Article Text

Abstract
Background and aims Peripancreatic fat necrosis occurs frequently in necrotising pancreatitis. Distinguishing markers from mediators of severe acute pancreatitis (SAP) is important since targeting mediators may improve outcomes. We evaluated potential agents in human pancreatic necrotic collections (NCs), pseudocysts (PCs) and pancreatic cystic neoplasms and used pancreatic acini, peripheral blood mononuclear cells (PBMC) and an acute pancreatitis (AP) model to determine SAP mediators.
Methods We measured acinar and PBMC injury induced by agents increased in NCs and PCs. Outcomes of caerulein pancreatitis were studied in lean rats coadministered interleukin (IL)-1β and keratinocyte chemoattractant/growth-regulated oncogene, triolein alone or with the lipase inhibitor orlistat.
Results NCs had higher fatty acids, IL-8 and IL-1β versus other fluids. Lipolysis of unsaturated triglyceride and resulting unsaturated fatty acids (UFA) oleic and linoleic acids induced necro-apoptosis at less than half the concentration in NCs but other agents did not do so at more than two times these concentrations. Cytokine coadministration resulted in higher pancreatic and lung inflammation than caerulein alone, but only triolein coadministration caused peripancreatic fat stranding, higher cytokines, UFAs, multisystem organ failure (MSOF) and mortality in 97% animals, which were prevented by orlistat.
Conclusions UFAs, IL-1β and IL-8 are elevated in NCs. However, UFAs generated via peripancreatic fat lipolysis causes worse inflammation and MSOF, converting mild AP to SAP.
- OBESITY
- PANCREATITIS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Peripancreatic necrosis may be associated with severe acute pancreatitis (SAP).
Cytokines and unsaturated fatty acids (UFAs) are increased in human and animal models of acute pancreatitis.
It is unclear whether these are markers versus mediators of SAP.
What are the new findings?
Pancreatic necrotic collections have higher interleukin (IL)-1β, IL-8 and UFA levels than pancreatic cystic neoplasms.
UFAs at concentrations less than in necrotic collections but not IL-1β and IL-8 cause necro-apoptosis.
Peripancreatic fat lipolysis causes multisystem injury independent of pancreatic necrosis.
How might it impact on clinical practice in the foreseeable future?
The distinction between a marker and mediator of SAP is made clear in this study.
It supports that targeting peripancreatic fat necrosis independent of pancreatic necrosis may improve SAP outcomes.
It supports that targeting lipolytic generation of UFAs, but not cytokines, may improve SAP outcomes.
Introduction
Agents elevated in body fluids of patients with severe acute pancreatitis (SAP) and animal models of acute pancreatitis (AP)1 ,2 include adipokines,3 ,4proteases like trypsin,5–8 unsaturated fatty acids (UFAs) such as oleic and linoleic acid (LA)1 ,9 ,10 or cytokines including interleukin (IL)-1β,11 IL-6,12–15 IL-8,12 ,15 ,16 monocyte chemotactic protein-1 (MCP-1)17 and tumour necrosis factor-alpha (TNF-α).13 Scoring systems to risk stratify AP include these.17–20 Clinically, however, SAP markers versus mediators are indistinguishable as causality is difficult to establish while studying human AP in isolation. Moreover, human SAP is independent of aetiology21–24 (with the exception of hypertriglyceridemic AP25–28), and SAP can occur with minimal pancreatic necrosis.29–31
In contrast, severity of animal models of AP is based on the aetiology (eg, bile salt vs caerulein32 ,33) causing severe necrosis alone or with worse outcomes,33 and therapies are considered relevant based on their efficacy in aetiologically different models. This rationale and lack of distinction between marker and mediators may partly explain the limited benefit noted in several clinical trials of AP, targeting proteases,34–43 reactive oxygen species44 and inflammatory mediators45 and mixed outcomes noted in the few case reports of anti-TNF therapy,46–50 though these targets seem scientifically sound based on animal models of AP. Interestingly, hereditary pancreatitis due to cationic trypsinogen (PRSS1) gene mutations, despite its high penetrance in initiating AP does not cause necrotising pancreatitis (NP) or AP-related mortality.51 Similarly, while cytokines are elevated in SAP, some studies show cytokines do not induce SAP-associated outcomes,52–56 and others have shown IL-657 and TNF-α58 to be protective in AP. These controversies support the need to look for alternate targets, which are mediators of SAP rather than its markers.
Several epidemiological studies suggest obesity59–66 or increased intra-abdominal fat is associated with SAP.63 ,67–69 These fat depots may undergo necrosis during AP and this has been linked to SAP for more than a hundred years.70 Peripancreatic fat necrosis is in the spectrum of necrotising pancreatitis,71 ,72 is a part of the revised Atlanta criteria,72 radiographic severity scoring systems (eg, Schroeder and Balthazar scoring)3 ,73 and correlates with worse outcomes during AP.74 ,75 However, it is unknown whether this has a causal role or is a mere marker of severity.
Triglyceride, which forms the bulk of adipocytes that are increased in obesity,76–78 is a good substrate for the lipases released basolaterally during pancreatitis79–82 as noted in human disease.83 ,84 In vitro studies by Mossner et al85 more than two decades ago showed a protective effect of lipase inhibition in vitro but not in vivo AP models.86 Thus, it remains unclear whether it is the exaggerated baseline proinflammatory state87–89 or some other acute mechanism that worsens AP outcomes in obesity.
We recently showed intrapancreatic lipase inhibition reduces necrosis and improves outcomes of biliary AP exacerbated by intrapancreatic fat.90 Thus, the observations that (i) AP outcomes in humans unlike rodent models are independent of aetiology, (ii) AP, associated with peripancreatic necrosis in humans, is severe for unclear reasons and (iii) cytokines have an unclear role in SAP outcomes prompted us to study the mechanisms of worse outcomes in human AP.
To do so, we initially determined the agents that are increased in human pancreatic necrotic collections (NC) that are known to have both pancreatic and peripancreatic necrosis.71 ,72 A PubMed search failed to pull up guidelines or an authoritative text on criteria that may be used to conclusively prove causality in human disease. We thus applied Koch's postulates (http://ocp.hul.harvard.edu/contagion/koch.html) of causality to determine mediators of SAP, whereby the mediator should (A) be present at high levels in an affected source (NC) compared with controls, that is, pancreatic cystic neoplasms (PCN); (B) be isolated from NCs in pure form; (C) convert mild AP in lean rodents to SAP when added in a pure form and (D) be re-isolated in this setting (equivalent to re-isolating the microorganism). We also went a step further to study the effect of preventing mediator generation on outcomes of AP.
We chose a pharmacological approach over a genetic approach since pancreatic lipases have a redundant role,91 dual lipase knockouts are embryonically lethal92 and us using of the mildest AP model, that is, caerulein AP in rats, to study exacerbating factors. Our results highlight the potential role of acute peripancreatic fat lipolysis in worsening outcomes of AP and help distinguish markers from mediators of SAP, along with suggesting a potential approach to improve SAP outcomes.
