature,38 osteomalacia and osteoporosis,39 liver disease,40 adverse pregnancy outcomes41 and lymphoma.42 Asymptomatic patients are typically diagnosed through screening. Screening may be initiated because the individual has a CD-associated disorder or has symptoms and is a first-degree relative to a patient with CD.
The diversity of the clinical presentation of CD emphasises the need for robust diagnostic criteria and careful disease work-up. It is therefore natural that over the years, several efforts have been made to define CD,1 ,7 ,43–45 and how it should be managed.8–16 While all these reviews discussed critical aspects of diagnostics, including the small intestinal biopsy and serology, there are scant reports discussing the genetic and immunological background,8–14 the role of human leucocyte antigen (HLA) testing,11 ,12 ,15 ,16 ,46 and even fewer have discussed issues such as QoL11 ,15 and patient support.15
During our previous review on definitions of CD and related concepts,7 we realised that there was a need for a consensus paper on modern management and diagnosis of CD. In this project, participants of the so-called Oslo group who first convened at the 2011 meeting on CD in Oslo7 collaborated with representatives of the BSG to write guidelines for the management of CD in adults. These guidelines will enable physicians, dietitian and other healthcare personnel to provide better care for their patients.
Genetics, immunology and trigger factors
Environmental factors are important in CD and ingestion of gluten is a prerequisite for the development of CD. Children breastfed at and beyond gluten introduction may be at lower risk of developing CD in childhood, although research is not consistent.47 Conversely, large amounts of gluten or gluten exposure without ongoing breastfeeding may increase the risk of future CD.48 ,49 Gastrointestinal infections, drugs, interferon α and surgery have also been implicated as trigger factors.47 ,50 ,51 The factors leading to the breakdown of tolerance to gluten are not known, but local pro-inflammatory changes are of paramount importance.52
A high prevalence (10%) among first-degree relatives of patients with CD53 and a greater concordance rate in monozygotic twins (∼75%)54 than in dizygotic twins indicates a strong genetic component.
CD shows a very strong association with a particular HLA variant termed HLA-DQ2.5.55 This molecule is encoded by the DQA1*05:01 and DQB1*02:01 genes in cis configuration on the DR3 haplotype. Thus most patients are DR3, DQ2 positive. A smaller subset of patients with CD express a very similar DQ2.5 molecule encoded by a combination of DR5 and DR7 positive haplotypes, in this situation by the genes in trans position. While the proportion of individuals with CD who are DQ2.5 positive varies in different geographical areas it is generally ≥90%. The majority of the remaining patients are HLA-DQ8 positive (DR4, DQ8 haplotype).56
Genome wide association studies have so far identified 40 loci outside of HLA with genes predisposing to or protecting against CD.57 ,58 Most of these genes have immunological functions and are related to B-cell and T-cell functions. Each of the non-HLA genes contributes little to genetic risk. With a population prevalence of 1% and a heritability of 50%, the 39 non-HLA loci account for 14% of the genetic variance whereas HLA in comparison accounts for 40%.57
The most striking morphological features of the active coeliac lesion are the infiltration in the epithelium of T-cell receptor α/β and γ/δ expressing CD8 positive T cells, natural killer cell like T cells, and the dense population in the lamina propria of plasma cells and activated antigen presenting cells.59 ,60 Although numerically not dominant, CD4 gluten reactive T cells (T-cell receptor α/β expressing)61 ,62 are crucial in the pathogenesis of CD and are not found in people without CD.63–65
The CD4, gluten-specific T cells in the lamina propria invariably recognise gluten presented by the disease-associated HLA-DQ2 and HLA-DQ8 molecules61–65 and recognise gliadin peptides that have been modified by tissue transglutaminase 2 (TG2). This modification (glutamine to glutamic acid deamidation process) introduces negative charges.66 These modifications produce peptides that bind with much higher affinity to the HLA-DQ2 and HLA-DQ8 molecules.67 Patients with CD who are untreated typically have high titres of antibodies against the endomysium antigen (EMA test), also shown to be the extracellular enzyme tissue TG2.68 On exposure to gluten, plasma cells produce antibodies to tissue transglutaminase (IgA-TG2)69 and deamidated gliadin peptides (IgA-DGP70 and IgG-DGP71).
Untreated and treated CD are characterised by an increase in γ/δ intraepithelial lymphocytes (IELs), although in treated disease, the IEL count is close to that in healthy individuals.72
Diagnostics
Diagnosis of CD is by serology and duodenal biopsy, ideally with the patient on a normal, that is, gluten-containing diet. Biopsy remains essential for the diagnosis of adult CD and cannot be replaced by serology. Exceptions are patients with coagulation disorders and pregnant women, in whom biopsy may not be feasible or should be postponed until postpartum.
To state definite diagnosis of CD, villous atrophy is required. However, lesser degrees of damage (≥25 IELs but no villous atrophy) combined with positive serology (IgA-EMA, tissue transglutaminase (TTG) or IgG-DGP) may also represent CD (‘probable CD’), and in these circumstances a trial with GFD may be considered to further support the diagnosis of CD. HLA status may also aid diagnosis. Differential diagnoses of lymphocytic duodenosis should be ruled out if there is no response to GFD (see table 2).
The diagnosis of CD is readily established in those who, while consuming a gluten-containing diet, have positive serology and a duodenal biopsy with obvious coeliac histology (increased intraepithelial lymphocytosis, crypt hyperplasia and villous atrophy; table 1). These patients can immediately initiate a GFD with confidence.
An algorithm for the diagnosis of coeliac disease
CD can also be suspected in patients with mild gastrointestinal symptoms, associated conditions or those at genetic risk.3 Such patients should be investigated initially with serology and if this is positive (or if there is still a high index of suspicion as among symptomatic first-degree relatives73), then undergo upper endoscopy and duodenal biopsies.
However, in some cases the diagnosis of CD may not be straightforward, for example, patients are already on a GFD and therefore antibodies are negative, biopsies were not oriented correctly (this could lead to false-negative or false-positive villous atrophy) or show solely intraepithelial lymphocytosis (lymphocytic duodenosis)74 without architectural changes. In these situations, the patient needs to be maintained on a gluten-containing diet and further evaluated with additional testing and, if necessary, referred to a centre or clinician with a specific interest in CD.
The diagnosis of CD is illustrated in table 1.
