Article Text

Abstract
Objective This phase II, randomised, double-blind, placebo-controlled clinical trial was designed to evaluate the efficacy and safety of PF-00547659, a fully human monoclonal antibody that binds to human mucosal addressin cell adhesion molecule (MAdCAM) to selectively reduce lymphocyte homing to the intestinal tract, in patients with moderate-to-severe Crohn’s disease (CD).
Design Eligible adults were aged 18–75 years, with active moderate-to-severe CD (Crohn’s Disease Activity Index (CDAI) 220–450), a history of failure or intolerance to antitumour necrosis factor and/or immunosuppressive agents, high-sensitivity C reactive protein >3.0 mg/L and ulcers on colonoscopy. Patients were randomised to PF-00547659 22.5 mg, 75 mg or 225 mg or placebo. The primary endpoint was CDAI 70-point decrease from baseline (CDAI-70) at week 8 or 12.
Results In all, 265 patients were eligible for study entry. Although CDAI-70 response was not significantly different with placebo versus PF-00547659 treatment at weeks 8 or 12, remission rate was greater in patients with higher baseline C reactive protein (>5 mg/L vs >18.8 mg/L, respectively). Soluble MAdCAM decreased significantly from baseline to week 2 in a dose-related manner and remained low during the study in PF-00547659-treated patients. Circulating β7+ CD4+ central memory T-lymphocytes increased at weeks 8 and 12 with PF-00547659 treatment. No safety signal was seen.
Conclusions Clinical endpoint differences between PF-00547659 and placebo did not reach statistical significance in patients with moderate-to-severe CD. PF-00547659 was pharmacologically active, as shown by a sustained dose-related decrease in soluble MAdCAM and a dose-related increase in circulating β7+ central memory T cells.
Trial registration number NCT01276509; Results.
- crohn’s disease
- pharmacotherapy
- integrins
- inflammatory bowel disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Crohn’s disease (CD) is an inflammatory bowel disease that is often not adequately controlled using current treatments.
Monoclonal antibodies that bind to integrins on lymphocytes and prevent their translocation can improve outcome in CD.
Inhibition of the mucosal addressin cell adhesion molecule (MAdCAM) limb at the endothelial surface of the α4β7-MAdCAM translocation system, which is responsible for efficient trafficking and retention of leucocytes in the intestine, increases circulating β7 + memory T lymphocytes in animals.
High placebo response rates in the Crohn’s Disease Activity Index endpoint can confound clinical trials in CD.
What are the new findings?
PF-00547659 did not meet the primary endpoint of clinical response in moderate-to-severe CD when compared with placebo.
In patients with greater evidence of inflammation by high-sensitivity C reactive protein or Simple Endoscopic Activity Score, posthoc analyses suggested the presence of a drug effect.
The human, anti-MAdCAM monoclonal antibody PF-00547659 was pharmacologically active, as shown by increased circulating β7+ central memory lymphocytes.
How might it impact on clinical practice in the foreseeable future?
Future clinical trials that address regression to the mean and identify patients with more evidence of inflammation are needed to determine the role of anti-MAdCAM therapy in the treatment of CD.
Introduction
Crohn’s disease (CD) is a chronic, transmural inflammatory disease of the gastrointestinal tract.1 It follows a relapsing and remitting disease course, but a majority of patients eventually progress to complications of stricture, fistula and abscess.2 Patients with moderate-to-severe CD who do not respond to conventional therapy with steroids or immunosuppressive therapy (azathioprine, 6-mercaptopurine, methotrexate)3 are often treated with biologic therapy, including anti-tumour necrosis factor (TNF) antibodies (infliximab, adalimumab, certolizumab pegol)4 and, more recently, anti-interleukin-12/23 monoclonal antibody (ustekinumab)5 and anti-α4β7 integrin antibody (vedolizumab),6 to induce and maintain clinical response and remission. However, patients who initially respond to anti-TNF or anti-integrin therapy can experience secondary loss of response, which is often resolved by switching to a drug within the same class.7 In addition, concerns remain about the safety of corticosteroid, immunosuppressive and anti-TNF therapies. Thus, additional biological therapies would have clinical value.