Methods
Human pancreatic fluid collections
Samples were collected as a part of protocols described previously.90 One of the original 15 NC fluid was too small in volume for the detailed analysis done in this study. Information collected included age, gender, body mass index (BMI), primary diagnosis, disease duration at the time of collection and modality of collection (ie, surgical or endoscopic). Sample collection predated the revised Atlanta criteria.72 For purposes of the study, the clinician classified samples as NCs (collections with solid debris, n=14) or pseudocysts (PCs) (fluid collections, n=11). In our study, all PCs were at least 3 months old. We chose PCN as the non-inflammatory control group (n=10); these included intra-pancreatic mucinous neoplasm, serous cystadenomas and mucinous cystic neoplasms. Minimum disease duration at intervention from the time of diagnosis of AP was 4 weeks, consistent with established guidelines.93 ,94
Animal work
Wistar rats (Charles River Laboratories, Wilmington, Massachusetts, USA) were acclimatised for at least two days prior to use. They were housed with a 12 h light/dark cycle, at temperatures ranging from 21 to 25°C, fed standard laboratory chow and allowed to drink ad libitum. Caerulein was purchased from Bachem (King of Prussia, Pennsylvania, USA). Other agents were procured as mentioned in relevant sections below.
Rat acinar cells were prepared fresh and used as described previously.95–97 Results reported for in vitro studies are from five experiments carried out in duplicates. For these in vitro studies, fatty acids were dissolved as described previously1 and cytokines (IL-1β and keratinocyte chemoattractant (KC)/growth-regulated oncogene (GRO); Peprotech Rocky Hill, New Jersey, USA) were dissolved as per the manufacturer's instructions. Caerulein pancreatitis was induced in rats with two doses of 20 µg/kg of caerulein administered intraperitoneally, for three consecutive days as described previously.97 ,98 This dose has been shown to reliably cause hyperamylasemia and induce oedema and inflammation of the pancreas.97 ,98 Equivalent doses to this have been used by different groups.99 ,100 Rats were sacrificed 3 days after the induction of pancreatitis. For caerulein pancreatitis with IL-1β and KC/GRO (the rat homologue of IL-8), these cytokines were dissolved as per the manufacturer's instruction and a total of six doses were administered at 200 ng/kg/dose and 6.0 µg/kg/dose, respectively, two times per day by tail vein for three consecutive days. These were given along with the caerulein injections (20 µg/kg/dose of caerulein administered intraperitoneally, two times per day, for 3 days). The daily doses are based on the mean serum concentrations of IL-1β (367 ng/L) and IL-8 (12 μg/L) in the severe glyceryl trilinoleate (GTL) model published previously.90 These doses cumulatively (ie, 1200 ng/kg and 36 μg/kg) exceed the median concentrations accumulated over several weeks in the NCs, which are 729 and 497 ng/L, respectively. Doses in these ranges have been used previously in rats53 ,101–103 These animals were sacrificed 2–4 h after the last injection. For caerulein pancreatitis in rats coadministered triolein or triolein+orlistat, a single dose of 3 mL (3% body weight) triolein (TCI, Philadelphia, Pennsylvania, USA) or triolein+orlistat (Cayman Chemical, Ann Arbor, Michigan, USA, 50 mg/kg dissolved in the triolein) was given intraperitoneally 2 h after the first dose of caerulein. Preliminary triolein dosing studies using 2% and 1% body weight were done on five animals per group (see online supplementary figure S3). Studies for this article had 8–12 animals in each group for the lung lavages and histology, with a total of 16–28 for mortality studies. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (Pittsburgh, Pennsylvania, USA) and the Mayo Clinic (Scottsdale, Arizona, USA).
Graphical depiction
Box and whisker plots were used unless there were <8 samples/group, in which case bar graphs were used. Box and whisker plots show the mean (dotted line), median (solid line), 25th and 75th centiles (two boxes) and 10th and 90th centiles (whiskers). Tukey–Kramer test when significant is depicted as ‘*’ in human studies. Summary statistics includes means±SEM or 95% CIs and/or medians and IQRs. All significance levels were evaluated at the two-tailed, p<0.05 level. For animal studies, all groups were compared by analysis of variance (ANOVA) to control. An ‘*’ indicates p<0.05 versus controls (Con) groups. † indicates p<0.05 between treatments with and without the lipase inhibitor orlistat. Other methods are detailed in the online supplementary section.
Results
NCs occur in obese patients and are enriched in UFAs, IL-1β and IL-8
Patients with NCs had a higher BMI than those with PCs (figure 1A). Biliary pancreatitis accounted for 13 of the 14 NCs. NCs had the highest DNA concentration (a marker of dead or exfoliated cells; figure 1B), UFAs (figure 1C, D) and SFAs (figure 1C, F). Of these, oleic (C18:1, 1235±412 μM), palmitic (C16:0, 613±214 μM), linoleic (C18:2, 586±185 μM) and stearic (C18:0, 170±46 μM) acids (figure 1E) were the main non-esterified fatty acid (NEFA). NCs also had the highest IL-1β and IL-8 (figure 2A, B), and resistin was higher in both NCs and PCs compared with PCNs (figure 2C). However, IL-6, MCP-1, nerve growth factor and HGF were similar among all groups (see online supplementary figure S1). The same agents as those increased in NCs (except IL-8) were elevated on combining all inflammatory collections (NCs and PCs) in comparison to non-inflammatory ones (PCNs) (see online supplementary figure S2).

Necrotic collections (NCs) occur in obese patients and are enriched in DNA and unsaturated fatty acids (UFAs). (A) Table showing biometric data, disease duration at the time of intervention and aetiology of pancreatic fluid collections. Patients with pancreatic cystic neoplasms (PCNs) were older than and those with NCs had higher body mass index (BMI) than those with pseudocysts (PCs). p Values for age and BMI (one-way analysis of variance) and gender (two-tailed Fisher's exact test) are mentioned. * indicates groups significantly different (p<0.05) from each other. Box plots showing (B) DNA, (C) proportion of UFAs, (D) amounts of UFA, (E) non-esterified fatty acid (NEFA), (F) SFAs in NC, PC and PCN. * indicates groups significantly different (p<0.05), and ‘NS’ those not significantly different from each other.

Necrotic collections (NCs) are enriched in interleukin (IL)-1β, IL-8 and resistin. Box plots showing (A) IL-1β, (B) IL-8 and (C) resistin levels in log scale being elevated in NC and pseudocysts (PC) compared with pancreatic cystic neoplasms (PCN). * indicates groups significantly different (p<0.05), and ‘NS’ those not significantly different from each other.