Serology in CD diagnosis
Serological detection depends on the presence of specific endomysial antibodies (EMAs, also called AEAs), IgA anti-tissue transglutaminase antibodies (IgA-TG2, also called a-TTG, TTA) and/or deamidated antigliadin antibodies (DGP, either IgA or IgG isotype).7 IgG-TG2 is primarily useful in patients with known IgA deficiency.75 ,76
There is continuing debate on the sole use of non-invasive tests to diagnose CD. Recently ESPGHAN proposed new guidelines for the diagnosis of CD in children. It suggests that in symptomatic paediatric patients1 in whom the IgA-TG2 level exceeds 10 times the upper limit of normal, EMA antibodies are positive on a separately taken blood sample, and HLA-DQ2 or HLA-DQ8 are positive, then biopsies do not need to be performed to confirm the diagnosis of CD.1 In adults, this strategy has also been proposed77; however, there are very strong arguments for retaining the biopsy as gold standard for the diagnosis of CD. A recent study from the (UK) National External Quality Assessment Service centre states that not all commercial IgA-TG2 kits are reliable and the ESPGHAN guidelines are therefore not translatable for use in all centres and should not be used in the UK.78 Also, 2% of patients with CD are IgA deficient (0.2% of the population in general) and as usual serology tests for IgA-TG2 and EMA are IgA based, this may lead to false negatives and a reduction in test sensitivity. If patients are known to be IgA deficient, IgG-TG2 or IgG-DGP antibodies can be used, or alternatively, such patients should proceed directly to biopsy.79
A combination of immunoassays offers the best sensitivity if either a positive IgA-TG2 or IgG-DGP is considered a positive detection test. The combination of IgG-DGP and IgA-TG2 is particularly useful as an addition to detection of patients with CD who are IgA deficient, IgG-DGP was able to detect a few more IgA-sufficient patients who were missed by IgA-TG2 alone.76
There are now several point-of-care tests commercially available, which allow both immediacy and the ability to use them in a physician office/primary care setting. However, there is a relative paucity of data on the sensitivity and specificity of such tests by comparison to the gold standard of duodenal biopsy.80 There is concern regarding the use of these tests as patients may start on a GFD without a firm diagnosis—which includes biopsy—and further studies should be performed before considering the use of these in everyday practice. One study utilising community health nurses demonstrated a lower than expected sensitivity for CD.81
Endoscopy in seronegative individuals
The prevalence of seronegative CD is 6–22% of all diagnosed cases.71 ,82 ,83 ,84 One study also found a high degree of variability in EMA values for sensitivity between laboratories,85 and upper endoscopy is generally well tolerated and safe.86 Individuals of white European, Middle Eastern, North African or North Indian origin who undergo upper endoscopy for anaemia, weight loss or diarrhoea should therefore have duodenal biopsies performed, irrespective of whether they have had serology for CD. These features may well indicate that CD or an alternative mucosal cause of malabsorption is present.87 In fact, it has been suggested that duodenal biopsy should be considered in any individual undergoing endoscopy, because CD is common and has many varied clinical manifestations, including reflux, a common indication for endoscopy.88
Recommendation
-
In individuals undergoing an upper endoscopy in whom laboratory tests or symptoms or endoscopic features suggest CD, duodenal biopsy should be considered. (Grade C)
Role of HLA in the diagnosis of CD
CD is associated with specific HLA types in virtually all populations in which this has been tested, and is associated with the carriage of the gene pairs that encode DQ2.5 and DQ8.46 ,89 The diagnostic value of HLA genotyping in patients who may have CD revolves around its high negative predictive value, meaning that patients who lack the appropriate HLA genotype pairs described above are very unlikely to have CD.90 However, the positive predictive value of the HLA genotyping for CD susceptibility is very low as a large proportion of individuals without CD carry either HLA-DQ2 or HLA-DQ8 (the prevalence of DQ2 in the general population varies between 0% and 40% while that of DQ8 varies between 0% and 20% between countries91). In family screening, DQ2-positive or DQ8-positive relatives (especially siblings) are at a higher risk of CD,53 ,92 with one study suggesting that DQ2 positivity was associated with a 16-fold increased risk of CD among first-degree relatives.53
Specific use of HLA typing
HLA genotyping may be used in patients with suspected CD but who fail to respond to a GFD. A negative test in this circumstance would indicate that patients are highly unlikely to have CD (<1% of patients with CD are negative for DQ2 and DQ893) and thus the clinician can direct diagnostic efforts elsewhere. HLA typing may similarly be used in patients who are self-treated on a GFD and never had appropriate testing for CD before changing their diet. HLA typing may have an adjunctive role to identifying individuals who are not genetically at risk of CD and in whom further evaluation for CD is not necessary, saving a large number of repeated tests for CD in patients who would otherwise have to undergo testing because they have symptoms and a first-degree relative with CD.94
Recommendations
-
HLA typing should be used to rule out CD. A positive DQ2.5 or DQ8 can never confirm the diagnosis. (Grade B)
-
HLA typing should be used in individuals who are self-treated on a GFD and never had appropriate testing for CD before changing their diet. (Grade B)
-
HLA typing can be used to rule out CD, and minimise future testing, in high-risk individuals with CD, for example, first-degree relatives. (Grade B)
Biopsy and endoscopy in CD
There are endoscopic markers of villous atrophy described—scalloping or reduction of duodenal folds and nodularity—but these are not sensitive enough to preclude a biopsy,95 and a normal endoscopic appearance may occur in the presence of villous atrophy.96–98 Therefore, the endoscopic appearance of the duodenum should not determine whether biopsy is performed.
Biopsy of the duodenum for a diagnosis of CD should be performed irrespective of the prior performance of serological tests, if the patient exhibits symptoms or signs of CD, such as diarrhoea, weight loss or anaemia. Biopsies can be mounted on fibre-free paper to aid orientation,99 or alternatively biopsies could be free floated in formalin. Consultation with the histopathology laboratory is recommended to agree on specimen presentation.
The villous atrophy may be patchy in CD; hence multiple biopsies from the bulb and the more distal duodenum are recommended. The taking of at least four biopsy specimens is associated with a doubling of the diagnostic rate compared with patients undergoing a lower number of biopsies (less than four).100 In patients with persistently positive coeliac serology but a normal mucosa, repeat small intestinal biopsy should be considered, including biopsies from the jejunum.101 Video capsule endoscopy may support a CD diagnosis in this setting.102
A diagnosis of CD has implications for family members, as overall, around 10% of first-degree relatives53 may be affected and there could be uncertainty in pursuing this diagnosis in the family if the index case does not have a definite CD diagnosis. Within an adult population the patient may have other indications for an upper endoscopy, for example anaemia, and thus exclusion of other diseases is essential. Upper endoscopy is generally well tolerated by adults and, in contrast to children, can usually be readily performed with mild or even no sedation. Finally histological appearance of the small intestinal mucosa may also predict the risk of certain future complications, such as lymphoma (patients with villous atrophy are at statistically significantly higher risk of future lymphoma than patients with a normal mucosa but positive coeliac serology).103
Nevertheless there are still adult patients who may be unable or unwilling to undergo an endoscopy. Under these circumstances, assessment of the serological assay and/or level of IgA-TG2 (if 10 times the upper limit of normal), positive DGP/EMA, or the use of capsule endoscopy may have a supportive role. The sensitivity of capsule endoscopy to detect CD is similar to that of conventional endoscopy when combined with biopsies. However, it is less invasive, with good specificity and may provide endoscopic images that can then be used to support the diagnosis of CD in conjunction with positive serology. This approach may also be taken in equivocal cases.104 ,105
Recommendations
-
The diagnosis of CD requires duodenal biopsy when the patient is on a gluten-containing diet and for the vast majority of adult patients also positive serology. (Grade B)
-
Duodenal biopsy should be retained as the mainstay for the diagnosis of adult CD and cannot be replaced by serology. (Grade B)
-
At endoscopy, if there is suspicion of CD, then at least four biopsy specimens should be obtained, including a duodenal bulb biopsy. (Grade C)
-
In serologically negative patients showing signs of malabsorption (such as anaemia or diarrhoea) or a family history of CD, a duodenal biopsy should be considered. (Grade C)
Histopathology diagnosis of CD
It is important that pathologists and clinicians appreciate that patients with CD benefit from a GFD regardless of the degree of damage in the small intestine and that minor degrees of histological change suggestive of CD should not be ignored.106
Marsh107 described a commonly used classification of this spectrum. This classification has been modified subsequently by Oberhuber108 and simplified by Corazza and Villanacci.109 Recently, Rostami and Villanacci110 defined microscopic enteritis (also called lymphocytic duodenosis,111 lymphocytic enteropathy111) and Villanacci et al112 published a practical classification with a user-friendly checklist for the histology report of CD.