Mucosal addressin cell adhesion molecule (MAdCAM) is expressed predominantly on the cell surface of high endothelial venules of organised intestinal lymphoid tissue, such as Peyer’s patches and mesenteric lymph nodes.8–10 It is not constitutively expressed in the central nervous system.11 MAdCAM plays a role in gut immune surveillance and also appears to facilitate excessive lymphocyte infiltration under conditions of chronic gastrointestinal inflammation.8–10 The α4β7 integrin is the recognised ligand for MAdCAM, and its expression on populations of CD4+ and CD8+ T cells—as well as on subsets of B cells—distinguishes it as a unique gut-homing lymphocyte. Selective inhibition of the interaction between MAdCAM and the α4β7 integrin appears to avoid the risk of central nervous system infections associated with the non-selective blockade of α4 integrins.12 13 Anti-α4β7 integrin antibody therapy with vedolizumab is effective for induction and maintenance of clinical remission in CD and no cases of progressive multifocal leukoencephalopathy have been reported with this treatment.6
PF-00547659 is a fully human monoclonal antibody that is expected to reduce gastrointestinal inflammation by binding to human MAdCAM and selectively reducing lymphocyte homing to the gut.14 The current phase II, dose-ranging, clinical trial was designed to evaluate the efficacy and safety of PF-00547659 in patients with moderate-to-severe CD who have a history of treatment failure or intolerance to immunosuppressive and/or anti-TNF agents.
Methods
Patients
This multicentre, randomised, double-blind, placebo-controlled, parallel-group trial was conducted at 103 centres in 15 countries (Austria, Belgium, Canada, France, Germany, Japan, Netherlands, Norway, Poland, Serbia, Slovakia, South Africa, South Korea, Spain, USA), beginning in April 2011 and was completed in October 2015. The protocol was approved by the institutional review board or ethics committee at each centre. (Protocol amendment information is provided in the online supplementary text.) All patients gave written, informed consent. All authors had access to study data and have reviewed and approved the final report. The study is registered on ClinicalTrials.gov (NCT 01276509).
Eligible patients were aged 18–75 years with moderate-to-severe ileal (terminal ileum), ileocolonic or colonic CD (Crohn’s Disease Activity Index (CDAI) score 220–450 and ulcers on colonoscopy performed and interpreted locally at the study site during or within 8 weeks prior to screening).15 Patients had failed or were intolerant to immunosuppressive therapies (ie, azathioprine, 6-mercaptopurine, or methotrexate) and/or anti-TNF agents and had a high-sensitivity C reactive protein (hsCRP) concentration >3.0 mg/L (the upper limit of normal) at the central laboratory. (Definitions of treatment failure and intolerance are provided in the online supplementary text.) Patients were excluded if they had received >20 mg/day prednisone or equivalent oral systemic corticosteroid dose within 2 weeks prior to randomisation, >6 mg/day oral budesonide within 2 weeks prior to randomisation or other biologics, including any anti-TNF agents, within 6 weeks from baseline/randomisation.
Study design
Eligible patients were randomly assigned in a 1:1:1:1 ratio and double-blind fashion to receive placebo or PF-00547659 22.5 mg, 75 mg or 225 mg (Pfizer, New York, New York, USA), administered by subcutaneous blinded injection at weeks 0, 4 and 8 and were followed through week 12. Randomisation was performed centrally according to a computer-generated scheme. Stratification factors were the status of treatment failure with or intolerance to immunosuppressive/anti-TNF therapies and concomitant immunosuppressive therapy type. Stable doses of azathioprine, 6-mercaptopurine and methotrexate were continued from screening through week 8; beginning at week 8, dosages of these prior immunosuppressive agents were tapered by approximately 25% per week so that the agents were discontinued by week 12. Doses up to 20 mg/day of prednisone (or equivalent) were permitted; no dose modification was allowed.