UFAs, but not other agents, enriched in NCs or PCs induce necrosis
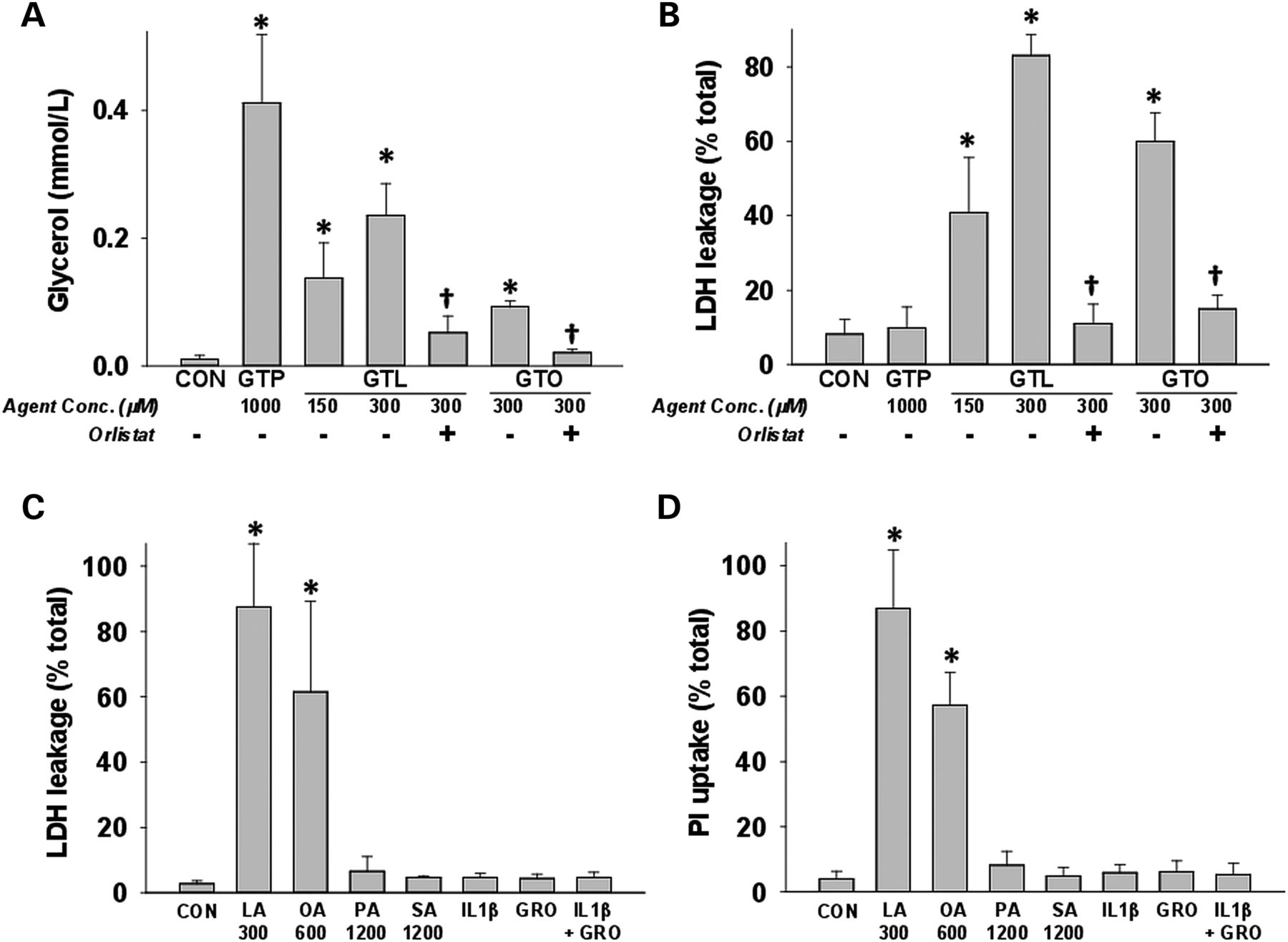
Lipolysis of triglycerides by pancreatic lipases was measured by glycerol generation after incubating acini with glyceryl tripalmitate (GTP), a saturated triglyceride, or the unsaturated triglycerides (UTGs), GTL or glyceryl trioleate (GTO) alone, or with the lipase inhibitor orlistat. Despite glycerol generation from GTL, GTO and GTP (figure 3A), significantly increased cell injury quantified by lactate dehydrogenase (LDH) leakage was only noted in the presence of GTL and GTO (figure 3B). Both lipolysis and cell injury were prevented by orlistat. To identify agents mediating acinar injury, we incubated pancreatic acini with agents increased in NCs or PCs at relevant concentrations and quantified LDH leakage or propidium iodide (PI, figure 3C, D) uptake after 5 h. The UFAs oleic acid (OA) and LA, at half the concentrations noted in NCs, caused acinar necrosis, while SFAs (palmitic acid (1.2 mM) and stearic acid (1.2 mM)), cytokines IL-1β and KC/GRO (the rat homologue of IL-8) alone or in combination, all of which were more than twofold concentrations in NCs, did not. The same was shown to be true for resistin recently.104

Unsaturated fatty acids generated via triglyceride lipolysis induce acinar cell death: (A) glycerol concentrations in medium from acini incubated with saturated ( glyceryl tripalmitate (GTP)) and unsaturated triglycerides (glyceryl trilinoleate (GTL)), glyceryl trioleate (GTO)) are reduced by orlistat. Only GTL and GTO cause cell injury quantified as (B)% lactate dehydrogenase (LDH) leakage, which is prevented by orlistat (50 μM). Acinar necrosis measured at 5 h as %LDH leakage (C) or as propidium iodide uptake (D) induced by various agents elevated in necrotic collections (NCs) and pseudocysts. Only oleic (OA) and linoleic (LA) acids induced necrosis at half their concentrations in NCs. Palmitic acid, stearic acid (SA) and inflammatory cytokines (10 ng/mL each) did not cause necrosis. These are more than two times the concentrations in NCs. * indicates groups significantly different (p<0.05) from control, and † indicates significant reduction of measured parameter upon lipase inhibition by orlistat versus without orlistat. Numbers on X-axis represent micromolar amounts.