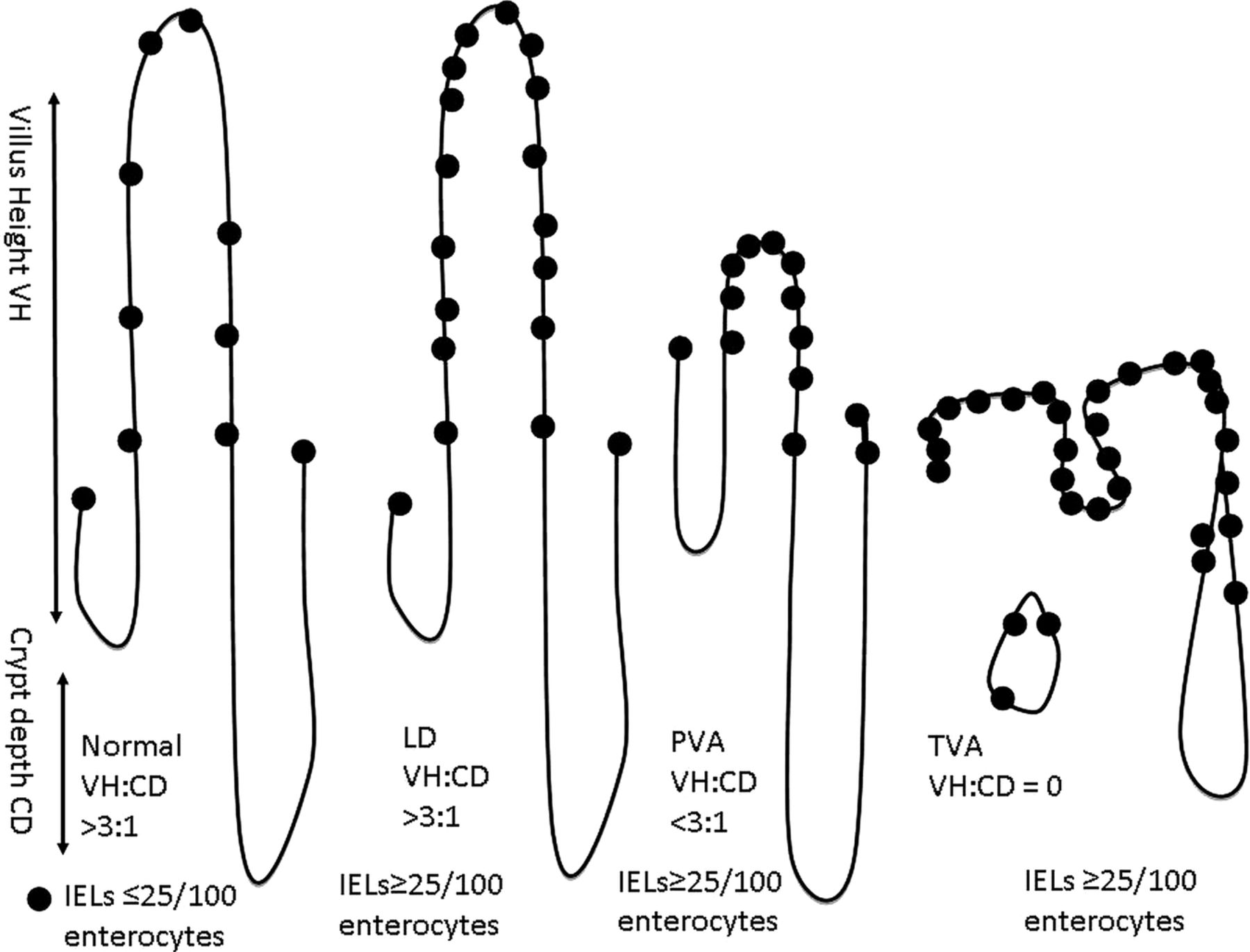
Some clinicians would prefer a descriptive report, and collaboration between pathologists and gastroenterologists as to the content of the report is valuable. It is important that if serology has not been performed prior to the biopsy, then this must be carried out by the requesting physician on receipt of a histology report suggesting a diagnosis of CD. A biopsy finding of villous atrophy is not specific for CD. Although CD is the commonest cause of villous atrophy, there are other causes (table 2); for this reason the addition of coeliac-specific serology seals the diagnosis. The biopsies must be properly oriented (usually by an experienced laboratory technician) as correct orientation is necessary for assessment of villous height crypt depth ratio (derived from the well oriented fields of the biopsies) (figure 1).
Histological mimics of CD in seronegative patients—conditions to be considered for investigation in an appropriate clinical context

Relationship between villous height and crypt depth. CD, crypt depth; IEL, intraepithelial lymphocyte; LD, lymphocytic duodenosis; PVA, partial villous atrophy; TVA, total villous atrophy; VH, villous height. The dots represent IELs.
The following features should be stated in the report:
-
Number of biopsies (including those from the duodenal bulb)113 and orientation.
-
The architectural features (normal, partial, sub-total or total villous atrophy).
-
Comment on the content of the lamina propria (in CD these are lymphocytes, plasma cells and eosinophils, and occasionally neutrophils, but cryptitis and crypt abscesses should suggest other pathology).
-
Presence of Brunner's glands.
-
Presence of crypt hyperplasia, villous height: crypt depth ratio (3:1).112 The absence of plasma cells suggests common variable immunodeficiency.
-
Evaluation of IELs (with immunocytochemical staining for T cells (CD3) in equivocal cases114) is vital. Counting IELs should be time efficient—simply counting IELs/20 enterocytes at the tips of five villi,115 ,116 or IELs per 50 enterocytes in two villi and summing these111 are both reliable and sensitive methods using H&E staining methods. The normal count has been variably cited; however, in evidence-based practice111 and in recent classifications,109 ,112 <25IELs/100 enterocytes should be taken as the norm.