Efficacy and safety assessments
Patients were evaluated at weeks 0 (baseline), 2, 4, 6, 8, 10 and 12 (or at the withdrawal visit). At each visit, electronic diary-card data were collected, a clinical assessment of CD and a physical examination were carried out, disease activity measured by CDAI, adverse events (AEs) and concomitant medications reported and samples taken for laboratory analysis. At each visit, patients underwent neurological assessment, which consisted of five tests: confrontational visual fields, timed 25-foot walk, nine-hole peg test, symbol-digit modality test and Multiple Sclerosis Neuropsychological Questionnaire. Neurology consultation was requested if significant unexplained changes were observed.
Pharmacokinetics and antidrug antibodies (ADA)
Blood samples for the assessment of PF-00547659 concentrations were collected and analysed at specified time points for the duration of the study using a validated assay. Analysis of ADA was conducted on serum samples using a multitiered approach, including screening assay, confirmatory assay and neutralising antibody assay. A positive anti-PF-00547659 antibody level was predefined as ≥4.64, independent of change from baseline, on at least one visit.
Efficacy evaluations and statistical analysis
The primary efficacy outcome was the proportion of patients who achieved a CDAI-70 clinical response (ie, a decrease from baseline in CDAI ≥70 points) at week 8 or week 12. The two time points were selected due to concerns about a potential increase in disease activity resulting from withdrawal of immunosuppressive therapy beginning at week 8. Secondary efficacy outcomes measured at all visits included the proportion of patients achieving CDAI-70 response, CDAI-100 response (ie, a decrease from baseline in CDAI ≥100 points), CDAI remission (ie, CDAI <150) and mean change from baseline in total CDAI.
Longitudinal data were analysed for both primary (CDAI-70 response) and key secondary (CDAI-100 response and CDAI remission) endpoints using a generalised linear mixed-effect model (GLIMMIX) with subject as a random effect; treatment, visit and visit-by-treatment interaction as fixed effects and previous status of treatment failure or intolerance to anti-TNF-α agents or immunosuppressive therapies, concomitant immunosuppressive therapy and baseline CDAI score as covariates. The GLIMMIX procedure incorporated the correlation between observations by modelling the random effects. The estimates (and SE) of each dose comparison with placebo were reported. For the CDAI-70 response endpoint, each dose for comparison of the Hochberg method was used to account for rejection of the null hypothesis if a treatment effect was detected at either of the two time points (week 8 or week 12). Because the CDAI-70, CDAI-100 and CDAI remission data were modelled, model estimates are reported along with the SEs. Change from baseline in CDAI was also analysed longitudinally, using a mixed-model repeated measures analysis of the data and the same variables as the GLIMMIX model. This model was used to estimate the difference between each dose and placebo at all postbaseline visits (week 2 through week 12). The family-wise error rate is controlled at one-sided 0.05 using the Bonferroni method for two time points (week 8 or week 12). Because this was a phase II study, the family-wise error rate was controlled at 5% rather than at the 2.5% that is typically used for phase III studies.
Posthoc analyses were performed to assess CDAI-70 response and CDAI remission in subgroups of patients with baseline hsCRP concentrations>5.0 mg/L and >18.8 mg/L (median) and patients with baseline median Simple Endoscopic Activity Score–Crohn’s Disease (SES-CD)16 >10 (25th percentile) and >17 (median). In most patients, the SES-CD was calculated using data from endoscopies performed at baseline; in patients who underwent endoscopy 8 weeks prior to randomisation, the SES-CD score was calculated using data from that procedure.
Other secondary outcomes were faecal calprotectin, hsCRP concentrations, β7+ central memory CD4+ T cells, soluble MAdCAM and mean change from baseline in these concentrations at weeks 4, 8 and 12 and PF-00547659 concentration by visit. The proportion of patients who tested positive for anti-PF-00547659 antibodies by visit (cumulative through week 12) was also assessed.