Peripancreatic UTG lipolysis, unlike cytokines increased in NCs, worsens AP independent of pancreatic necrosis and acute inflammatory cell infiltration
We have recently shown intrapancreatic UTG lipolysis to cause severe pancreatic necrosis.90 To understand the role of peripancreatic fat and discriminate the role of this acute lipotoxicity from that of cytokines in AP outcomes, we compared caerulein pancreatitis outcomes in lean rats coadministered IL-1β and KC/GRO with those coadministered triolein alone or with orlistat. The triolein dose (3% body weight) was based on (A) fat exceeding 30% body weight in obesity,105 (B) visceral fat averaging >3% body weight in obese humans106 ,107 and (C) preliminary dosing studies on mortality and organ failure (see online supplementary figure S3). The cytokine doses chosen, as detailed in the methods, are relevant to those in NCs and the serum cytokine concentrations found in the severe GTL model published previously.90
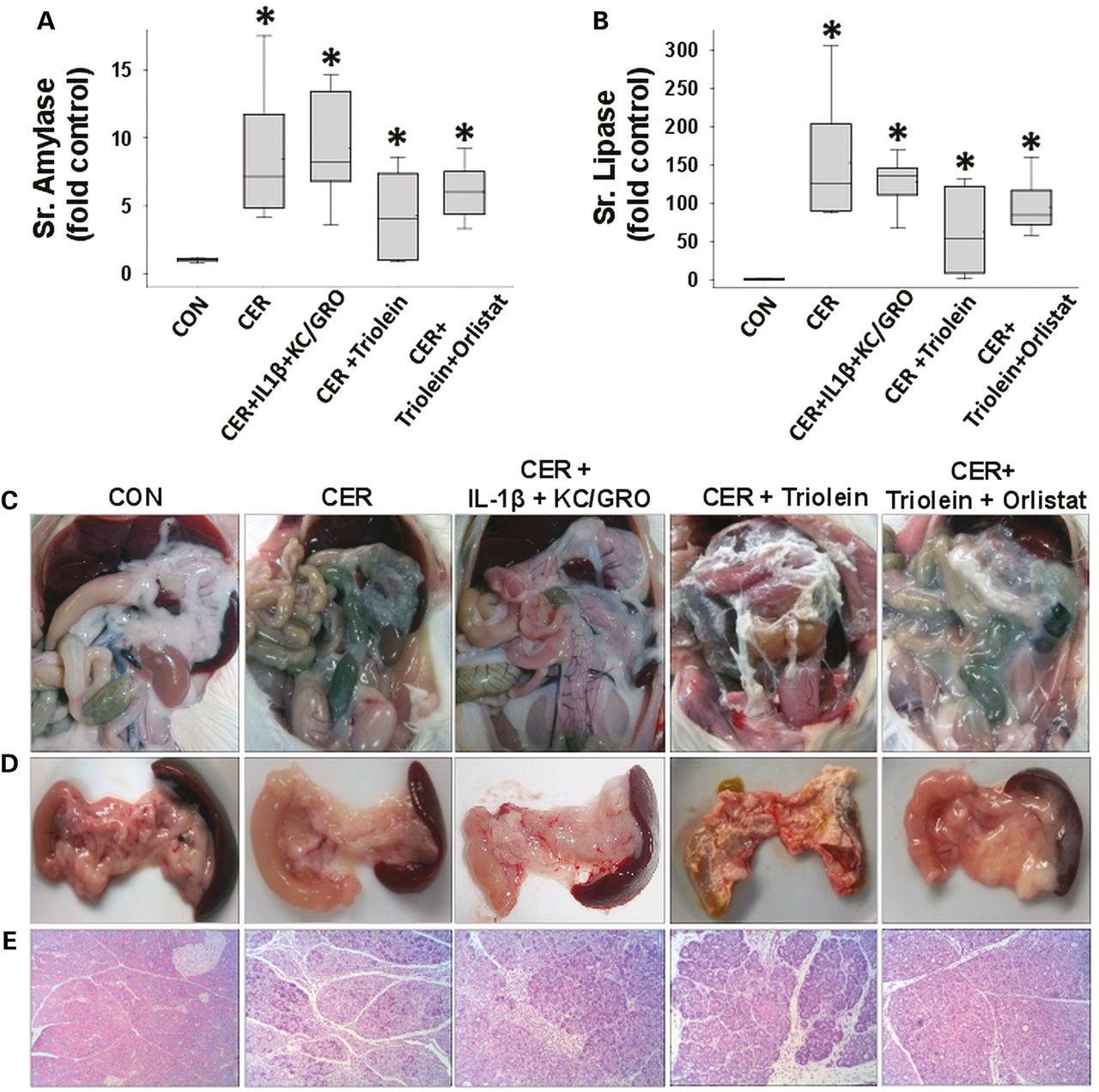
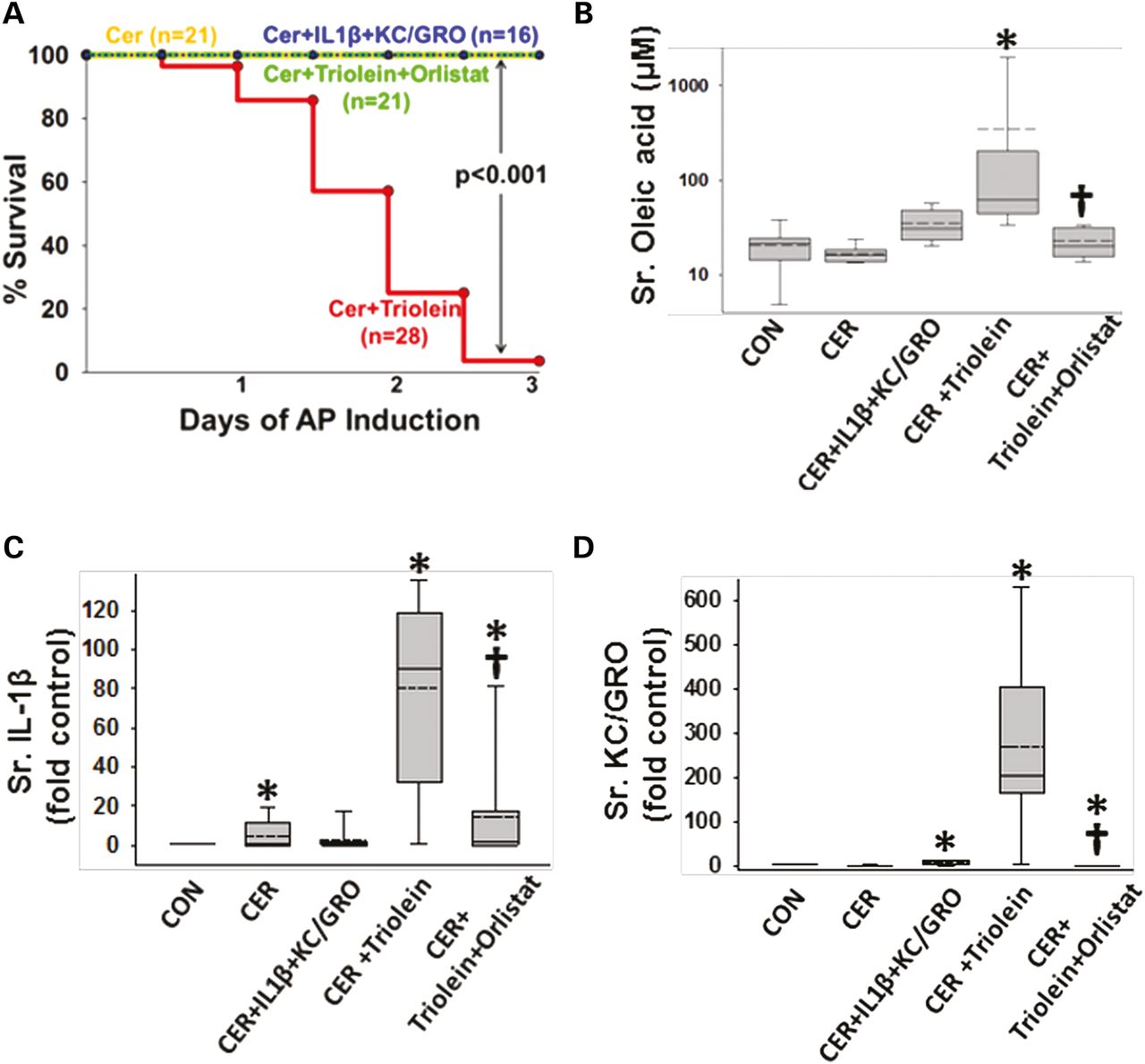
Administration of IL-1β and KC/GRO with caerulein caused a febrile response (38.5±0.5°C vs37.2±0.4°C in controls, p<0.001) consistent with the effects of IL-1β described previously.53 ,102 ,103 The parameters of induction of pancreatitis, that is, increase in serum amylase or lipase more than threefold above controls used in humans108 were similar in all groups (figure 4A, B). However, lipase activity in the ascites of rats with pancreatitis was increased, which was prevented in the orlistat-treated group (see online supplementary figure S4), supporting a local effect on peripancreatic lipolysis. At the time of necropsy, the triolein-treated group had strands of saponified fat in the peritoneal cavity and around the pancreas (figure 4C, D), while those coadministered triolein with orlistat had a thick whitish fluid coating, consistent with the appearance of unhydrolysed triolein. On histology, there was no evidence of pancreatic necrosis histologically among any of the pancreatitis groups (figure 4E). While cytokine coadministration did increase infiltration of myeloperoxidase (MPO)-positive cells into the pancreas (figure 5A3, A6) and lungs (figure 5B3, B6) compared with caerulein alone (figure 5A2, B2), it did not cause mortality in caerulein AP (figure 6A). In contrast rats coadministered triolein had 97% mortality, increased serum oleate, IL-1β and KC/GRO (Figure 6A–D). All these were dramatically improved in the group coadministered orlistat along with the triolein. Triolein administration alone did not result in induction of pancreatitis or any of the changes described above (see online supplementary figure S5).