-
Use of a simple classification system greatly enhances intra-observer agreement.117
Other causes of lymphocytic duodenosis and villous atrophy
Lymphocytic duodenosis74 is a common condition (3.8% of a population negative for coeliac serology111) seen in association with infection (particularly Helicobacter pylori), altered immune states, for example, common variable immunodeficiency, autoimmune and chronic inflammatory disorders, drugs and neoplasia.16 ,118–120 The villous architecture is normal, typically there is no crypt hyperplasia and IELs are ≥25/100 enterocytes. Of note, in a single study 16% of cases of lymphocytic duodenosis were found to have CD, and in 66% a known association was found by further investigation.121 Similarly there are other causes of villous atrophy in duodenal biopsies, including immune disorders and deficiency, food hypersensitivity, infection, drugs, neoplasia and miscellaneous disorders120 ,122 (table 2). In a study of non-coeliac enteropathy, 70% of patients with this condition were initially diagnosed as having CD.123
Novel diagnostic methods
While the current standard tests of serology and conventional histology are usually adequate to reach a diagnosis of CD, there are patients whose tests are equivocal and diagnostic uncertainty remains. Several novel diagnostic approaches have been undertaken. The deposition of IgA antibodies in close proximity to TG2 in the small intestine has shown promise as a way of defining early or potential CD in patients who are seropositive but lack any of the usual histological markers for CD. Recent work from Finland on IgA-TG2 autoantibody deposition in the small intestine in such patients shows promise in delivery of an early prediction of development of CD. However, this is currently experimental and the methodology requires tissue sections frozen in liquid nitrogen.124 Another diagnostic method meriting further evaluation is EmA assay in the culture medium of small intestinal biopsies.125 ,126 Other investigators have reported their findings using new techniques associated with endoscopy to enhance the diagnosis of CD. These include confocal microscopy, high-resolution magnification endoscopy, optical band imaging127 and optimal coherence tomography. These novel techniques are still limited by availability, tolerability and cost.98 However, the immersion technique and dye enhancement in which the endoscopist instills water or a contrast dye (for example, indigo carmine or methylene blue) into the bowel lumen, with or without the assistance of magnification endoscopy, enhancing the visualisation of the villus128 can be readily used and improves visualisation of villi, thus increasing the sensitivity for detection of villous atrophy.98
Dermatitis herpetiformis
Dermatitis herpetiformis (DH) is the cutaneous manifestation of gluten-sensitive enteropathy precipitated by exposure to dietary gluten.129 It is characterised clinically by herpetiform clusters of intensely itchy urticated papules and small blisters distributed on the extensor aspects of the elbows and knees and over the buttocks and on the scalp. The commonest age of onset is between the third and fourth decade, though the condition may occur at any age after weaning. Male patients are affected twice as often as female patients. For the majority of patients the disease is lifelong with varying periods of activity, potentially due to varying degrees of dietary adherence.
The major diagnostic criterion for diagnosis is the presence of granular IgA deposits in the dermal papillae of uninvolved perilesional skin as shown by direct immunofluorescence, and the diagnosis should not be made unless this has been confirmed.130
Less than 10% of patients with DH have symptoms or signs of malabsorption but most have evidence of CD that responds to a GFD and relapses on gluten challenge. Patients with DH present with their skin manifestations and are not usually troubled by the underlying small bowel problem at the time of presentation.129 ,131 Abnormality of the small intestinal mucosa with either total or subtotal villous atrophy is found in approximately 70% of patients with DH.132 A further 25% have normal villous architecture with increased IELs.
DH shares with CD an increased risk of developing lymphomas but this seems to be confined to those with severe gut involvement. The risk similarly declines with time on a strict GFD.133
Due to rash and itch, dapsone is often initiated. More than 70% of patients on a strict GFD are however able to slowly wean off dapsone over a period of 24 months.131
Follow-up
There is a paucity of data pertaining to adherence to a GFD being improved by follow-up in patients with CD.17 ,134–138 Only one previous historical study has assessed the impact of regular follow-up (annual review) at a dedicated doctor-led coeliac clinic.136 The investigators suggested that adherence was improved by having access and regular follow-up within the setting of a specialist coeliac clinic (improvement in adherence was 97.5% for those under clinic follow-up vs 40.4% for those no longer under follow-up); however as this was an observational study and there were likely to be marked biases in referral of cases, it is not possible to be confident that the associations seen were causal. There are no published data assessing the value of this approach or whether adherence to a GFD, QoL, avoidance of complications, or satisfaction with the service is improved by offering a dietitian-led coeliac clinic. One of the key factors relating to adherence is dietetic input and regular follow-up.17 ,134 Optimally, the clinic should have gastrointestinal and dietetic expertise.139 Patients should be encouraged to join disease-specific patient support groups if applicable.140
Once the disease is stable and the patients manage their diet without any problems, annual follow-ups should be initiated. The physician should check on intact small intestinal absorption (full blood count, ferritin,32 serum folate,141 vitamin B12,142 calcium,143 alkaline phosphatase144), associated autoimmune conditions (thyroid-stimulating hormone and thyroid hormone(s),145 and serum glucose),146 ,147 liver disease (aspartate aminotransferase/alanine aminotransferase)40 and dietary adherence (anti-TG2 or EMA/DGP), although the sensitivity and specificity of the latter148 ,149 cannot substitute for structured dietary interview.
In follow-up of CD, the key endpoints are normalisation of the health of patients judged by an absence of symptoms, and mucosal healing.17 A lack of symptoms72 ,150 ,151 or negative serological markers are not reliable or responsive surrogates of mucosal response to diet. Dickey et al reported that among 32 patients with CD and persistent villous atrophy, EMA had normalised in 27 (84%); while another British study found that 7/16 (44%) individuals with persistent villous atrophy at follow-up biopsy had a normalised TTG.149 ,152–154
The proportion of patients who do not achieve full histological recovery on diet varies, with most reports suggesting mucosal healing in 57–76%.150 ,155–159
Some experts favour repeat intestinal biopsy after 1 year of dietary therapy; others, however, do not believe a repeat biopsy is essential for coeliac management in typical cases. It is universally acknowledged that there is little evidence to address whether clinical outcomes are significantly altered as a result of re-biopsy and that the cost–benefit analysis of such an approach has yet to be fully established.
Recommendations
-
Follow-up biopsies may be considered in patients with CD, and are potentially helpful in identifying patients at increased risk of lymphoma. (Grade B)
-
Follow-up biopsies are not mandatory if the patient with CD is asymptomatic on a GFD, and has no other features that suggest an increased risk of complications. (Grade C)
-
Follow-up biopsies should be undertaken in patients with CD whose condition does not respond to a GFD. (Grade C)
Assessing adherence to the GFD
Whilst the panel of experts agree adherence to the GFD is most important for the health of a patient with CD, there are no evidence-based grade A recommendations regarding the most useful way to assess this. Dietary adherence should guarantee mucosal healing and at least remission of most gastrointestinal symptoms. At present there are no non-invasive biomarkers that indicate complete mucosal recovery and a number of studies indicate a high prevalence of villous atrophy in adult patients with CD who appear to be adherent (see table 3). When specifically questioned, the experts agreed there is a difference between the first-year follow-up of a newly diagnosed patient and long-term follow-up of an adherent patient with stable disease. An adherent patient with stable disease needs less follow-up and testing than a patient with newly diagnosed CD in whom the GFD has just commenced, and neither the mucosa nor biochemical aberrations have yet normalised.
Histological recovery of duodenal mucosa in CD
There are four steps to assess dietary adherence: clinical assessment of symptoms, dietetic review, serum antibodies and follow-up biopsy.