The β7+ central memory CD4+ T cells, defined as β7+ CD27+CD45RO+CD4+ lymphocytes,17 were measured by fluorescence-activated cell sorting (FACS) from blood samples taken at baseline, week 8 and week 12. Soluble MAdCAM was measured at baseline and week 12. (Details of flow cytometry methods are provided in the online supplementary text.) The FACS parameters were log transformed and analysed using a linear mixed model, with change from baseline as response; treatment, anti-TNF experience, concomitant immunosuppressive therapy, baseline (log transformed), visit and treatment by visit interaction as fixed effects and patients as random effects.
Determination of sample size was based on an assumed CDAI response rate of 35% for the placebo group and 60% for at least one PF-00547659 dose at either week 8 or week 12. A study population of 240 (60 in each treatment group) was estimated to provide at least a 78% probability to detect a statistically significantly greater CDAI-70 response rate with PF-00547659 compared with placebo.
Analyses of efficacy, biomarker and pharmacokinetic outcomes were based on findings from the modified intent-to treat population, defined as all randomised patients who received at least one dose of investigational product.
Results
Patients’ disposition and baseline characteristics
Of 494 screened patients, 265 were eligible for study entry and randomised (figure 1): 64 patients to placebo and 68, 65 and 68 patients to PF-00547659 22.5 mg, 75 mg and 225 mg, respectively. The most common reasons for patient ineligibility were failure to satisfy inclusion criteria for hsCRP concentration (71/229 (31%)), CDAI (24/229 (11%)) and ulcers on colonoscopy (17/229 (7%)). Three patients were randomised but not treated. (Details on patient disposition during the anti-TNF washout period and during the period between anti-TNF discontinuation and colonoscopy are provided in the online supplementary text.)

Disposition of patients. *Three patients were randomised but did not receive treatment (placebo, n=1; PF-0547659 22.5 mg, n=1; PF-0547659, 75 mg, n=1).
Treated patients had a mean age of 35.5 years and a mean CDAI total baseline score of 315.3 (standard deviastion (SD), 65.2). As shown in table 1, treatment groups were similar with respect to age, race, weight, fistulising disease, prior anti-TNF-α exposure, concomitant immunosuppressive therapies and CDAI score at baseline. Of the 262 patients who received treatment, 227 (87%) completed the week 12 study visit. The majority had been treated previously with anti-TNF-α therapies (95%) and were not receiving concomitant immunosuppressive treatment (61%) at the time of randomisation. Only 13 (5%) patients were anti-TNF-α-naïve, all of whom were intolerant to or had failed immunosuppressive therapy. Twenty (8%) patients had their last dose of anti-TNF therapy within 8 weeks of treatment; 59 (23%) had their last dose within 12 weeks of treatment. Distribution of prior anti-TNF experience (failure vs intolerance) was similar among groups, as was prior treatment with immunosuppressive agents.
Baseline characteristics
Primary endpoint
The proportions of patients who achieved a CDAI-70 response over time in the four treatment groups are shown in figure 2. At week 8, 47.7% of patients in the placebo group had a CDAI-70 response versus 52.7%, 60.1% and 62.7% of patients in the PF-00547659 22.5 mg, 75 mg and 225 mg groups, respectively. Corresponding proportions of CDAI-70 responders at week 12 were 58.6% in the placebo group and 62.0%, 64.7% and 57.5% in the PF-00547659 22.5 mg, 75 mg and 225 mg groups. The Hochberg-adjusted p values showed no significant difference between placebo and any dose of PF-00547659 at either week 8 or 12.