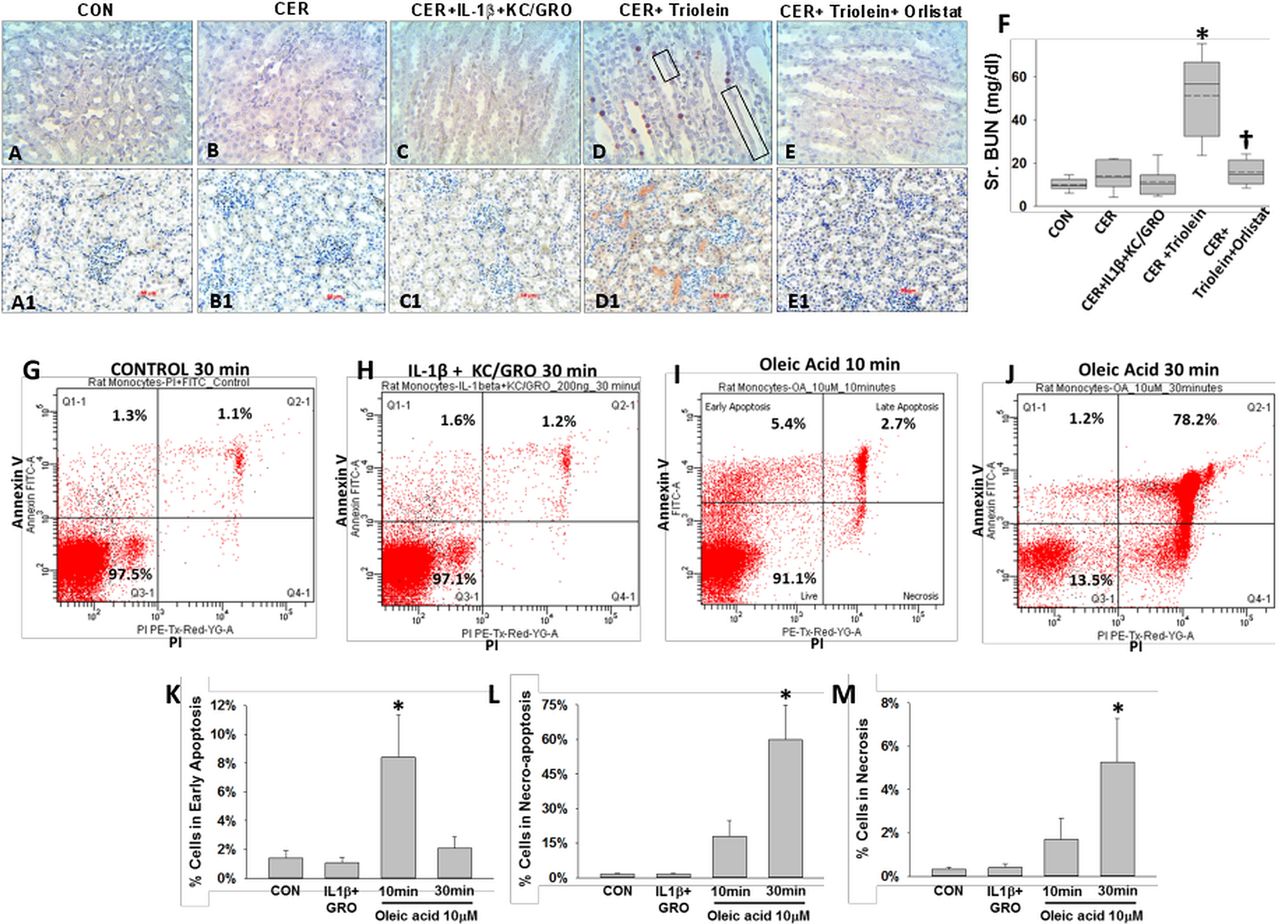
During caerulein pancreatitis, coadministration of interleukin (IL)-1β with keratinocyte chemoattractant (KC)/growth-regulated oncogene (GRO), or intraperitoneal triolein alone or with orlistat does not affect pancreatitis initiation or cause pancreatic necrosis: serum amylase (A) and lipase (B) were similarly increased more than threefold above normal in all groups with caerulein pancreatitis at 1 day. Fat stranding in the peritoneal cavity (C) and surface of pancreata (D) in animals coadministered triolein with caerulein (CER) was prevented by orlistat, with remnant unhydrolysed triolein appearing as a thick whitish liquid coating. (E) Histologically there was no evidence of necrosis in any group. Please note ductal metaplasia-like appearance in survivors up to 3 days.

Interleukin (IL)-1β+keratinocyte chemoattractant (KC)/growth-regulated oncogene (GRO) and triolein coadministration results in worse local and systemic inflammation during caerulein (CER) pancreatitis. Immunohistochemistry images for myeloperoxidase (MPO) in the pancreas (A1–5) and lungs (B1–5), with each group mentioned at the bottom of the image. While caerulein pancreatitis increased MPO-positive cells compared with controls (A2, A6*), IL-1β+KC/GRO coadministration caused a further increase (A3, A6 †), compared with caerulein alone. Systemically, in the lungs, IL-1β+KC/GRO coadministration (B3) and triolein coadministration (B4) significantly increased MPO-positive cells compared with caerulein pancreatitis alone (B2) or other groups (B1,4,5). Orlistat treatment significantly reduced (B6, †) the lung MPO increased noted in the CER+ triolein group.