Symptoms
A meta-analysis of seven studies including more than 3000 subjects showed that the presence of gastrointestinal IBS-like symptoms is common in CD. IBS-type symptoms are more common in patients with CD who are not adherent to a GFD (OR 2.69; 95% CI 0.75 to 9.56).161 However, patients with CD who are also adherent to a GFD are more likely to experience (persistent) symptoms than controls.161
Dietetic review
The second step is a careful dietetic review conducted by a dietitian or dedicated physician. Apart from a visual analogue score scale which consists of an unmarked line with the anchor sentences ‘I never adhere to my diet’ and ‘I always adhere to my diet’ at each end,162 there are a number of questionnaires evaluating self-reported GFD adherence and food frequency in the English language163–166 that are also available in other languages.167–171 These questionnaires should be augmented by a dietetic review, which is a useful tool to tease out inadvertent gluten intake and to provide education for a balanced and adequate nutrient intake. There is no standard or quality control for dietetic review because local diets and habits require a specific structured interview, which is related to the quality of the diet. Currently, no data are available on GFD review outcomes in different countries, and there is no evidence that a careful review can substitute for other tools (eg, biopsy) to predict mucosal damage.
Studies report that poor dietary adherence due to occasional lapses is frequent and it is influenced by a number of factors, such as age at diagnosis, knowledge of disease and psychological factors.172 ,173
Serology
The third step in the first year is to check the IgA-TG2 or appropriate serology. Despite contradictory results,148 ,149 it is reasonable to assume that positive antibody titres correspond to some gluten intake and there is also some evidence that low TTG titres do not accurately predict mucosal recovery.148 ,149 Tursi et al148 reported that out of 17 patients with persistent villous atrophy 1 year after diagnosis, only 1 (6%) was anti-TG2-positive and 3 (18%) EMA-positive. Vahedi et al149 reported a substantially higher sensitivity for persistent total villous atrophy (73% and 91% for IgA-TG2 and EMA, respectively), but reported no data for partial villous atrophy.
Follow-up biopsy
The last step is the follow-up biopsy. Some authors suggested that it is important to perform a duodenal biopsy to assess the recovery of intestinal mucosa and to exclude RCD and malignancies. However, one recent study of 7648 individuals failed to show that overall mortality was increased in patients with CD with persistent villous atrophy at follow-up biopsy in patients with a median follow-up of more than 11 years.174
In many cases 1 year is too brief a timespan to obtain complete recovery of duodenal mucosa. Tuire et al72 found that IELs were more frequent even 2–5 years after coeliac diagnosis compared with thereafter. Some experts do not routinely perform a follow-up biopsy in asymptomatic patients with negative serology and good adherence. Currently there are no studies indicating an absolute necessity of follow-up biopsy for all patients, but of eight major studies, five150 ,151 ,159 ,160 ,175 examined follow-up biopsies at roughly 2–5 years (table 3). This may be related to costs (economic and psychological) of performing a procedure that in theory should be repeated many times over the years of follow-up as dietary adherence may vary over time.
The authors of this review underline the necessity of distinguishing asymptomatic patients in whom clinical improvement, negative serology, and potentially a follow-up biopsy and good adherence assessed by dietetic review are considered sufficient from symptomatic patients in whom repeated biopsies are needed to rule out RCD or malignancies. Studies report that poor dietary adherence due to occasional lapses is frequent and it is influenced by a number of factors, such as age at diagnosis, knowledge of disease and psychological factors.172 ,173
Histological recovery of duodenal mucosa in adult patients with CD
In adults, neither symptoms72 ,150 ,151 ,156 ,158–160 nor serology149 is reliable to predict small intestinal damage;149 assessing mucosal healing by biopsy is the key. Serum antibodies have poor sensitivity for persistent villous atrophy, especially 1 year or more after diagnosis and institution of a GFD.
Lymphocytic duodenosis is commonly seen on biopsy of follow-up patients. It is rarely symptomatic, although it may also correlate with transgression of adherence from the GFD.72 Early biopsy (at 6 months) is not considered to be optimal.156
Recommendations
-
When adherence is questioned, it should be reviewed by a dietitian. (Grade C)
-
Symptomatic patients should be evaluated more thoroughly than asymptomatic patients. (Grade C)
Gluten challenge
To perform a gluten challenge, a recent study recommends a 14-day gluten intake at ≥3 g of gluten/day (two slices of wheat bread per day) to induce histological and serological changes in the majority of adults with CD.176 The challenge can be prolonged to 8 weeks if serology remains negative at 2 weeks (in the Leffler et al176 study, serology was negative after 2 weeks in all cases, but positive after another 2 weeks).
Medical management during follow-up
Long-term follow-up can be in secondary care clinics or in primary care as long as the expertise is available.177 However, prompt access to specialist centres or secondary care is recommended if any problems arise, and it should be noted that the need for long-term follow-up is controversial.17 ,178
The risk of osteoporosis144 ,179–183 and bone fracture184–190 is increased with CD,2 with one Swedish study showing an excess risk of any fracture of 481/100 000 person-years in adults with CD189 and a British study (13% of individuals were children) 320/100 000 person-years.184 The excess risk is reduced with good dietary adherence and reduction in intestinal villous atrophy, and bone density increases during the first year of GFD adherence.191–195 However, one population-based study found a similar excess risk for fractures before and after coeliac diagnosis (eg, the incidence ratio 5–10 years before CD diagnosis was 1.8 compared with 2.2 some 5–10 years after diagnosis).189
On the basis of current evidence, the suggestion should therefore be to measure calcium, alkaline phosphatase and vitamin D levels (and parathyroid hormone for compensatory increase) at diagnosis and replace as necessary. Calcium intake should be maintained at or above 1000 mg per day.196 Bone density should be measured in those at high risk of osteoporosis; appropriate criteria for judging this are given by the BSG (http://www.bsg.org.uk/images/stories/clinical/ost_coe_ibd.pdf). Repeat bone density investigations (generally after an interval of ≥2 years) should otherwise be considered in patients who have low bone density on index measurement following initiation of appropriate treatment, or who have evidence of ongoing villous atrophy or poor dietary adherence. Postmenopausal women with CD may require supplementation in addition to the GFD.197 Loss of bone density at a greater than expected rate should prompt measurement of vitamin D levels, dietary review of adherence, consideration of repeat intestinal mucosal biopsy and review of additional risk factors such as hypogonadism.