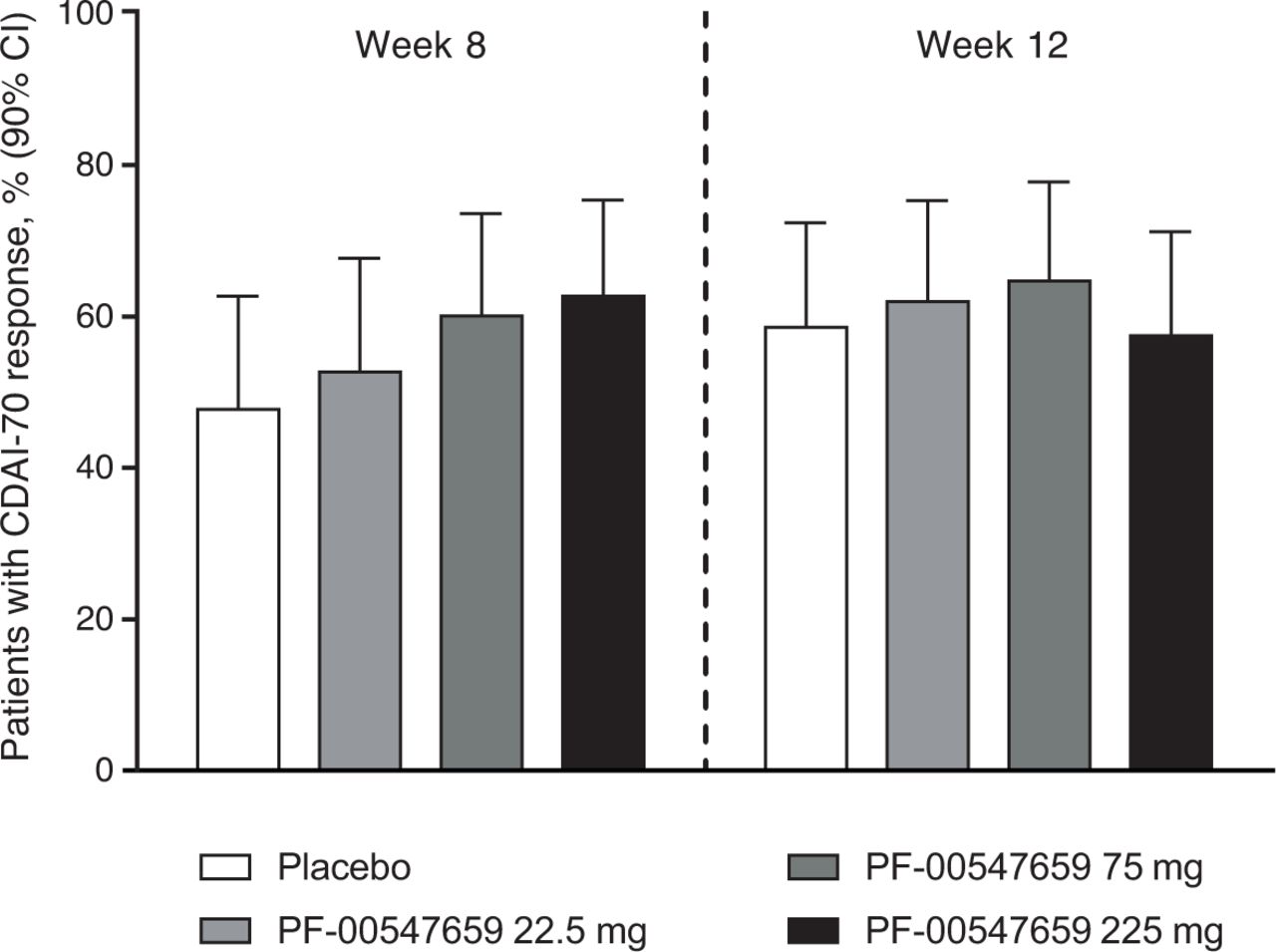
Proportions of patients who achieved a CDAI-70 response (90% CI) with PF-00547659 22.5 mg, 75 mg and 225 mg versus placebo at week 8 and week 12, modified intention-to-treat group. CDAI-70, Crohn’s Disease Activity Index 70-point decrease from baseline.