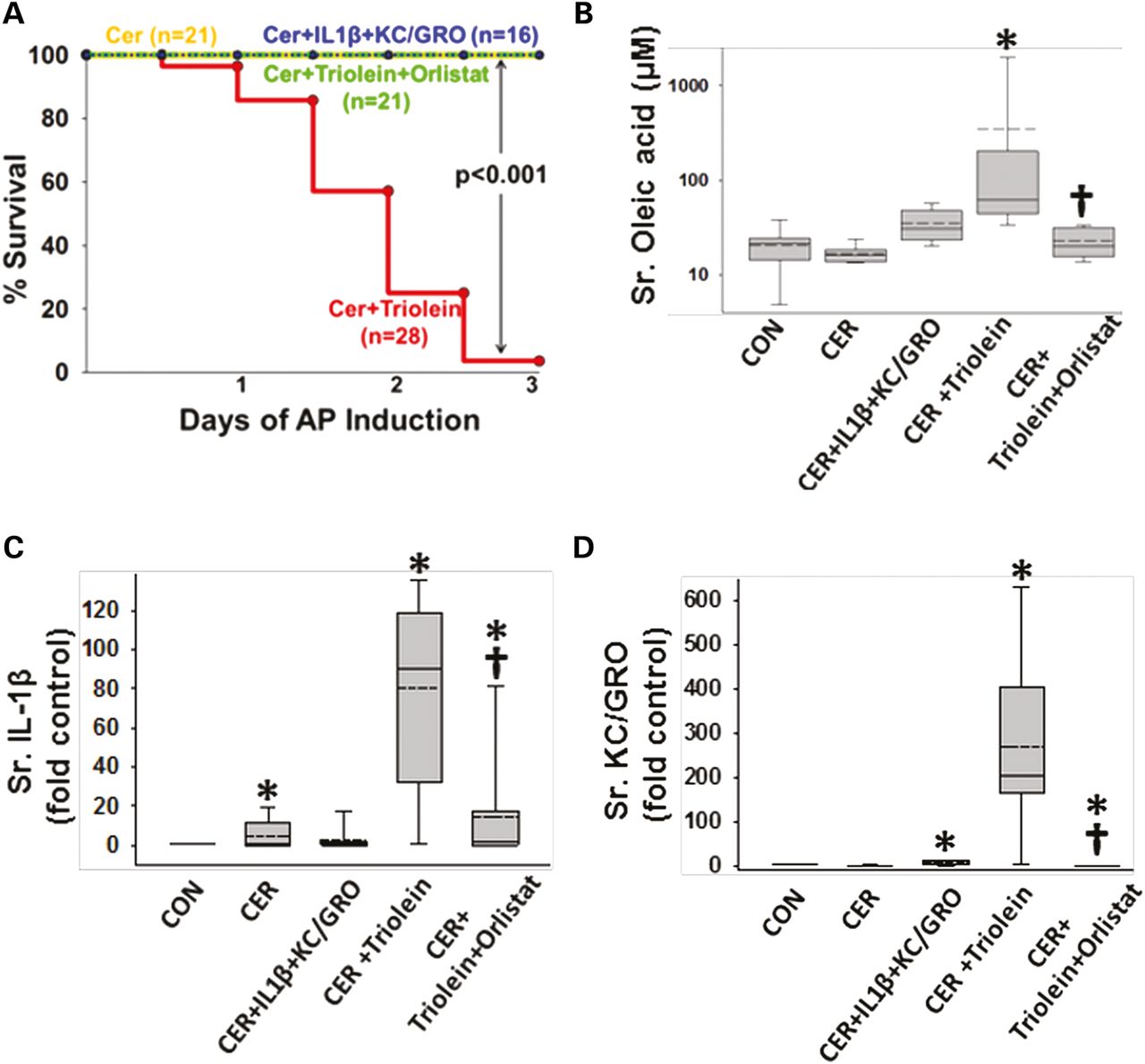
Lipolysis of triolein unlike interleukin (IL)-1β+keratinocyte chemoattractant (KC)/growth-regulated oncogene (GRO) coadministration results in a worse cytokine response and mortality in caerulein (CER) pancreatitis. (A) Kaplan–Meier curves showing time course of mortality in the triolein coadministered group (red), which is prevented by orlistat (green) and is absent in other groups. Number per group is mentioned in parenthesis. Serum oleate (B), IL-1β (C) and KC/GRO (D) are significantly increased in the acute pancreatitis (AP) group coadministered triolein and reduced by orlistat. Multiple comparisons were done using one-way analysis of variance. Significant (p<0.05) difference from controls is denoted with an *; † over the CER+triolein+orlistat group indicates significant difference from CER+triolein.
Lipotoxic cell death mediated by UFAs results in multisystem organ failure
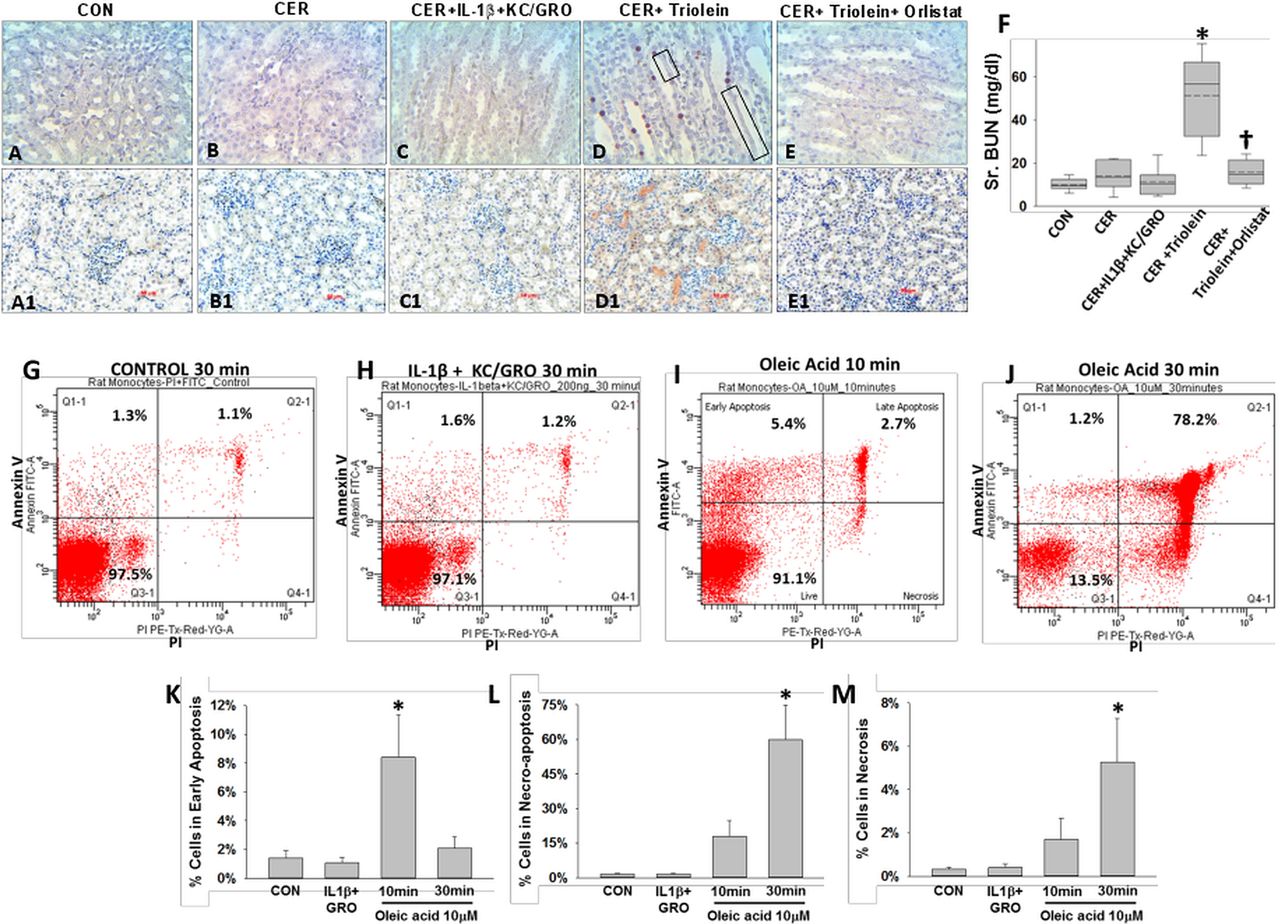
Mortality from SAP in humans can occur with multisystem organ failure (MSOF) independent of necrosis.72 ,74 Thus, we looked for evidence of kidney failure and lung injury. Rats with pancreatitis coadministered triolein had respiratory failure evidenced by a reduction of oxygen saturation from 97±1% at baseline to 89.3±0.9% (p<0.01) 2 h prior to sacrifice. This was associated with an increase in terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)-positive cells in the lungs (figure 7A–F), as previously shown in lipotoxic acute respiratory distress syndrome.1 ,109–112 The bronchoalveolar lavage of the triolein coadministered group had an increase in fluorescein isothiocyanate leakage dextran, LDH leakage, protein and PI-positive cells (* in figure 7G–J). All these detrimental changes in parameters of lung function and lung injury were absent in all other groups and completely prevented in the rats coadministered orlistat († in figure 7E–J). The kidneys of the caerulein+GTO group had kidney injury evidenced by TUNEL-positive tubular cells, loss of tubular cells (boxes in figure 8D), kidney injury molecule-1 (KIM-1) positivity in the renal tubules (figure 8D1) and a higher serum blood urea nitrogen (figure 8F), consistent with renal failure. These adverse outcomes were prevented by orlistat (figure 8E, E1, F [†]). The administration of triolein alone (see online supplementary figure S5) or the cytokines along with caerulein (figures 7C and 8C, C1 and graphs in figures 7 and 8) did not result in these adverse outcomes. Peripheral blood mononuclear cells (PBMCs) treated with a fraction of serum OA concentrations (10 μM, figure 8I, J) had a large increase in apoptosis initially, which progressed to predominantly necro-apoptosis. IL-1β and KC/GRO at more than two times serum concentrations did not cause this (figure 8H). These observations suggest that acute lipolytic generation of UFAs from peripancreatic fat worsens outcomes of AP by UFA-mediated necro-apoptotic distant organ injury.

Lipolysis of triolein unlike interleukin (IL)-1β+keratinocyte chemoattractant (KC)/growth-regulated oncogene (GRO) coadministration results in lung injury during caerulein (CER) pancreatitis: images of terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) of lungs (A–E) with each group mentioned at the bottom of the image. There was significantly more positive staining in the lungs (D) of CER+triolein group (*), which is prevented by orlistat (E and F), depicted as †. Bronchoalveolar lavage (BAL) fluid was analysed for fluorescein isothiocyanate (FITC) fluorescence (G), lactate dehydrogenase (LDH; H), total protein (I) or stained with propidium iodide, and the fluorescent cells were counted (J). Significant (p<0.05) difference from controls is denoted with an *; † over the CER+triolein+orlistat group indicates significant difference from CER+triolein.