Hyposplenism198 associated with CD may result in impaired immunity to encapsulated bacteria, and an increase in such infections has been demonstrated in CD.199–201 Hyposplenism does not seem to correlate with duration of GFD.198 Vaccination against Pneumococcus is therefore recommended.202 However, it is unclear whether vaccination with the conjugated vaccine is preferable in this setting and whether additional vaccination against Haemophilus, Meningococcus and Influenza203 should be considered if not previously given.204 It should also be noted that patients with CD may have a weaker response to hepatitis B vaccination than normal.205 ,206
Recommendations
-
Newly diagnosed patients should have vaccination for Pneumococcus. (Grade C)
-
Bone density should be measured after 1 year of diet in patients who have additional risk factors for osteoporosis or if over the age of 55 years. (Grade D)
-
Adult patients with CD should have a calcium intake of at least 1000 mg per day. (Grade D)
-
Patients with CD require follow-up by a dietitian and/or clinician with an interest or expertise in this field. (Grade D)
-
Patients should have annual haematological and biochemical profiles. (Grade D)
-
A GFD is the core management strategy for prevention of osteoporosis. (Grade D)
Screening for CD
Table 4 lists the WHO criteria for general population screening. CD fulfils many of these criteria but not all of them as yet. Our recommendation is active case finding but not mass screening. Given that CD is a common disease (about 1% of the western population22 ,24) and that there is a therapy (GFD) that most often relieves symptoms, and may even have an effect on the risk of future complications, any patient with signs or symptoms of CD should undergo testing.
CD as a candidate for general population screening
In addition, testing should be carried out in high-risk groups such as those with iron deficiency anaemia,224 Down's syndrome,225 type 1 diabetes mellitus,147 osteoporosis184 ,189 ,226 and IBS when CD is suspected. The coeliac prevalence in these groups typically varies between 2% and 5%.144 ,227 ,228
Recommendations
-
There is insufficient evidence to recommend population screening for CD, however there should be a low threshold for case finding in clinical practice as per National Institute for Health and Care Excellence guidelines.3 (Grade B)
-
Symptomatic first-degree relatives of patients with CD should undergo CD testing. (Grade C)
Quality of life
There has been a growing interest in how patients with CD perceive the impact of their diagnosis, how their psychological state is influenced by the disease and the effect of GFD. Several studies report that patients with CD have lower QoL scores than the general population.229–232
Though some studies have found improved QoL with GFD treatment in symptomatic and screening-detected patients,222 ,233 others suggest that any benefit of diet is restricted to those presenting with symptomatic disease.214 ,220 ,221 Since mood disorders such as anxiety, depression and fatigue are often linked with CD, before and after diagnosis, it is likely that they may contribute to the effect upon QoL. A recent meta-analysis234 suggested that depression, but not anxiety, is more common in adults with CD. Also fatigue has been linked to undetected CD.235
Gluten-free diet
The mainstay of treatment of CD and DH is a GFD. The term gluten should be used to indicate not only wheat-based proteins (gliadins), but it also includes those from barley (hordeins) and rye (secalins), and cereal hybrids such as triticale.236 Originally oats were also avoided in the GFD. Earlier research indicates that oats uncontaminated by gluten are probably safe for patients with CD.237 ,238 This is important because oats contain soluble fibre, are able to lower blood glucose and attenuate insulin response.239 The appearance of symptoms related to introducing oats might be due to cross contamination. Also a small percentage of patients with CD may be sensitive to oats240 and develop symptoms or even mucosal damage.241 ,242 Patients with CD should be educated to avoid cereals and food containing gluten (breakfast cereals, flours, pasta, cakes, biscuits, sauces etc) derived from wheat, barley or rye and food made from gluten-contaminated oats, and encouraged to eat naturally occurring gluten-free foods and alternative sources of starch (corn, rice, potatoes etc). Levels of susceptibility to gluten contamination of food vary among patients with CD. Although it has also been suggested that the acceptable threshold for gluten content in gluten-free products can from the clinical point of view be set at 100 ppm (¼ mg/kg),243 the Codex Alimentarius Commission of the WHO issued new guidelines for gluten content of processed food in 2008 and a law from the European Commission (EC41/2009), effective since January 2012, stipulated that foods labelled as ‘gluten free’ should contain ≤20 parts per million of gluten, and that this gluten content is safe for the coeliac population. The 20 ppm threshold for gluten-free food is also accepted by the US Food and Drug Administration, effective since August 2013. Newly diagnosed patients should be referred to a dietitian to discuss dietary management.243 It is important that they are educated not only to avoid gluten but also to have a sufficient intake of nutrients, vitamins, fibre and calcium present in their GFD.244 Recent data also indicate that a strict GFD might be of help in reaching ideal body weight, whether an individual is underweight or obese at diagnosis.245 Data suggest that adherence to the GFD is better achieved when the patient is well educated and supported by carers and families. However, there is a wide variation in provision of dietary consultation services for patients diagnosed with CD in the world. A survey indicates that dietetic support may be underprovided in the UK.246
Safe gluten intake?
As shown previously, diagnosis and advice on a gluten-free diet often lead to mucosal recovery, while poor adherence to diet slows or hinders complete recovery.
Whilst contamination of the diet by gluten may be unavoidable and increased IEL counts are associated with less severe nutritional and metabolic consequences than classic CD with villous atrophy, patients may have significant signs and symptoms related to this disorder. A clinical response and mucosal recovery can be achieved by strict adherence to a gluten-free diet. On balance, data support treatment of patients with CD regardless of the degree of mucosal damage.247
A recent review on ‘safe’ gluten levels argues that daily intakes of <10 mg have no effect on mucosal histology,248 whereas definite alterations are caused by a daily intake of 500 mg and observable alterations by 100 mg. A calculated daily intake of 30 mg seems not to harm the mucosa. Therefore, at present, a safe limit could be set between 10 and 100 mg.249 The most comprehensive systematic review (35 studies) suggests that while the amount of tolerable gluten varies among people with CD, a daily gluten intake of <10 mg is unlikely to cause significant histological abnormalities.248
There is extensive research on GFD and how this may influence the clinical course. While a recent study of more than 7000 individuals undergoing follow-up biopsy found no association between persistent villous atrophy (likely to signal poorer dietary adherence) and overall mortality,174 this does not rule out that poor dietary adherence is negative for specific health outcome, such as autoimmune disease,250 ,251 pregnancy outcome (two studies have found poor foetal outcome in pregnant women with undiagnosed CD but not in diagnosed CD41 ,252), and especially the risk of lymphoma. Most studies on lymphoma risk according to dietary adherence point towards a protective effect from GFD but the studies have so far been small in size,212 ,253–255 preventing firm conclusions. However, recently Lebwohl et al175 found a statistically significantly increased risk of lymphoma in patients with CD with persistent villous atrophy compared with those with mucosal healing.
A frequent complaint of patients with CD is that they experience limitations in their social life because of difficulty accessing gluten-free meals or concern about the safety of food when eating out. The patients’ worries are justified as a survey shows that chefs’ knowledge about CD is lower than that of the general public.256 Therefore, education about a GFD needs to be directed to catering personnel. Another criticism is that the availability of gluten-free food is clearly limited in more rural areas and shopping for gluten-free food is time consuming. In most countries, high-quality gluten-free products are available in supermarkets or in special health food stores and on the internet, but the cost of gluten-free food is much greater than the equivalent wheat-based foods.257 ,258
Recommendations
-
Patients should adhere to a GFD and have an intake of less than 10 mg gluten per day. (Grade B)
-
Gluten challenge is not recommended in the ordinary patient with CD, but in patients in whom the diagnosis remains unclear despite a follow-up biopsy, gluten challenge should be performed. (Grade C)
-
Patients may commence gluten-free oats at diagnosis. (Grade D)
-
A GFD is recommended to decrease the excess risk of adverse foetal outcome and of lymphoma among patients with CD. (Grade C)
Patient information and support
Patient support should not be a monologue by a physician, but instead a two-way communication involving the patient, and his/her family. Ideally, collaboration between the patient, the patient's family, an expert dietitian and interested physician should be the setting in which the GFD should be initiated. Family involvement is very important as the disease (including GFD) will inevitably affect family members, but also joining a national coeliac support group can help patients cope with their disease.