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lipolysis of triolein unlike interleukin (IL)-1β+keratinocyte chemoattractant (KC)/ growth-regulated oncogene (GRO) coadministration results in necro-apoptotic injury and renal failure during caerulein (CER) pancreatitis: images of terminal deoxynucleotidyl transferase dUTP nick end labelling (A–E) and immunohistochemistry for kidney injury molecule-1 (A1–E1) in kidneys, with each group mentioned above the image. There was positive staining in the CER+triolein group, sloughing of renal tubular cells (black boxes) along with renal failure evidenced by an increase in serum blood urea nitrogen (BUN) (F, *), which is prevented by orlistat depicted as †. Rat peripheral blood mononuclear cells from the control group (G), those treated with IL-1b+KC/GRO (200 ng/mL each for 30 min; H), 10 μM oleic acid (OA) for 10 min (I) or 30 min (J) were stained for annexin V and propidium iodide (PI) and analysed by flow cytometry. OA significantly increased (*) early apoptosis at 10 min (K), which progressed to predominantly necro-apoptosis by 30 min (L) with progressively increasing necrosis (M).
Discussion
In this study, we initially found NCs to have high levels of UFAs, IL-1β, IL-8 and resistin, which remained significant after adjusting for age, gender and BMI. Peripancreatic necrosis is a part of necrotising pancreatitis.71 ,72 Having previously shown that intrapancreatic UTG lipolysis worsens pancreatic necrosis,90 in this study we tested whether lipolysis of extra-pancreatic UTG or cytokines caused MSOF independent of pancreatic necrosis. Applying Koch's postulates to sort out which agent most closely fulfils these by worsening AP to SAP, we identified UFAs (with oleate as the prototype, online supplementary figure S6) as being the best candidates for being SAP mediators. The acute conversion of caerulein AP to lethal disease by peripancreatic UTG lipolysis in lean rats suggests acute UFA toxicity worsens AP and not the baseline or exaggerated proinflammatory response of obesity. Using the mild caerulein model, we also learn that like in human disease,21–24 outcomes of AP can be unrelated to aetiology, with a modifier such as lipotoxicity. Lastly, we learn that peripancreatic fat necrosis can worsen outcomes independent of pancreatic necrosis by distant organ injury. Recent clinical literature 3 ,74 ,75 ,113 and the revised Atlanta criteria72 accept peripancreatic necrosis as a risk factor for SAP. These parallels we note have mechanistic, translational and therapeutic relevance, and are discussed below.
Peripancreatic fat necrosis causes SAP in the absence of pancreatic necrosis
Early mortality in human SAP may occur with minimal pancreatic necrosis.29 ,30 ,31 Traditional animal models such as the taurocholate model however relate severity to local necrosis.33 Similarly, the mouse caerulein AP is regarded as severe compared with rat AP114 since mice have more necrosis.33 In the current study, we note peripancreatic lipolysis of triolein to cause mortality in caerulein pancreatitis without pancreatic necrosis. This phenomenon, which replicates the picture in human SAP, is prevented by lipase inhibition.
Unsaturated fatty acid lipotoxicity due to visceral fat lipolysis mediates MSOF in SAP
We note the UFA OA to be the most abundant fatty acid in NCs. UFAs inhibit mitochondrial complexes I and V.1 While we noted early caspase 3/7 activation in acini, this was not sustained (results not shown), perhaps due to concurrent ATP depletion104 ,115 ,116 triggering the necrosis noted at 5 h. Noting this, and the dual annexin V and PI staining of PBMCs, TUNEL positivity in lungs and renal tubules (which are also KIM-1 positive), the likely mode of injury induced by UFAs is necro-apoptotic.
Serum oleate concentrations in rats coadministered triolein and caerulein (350±294 μM) are similar to patients with SAP with complications (614±146 μM).117 An increase in serum C18 UFAs (mainly oleate), similar to what we note, was reported in dogs with AP.118 Patients with SAP with complications also had serum free fatty acid >1400 μM,117 which is similar to what we noted in our study (1421±851 μM). We have previously shown elevated UFAs in biliary SAP90 and IL-12, 18 induced AP.1 UFAs cause organ failure including acute lung injury,109–112 renal tubular toxicity,119 ,120 renal failure110 ,121 and hypocalcaemia.121 This spectrum of endpoints secondary to UFAs and the findings of this study support UFA generation as a target to improve SAP outcomes. The published evidence mentioned above, along with this study showing that MSOF in SAP results from triolein hydrolysis to oleate and not triolein alone (see online supplementary figure S5), brings this two-step phenomenon (ie, increased UTG (in fat)+lipase (in AP)→high UFA→AP with MSOF (ie, SAP)) to closely replicate the third postulate of Koch as shown in online supplementary figure S6. Such two-step fulfilment would be relevant to therapy in diseases such as cholangitis, where neither biliary obstruction alone nor bacteria entering the biliary tract alone (such as post choledochojejunostomy) would cause cholangitis, which would only happen when the two are combined such as after recurrence of a stricture.
The same mode of initiating pancreatitis may have different outcomes
While severity of traditional animal models is attributed to the aetiology,33 outcomes in human AP are unrelated to the aetiology.21–24 ,93 While all groups in our study fulfil the diagnostic criteria for AP,108 the improved outcomes in the triolein+ orlistat group with AP were unrelated to serum amylase or lipase, but associated with reduced lipase activity in ascites, supporting the notion that improved outcomes may result from targeting the modifier, that is, lipase-mediated UFA generation from the hydrolysable peripancreatic UTG in visceral fat and not the primary insult. Interestingly, this resonates with hypertriglyceridemic (HTG) pancreatitis being the exceptional AP aetiology that is typically severe,25–28 ,122 though excluding the patients with HTG from our analysis in figures 1 and 2 did not change the results.
Serum cytokines are markers of AP severity
Predictive models for SAP often use serum cytokines.11–17 ,123 In agreement with the trends noted in patients and consistent with UFAs being proinflammatory,111 ,124 ,125 cytokines were higher in NCs (also enriched in UFAs) and in rodents administered triolein during AP. These cytokines were reduced by lipase inhibition consistent with previous studies showing cytokine upregulation occurs secondary to UFAs.1 This observation, the lack of cell death in acinar cells and PBMCs treated with IL-1β+KC/GRO, the mild course in the IL-1β+KC/GRO groups (despite higher MPOs and a febrile response), suggests that the increased inflammatory response may be a marker but is not a mediator of the adverse events resulting in SAP. This is supported by previous studies showing cytokine levels follow the course of lipotoxicity and normalise on lipase inhibition.1 ,90 Similarly, mice administered IL-6 do not develop organ failure126 and obesity-associated SAP is unaffected in IL-6 knockout (KO) mice.126 Additionally, the elevation of IL-6 and TNF-α in response to intravenous UFAs111 supports UFAs as drivers of inflammation. The cytokines themselves may potentially have a protective role in AP as shown in AP models using IL-6 KO mice57 or on neutralising TNF-α.58 Similarly, IL-8/KC/GRO/CXCL1 mediated neutrophil influx is protective in other models of lung injury.127–129
The human part of this study is limited by its small size, analysis of a limited number of cytokines/chemokines and the lack of data on agents degraded over the course of the illness before intervention was clinically indicated. In addition, the endoscopist or the surgeon characterised collections as NCs or PCs, and since sample collection predated revised Atlanta criteria, we were unable to apply the definitions detailed in the revised criteria. However, this is unlikely to influence our conclusions since all NCs were walled off at the time of intervention (consistent with the definition of walled-off pancreatic necrosis) and grouping NCs and PCs together as being postinflammatory showed similarly high UFAs and cytokines vs PCNs (see online supplementary figure S2), the causality of which was explored in animal and in vitro studies. While we do not have information on underlying chronic pancreatitis in patients with PC, all PCs in our study were present for at least 12 weeks. Thus, the potential misclassification of a small proportion of these collections is unlikely to have significantly influenced our primary finding of UFA-induced lipotoxicity since both NCs and PCs alone or together had higher UFAs compared with PCNs and both IL-1β and IL-8 were analysed mechanistically, even when IL-8 was not significantly different in PCs from PCNs. Lack of information on which fluid collections were infected is also a limitation of this study. Infections are likely to predominantly affect cytokine concentrations. However, the cytokines increased in PCs and/or NCs did not worsen outcomes in our in vitro or animal experiments. Conversely, microbes infecting NP do not produce pancreatic lipases, and UFAs as a consequence, which result in acinar necrosis. Therefore, our observations regarding the role of UFA-induced lipotoxicity are unlikely to be affected by infections.
In summary, human pancreatic necrosis collections are enriched in UFAs and cytokines. Peripancreatic fat necrosis resulting in UFA generation causes MSOF and mortality, converting mild AP to SAP independent of pancreatic necrosis. Therefore, targeting UFA-mediated lipotoxicity, rather than cytokines such as IL-1β or IL-8, may be a better option to improve AP outcomes, especially in the context of obesity.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
- Data supplement 5 - Online figure 4
- Data supplement 6 - Online figure 5
- Data supplement 7 - Online figure 6
Footnotes
PN and KP contributed equally to this work and share first coauthorship.
Contributors VPS and SN designed and conceptualised the study. Acquisition of data was carried out by PN, KP, CD, RNT, CDO, SN, KL, RB, JC, AS, GIP, AK, DCW, JPD, RAC, CA, DJ, FMM and DY. Analysis and interpretation of data was done by PN, KP, CD, SN, JPD, MDC, RP, RAC, CA, DJ, FMM, DY and VPS. Manuscript was drafted by AS, SN, MDC, RP, DY and VPS, while AS, MDC, RP, AK, DCW, FMM, DY, SN, VPS performed critical revision of the manuscript for important intellectual content. Statistical analysis was done by PN, KP, RNT, MDC, DY and VPS. Funding was obtained by SN and VPS, and the entire study was supervised by VPS
Funding This project was supported by grant number RO1DK092460 (VPS) and the Clinical Translational Science Institute (CTSI) supported by the National Institutes of Health through grant numbers UL1RR024153 and UL1TR000005 (VPS, SN). This project used the UPCI Cancer Biomarkers Facility: Luminex Core Laboratory that is supported in part by award P30CA047904. Funding was also provided by a startup package from the University of Pittsburgh, Department of Medicine (VPS).
Competing interests None.
Ethics approval Institutional Review Board at the University of Pittsburgh Medical Center (IRB number PRO10120148) and Washington University Medical Center and the Committee for Oversight of Research Involving the Dead at the University of Pittsburgh Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.