Patients need information, reassurance and the opportunity to learn at their pace about the rather challenging demands of a GFD. They particularly need to be encouraged and motivated to adapt to and maintain a GFD. Continued professional engagement in their follow-up care is likely to help sustain that motivation. Unfortunately, many patients report that they are not satisfied with the amount and quality of the information offered by their physician.216 Physicians should inform patients even before the CD diagnosis, offering information about serological testing, and what it means to undergo a small intestinal biopsy. Worries are common in patients awaiting a biopsy259 and the physician should address this anxiety.
Patients may want information about when and where CD occurs, its aetiology and whether it is a common disease or not. Understanding the vital role of GFD in the management of CD is important for dietary adherence.140 Attaining dietary adherence may be particularly difficult in patients identified through screening. Patients with CD often report reduced QoL because of dietetic restrictions and this must be taken into account.260–263 Part of the reduced QoL may be due to social restrictions, such as not being able to eat out with friends, and the economic burden from the more expensive GFD.258 ,264 Patients should also be informed that while most symptoms are likely to go away on a GFD, some symptoms may persist (see section on ‘Non-responsive CD’).
Recommendation
-
At diagnosis, patients should be encouraged to join their national coeliac support group. In the UK, patients should be advised about gluten-free items on prescription (FP10), details of which are available from http://www.coeliac.org.uk.
Non-responsive CD
After adoption of the GFD, 4–30% of patients with CD report persisting symptoms and are considered to be affected by non-responsive CD (NRCD).265 Once the initial diagnosis of CD has been confirmed, adherence to the GFD should be assessed by an expert dietitian as inadvertent or deliberate gluten exposure is the most frequent cause of NRCD.265 ,266
After these initial steps, evaluation should be individualised; however assessment for ongoing enteropathy plays a central role, therefore a follow-up biopsy is needed. Small bowel imaging should be performed in any patient with abdominal pain, persisting fever, obstruction, anaemia, gastrointestinal bleeding or unexplained weight loss.267 If duodenal biopsy does not reveal a persistent enteropathy, symptoms are likely to be due to a second condition.265 ,268
While the GFD is efficacious at controlling the signs and symptoms of CD and improving intestinal histology in most patients, treatment of CD is currently imperfect. Over time, virtually all individuals with CD will have symptomatic exacerbations due to gluten exposure and up to 30% of patients will have symptoms severe or chronic enough to visit their treating physician265 ,266
Comprehensive monitoring of patients with CD requires assessment for NRCD and efficient evaluation for potential aetiologies. NRCD may be considered in three categories.265 ,266 ,269 First and most commonly, NRCD is due to continued dietary exposure to gluten.265 ,266 ,269 Second, it may be due to a pre-existing or coincidental condition causing symptoms that resemble CD which may have led to the detection of otherwise asymptomatic CD—this includes IBS33 or colonic malignancy causing anaemia.270 Finally, it may be due to conditions associated with CD,268 such as secondary lactose intolerance,265 pancreatic exocrine insufficiency,271 small bowel bacterial overgrowth, microscopic colitis272 and cow's milk protein sensitivity.
Very occasionally a state refractory to gluten withdrawal occurs, referred to as RCD. Figure 2 suggests an outline for investigating patients with NRCD.

{kind=link}
{kind=link}
Investigation of the patient with non-responsive coeliac disease (NRCD). Based on a figure by Mooney et al.305 FODMAPs, fermentable oligosaccharides, disaccharides, monosaccharides, and polyols; GI, gastrointestinal; HLA, human leucocyte antigen; RCD, refractory coeliac disease; SIBO, small intestine bacterial overgrowth.
In symptomatic patients with ongoing enteropathy and RCD, coeliac-related malignancies and disorders that mimic CD (table 2) must be excluded. RCD is defined as persistent or recurrent malabsorptive symptoms and/or signs with villous atrophy despite a strict GFD for more than 12 months in the absence of other causes of villous atrophy or malignant complications and after confirmation of CD.7 RCD is subdivided into type I (RCDI) and type II (RCDII). The most important aspect of differentiating RCDI and RCDII is demonstration of a monoclonal population of T cells or aberrant T cells in the latter. There are different methods available for this, including genetic analysis of T-cell receptor clonality, immunohistochemistry and flow cytometry. Most laboratories will employ at least two methods but their relative contributions remain uncertain. Currently there is variation in the criteria used to diagnose RCDII. The two factors that support the diagnosis of RCDII include loss of normal surface markers CD3 and CD8 with preserved expression of intracytoplasmic CD3 and detection of monoclonal rearrangement of T-cell receptor chain.120 Areas of uncertainty include the use of immunohistochemistry versus flow cytometry for IEL classification and the relative prognosis of patients with discordant IEL and T-cell studies.
Patients with normal CD3 and CD8 expression and no evidence of T-cell monoclonality have RCDI with a good prognosis. Patients with RCDII have a poorer prognosis, due predominantly to nutritional complications and transformation into enteropathy-associated T-cell lymphoma (EATL).273 ,274 Ulcerative jejunoileitis (UJI) is a rare condition characterised by inflammatory ulceration of the small bowel that arises in RCD.274–276 The finding of UJI should raise suspicion for lymphoma.
There is no standard treatment for RCD. Elemental diets, systemic steroids, oral budesonide, oral thioguanines including azathioprine are used in RCDI (and sometimes are beneficial), but have limited benefit in RCDII.277 ,278 In RCDII, cyclosporine, cladribine and high-dose chemotherapy with autologous stem cell support have been reported; however, therapy must be individualised and include surveillance for EATL.120
EATL is a rare lymphoma strongly associated with RCDII,273 which carries a poor prognosis with a cumulative 5-year survival of less than 20%.279 ,280 Currently, two groups of EATL are recognised281: EATL type I accounts for 80–90% of all cases and is a large cell lymphoma exclusively associated with CD.282 In contrast, EATL type II has not been associated with CD.281 ,283
The poor prognosis of EATL is determined by extent of disease at diagnosis, multifocal small bowel involvement, poor general health and presence of complications including perforation that preclude chemotherapy.120 Presence of RCDII is associated with a poor prognosis compared with isolated EATL in CD without RCDII. There is no proven effective treatment in RCDII, though a number of strategies have been proposed, and patients should be referred to a tertiary centre to optimise their management.
Finally, numerous studies have confirmed the association between CD and B-cell lymphoma284–286 and CD and small intestinal adenocarcinoma.42 ,254 ,287–289
Recommendations
-
Patients with persistent symptoms despite a GFD should have a follow-up biopsy. (Grade B)
-
In symptomatic patients with ongoing enteropathy and RCD, coeliac-related malignancies and disorders that mimic CD must be excluded. (Grade C)
-
Small bowel imaging should be performed in any patient with abdominal pain, fever, obstruction, anaemia, gastrointestinal bleeding or unexplained weight loss. (Grade D)
-
Patients with RCD should be referred to a tertiary centre to optimise their management. (Grade D)
Novel treatment
The role of non-dietary therapies—as an adjunct or as an alternative to the GFD—has yet to be ascertained. Of the candidate approaches, immunotherapy (including hookworm exposure)290 ,291 is currently explored as an alternative to GFD but, even if successful, is unlikely to benefit all patients. The role of glutenases (propyl endopeptidases) and tight junction regulators (table 5) are likely to be in reducing the threshold response and optimising the benefits of gluten restriction rather than allowing a normal gluten-containing diet.
Novel treatment in CD
Table 5 contains a list of potential novel treatments. None of the available novel treatments can as yet (January 2014) be recommended for use outside clinical trials.
Recommendation
-
None of the available novel treatments can as yet be recommended for use outside clinical trials. (Grade D)
Discussion
We recommend testing for CD in those with suggested symptoms or syndromes, especially if they have a first-degree relative with CD.53 There is not yet sufficient evidence to support indiscriminate general population screening. Specific serology such as IgA-TG2 and IgG-DGP with or without a strategy for determination of total IgA level should be the preferred serologic strategy for detection of CD. Ideally a combination of serology and biopsies done on a gluten-containing diet will then provide the most robust diagnosis of CD. Additional testing may be necessary for those with less than clear-cut results.
We recommend a duodenal biopsy before the diagnosis of CD. This contrasts with the recent ESPGHAN recommendations where a duodenal biopsy is optional in symptomatic paediatric patients in whom the IgA-TG2 level exceeds 10 times the upper limit of normal, EMA antibodies are positive on a separately taken blood sample and HLA typing is positive for DQ2 or DQ8.1 CD has been linked to a large number of symptoms (table 1 in the ESPGHAN review on paediatric CD lists 16 such symptoms and signs with additional symptoms in the text).1 There is a risk that all symptoms, independent of their origin, may be taken as a sign of CD when the sensitivity and specificity of even gastrointestinal symptoms are moderate in CD.304 Other reasons to require a small intestinal biopsy prior to diagnosis is that not all commercial IgA-TG2 kits are of high quality,78 and that alternative diagnoses may be more common, and sometimes serious, in adults with suspected CD. Finally, an initial biopsy is important for the follow-up of patients, especially those whose condition is non-responsive to a GFD.
Adequate (more than four) biopsies should be taken, from the distal duodenum and the duodenal bulb to maximise diagnosis. The threshold for abnormal IELs is ≥25/100 enterocytes, but for a definite diagnosis villous atrophy is required. However, lesser degrees of damage (≥25 IELs but no villous atrophy) combined with positive serology (IgA-EMA, TTG or IgG–DGP) may also represent CD (‘probable CD’),
The treatment of CD is a lifelong and strict GFD. The goal of treatment is to relieve symptoms, achieve mucosal healing, avoid complications of CD, and have a good QoL with a nutritionally complete GFD. This is best achieved when patients are motivated and receive expert information in a collaborative way, with resources including expert dietitians and interested medical care. Follow-up of CD is needed to ensure response to symptoms, prevention of consequences, and continued maintenance of motivation to remain gluten free.
Discussions upon the issue of repeating duodenal biopsies were intense in our group. There is no conclusive evidence of the benefit of universal follow-up biopsy, and we were unable to reach a consensus. Some would undertake follow-up biopsies in all patients with CD after 2–5 years on a GFD (table 3). There are others who reserve follow-up biopsies for those in whom there are persistent and recurrent symptoms or those for whom the follow-up biopsies are necessary to help confirm the diagnosis in the setting of continued diagnostic uncertainty.
There are novel techniques that may enhance the sensitivity of endoscopic examination, of which the immersion technique is probably the most feasible currently. An understanding of the precise value of various serologic strategies in the detection of CD is continuing to evolve, as are advances in therapies that may ultimately provide some mitigation of the impairment of the QoL that is inherent in a strict GFD.
Ultimately, this review should not be regarded as fixed. Instead it represents our understanding of what is best in adult CD management according to current knowledge. It is likely, and indeed we hope, that there will be substantial progress in diagnosis, evaluation and management of CD to reduce the burden on the patient and society. We anticipate that future updated versions of these guidelines will be based on evolving published literature that may have an audit or research base relevant to adult CD. Data in this review were mostly retrieved through searches of PubMed. We acknowledge that there are other medical databases and we cannot rule out that had we searched more than one database we may have identified additional relevant studies that have now been left out of this review.
Areas for future research
The panel of experts recognise that the main challenge in the future is to allocate available resources effectively to reduce the burden of disease from CD.
Future research should focus on the following areas:
-
how to induce long-term remission without a GFD, that is, novel therapies and vaccine;
-
better understanding of the disease processes, including genetics and antigen presentation;
-
prevent and cure extra-intestinal manifestation and complications, including infections;
-
be able to assess tolerable amount of gluten for individual patients;
-
define the role of duodenal biopsy, serology and point of care testing at diagnosis and follow-up;
-
find a robust and valid blood marker for diagnosis and monitoring of the disease;
-
understand the pathogenesis of RCDI and II.
Acknowledgments
We thank all the patients involved in the related focus groups and discussions.
References
Footnotes
-
Contributors JFL and DSS initiated the study. JFL coordinated the project and conducted the web surveys on CD. DSS supervised the project and served as the representative of the British Society of Gastroenterology (BSG). All authors contributed to the literature searches, contributed to the writing of the manuscript, and approved the final version of the manuscript.
-
Funding JFL was supported by the Swedish Research Council (522-2A09-195) and the Swedish Society of Medicine while writing the draft of this paper. JCB was supported by Consejo en Investigacion en Salud GCABA. KK was supported by The Academy of Finland, the Competitive Research Funding of the Tampere University Hospital (9N062), and The Sigrid Juselius Foundation. JAM was supported by DK 57892. DSS has received an educational grant from Biocard and Simtomax to undertake an investigator-led research studies on CD and/or gluten sensitivity, and an educational grant from Dr Schär (a gluten-free food manufacturer) to undertake an investigator-led research study on gluten sensitivity.
-
Competing interests DAvH, TRC: received grant support from Coeliac UK. PHRG: scientific advisory board of Alvine Pharmaceuticals and ImmusanT. DAL: Ironwood Pharmaceuticals, Alvine Therapeutics, Alba Pharmaceuticals, Shire Therapeutics, In Nova Diagnostics and CRICO Risk Management Foundation. JAM: consultant for Alba Pharmaceuticals, Alvine Therapeutics, Flamentera, 2GPharma Inc and ImmusanT. KEAL: ImmusanT, Regeneron and Alvine Pharmaceuticals. DSS: received educational grants from Coeliac UK, Biocard, Simtomax and Dr Schär (a gluten-free food manufacturer) to undertake an investigator-led research study on CD and/or gluten sensitivity.
-
Ethics approval British Society of Gastroenterology.
-
Provenance and peer review Not commissioned; externally peer reviewed.