Secondary endpoints
Efficacy analyses for the key secondary endpoints supported the findings of the primary endpoint analysis (table 2). At week 8, 41.4% of patients who received placebo compared with 50.5%, 48.3% and 57.0% of those who received PF-00547659 22.5 mg, 75 mg and 225 mg, respectively, achieved a CDAI-100 response. Corresponding proportions of CDAI-100 responders at week 12 were 44.4%, 56.0%, 47.7% and 53.8%.
Proportions of patients who achieved a CDAI-100 response and CDAI remission at week 8 and week 12
CDAI remission was achieved at week 8 in 16.7% of patients in the placebo group compared with 29.1%, 23.8% and 26.9% of those in the PF-00547659 22.5 mg, 75 mg and 225 mg groups, respectively. At week 12, 23.0%, 26.8%, 28.5% and 29.6% of patients in these respective groups were in remission.
Although greater proportions of patients in all of the PF-00547659 dose groups achieved a CDAI-100 response and remission than in the placebo group, the differences between placebo and each of the PF-00547659 doses at week 8 and week 12 were not statistically significant. Change in CDAI from baseline, analysed using the mixed-effect repeated measures model, was consistent with the CDAI-70 response rate analysis.
Posthoc analyses
In analyses of CDAI-70 response, no treatment effects were observed at week 8 or week 12 in subgroups of patients with baseline hsCRP >5 mg/L or >18.8 mg/L. The proportions of patients who achieved CDAI remission were greater in all PF-00547659 dose groups than in the placebo group at both weeks 8 and 12 in both hsCRP subgroups, but the between-group differences did not reach statistical significance (figure 3). Although treatment effects were not seen in analyses of CDAI-70 response at either time point in subgroups of patients with SES-CD >10 or >17, significant differences in the proportions of patients achieving CDAI remission were observed at both weeks 8 and 12 in patients with baseline SES-CD >17 who received 75 mg and 225 mg of PF-0547659 (figure 3).

Proportions of patients who achieved CDAI remission (90% CI) with PF-00547659 22.5 mg, 75 mg and 225 mg versus placebo in subgroups with baseline median hsCRP concentration of (A) >5 mg/L and (B) >18.8 mg/L and with baseline median SES-CD of (C) >10 (25th percentile) and (D) >17. *p<0.05, PF-00547659 versus placebo. CDAI-70, Crohn’s Disease Activity Index; hsCRP, high-sensitivity C reactive protein; SES-CD, Simple Endoscopic Activity Score–Crohn’s Disease.
Exclusion of patients who had their last dose of anti-TNF therapy within 12 weeks of randomisation did not affect findings of the analyses of response or remission.
Biomarker endpoints
Faecal calprotectin concentrations varied widely across groups at baseline (table 1). Geometric mean per cent change from baseline in faecal calprotectin concentrations was reduced by week 8 in all active dose groups; concentrations continued to decline in the PF-00547659 22.5 mg and 75 mg groups but increased in the 225 mg group (online supplementary figure S1). Concentrations from baseline were also reduced in the placebo group at week 4 but increased by week 8 and remained stable at week 12. At week 12, reductions (geometric mean) of 6.5%, 36.4%, 29.4% and 10.5% were observed in the placebo and PF-00547659 22.5 mg, 75 mg and 225 mg groups, respectively.
Serum hsCRP concentrations markedly declined from baseline in all three PF-00547659 groups at week 4, continued to decline in the 22.5 mg and 75 mg groups and remained stable in the 225 mg group at week 12 (online supplementary figure S2). Concentrations of hsCRP in the placebo group also declined from baseline to week 4 but subsequently increased between week 8 and week 12. At week 12, a 5.6% increase (geometric mean) from baseline in hsCRP concentration was observed in the placebo group compared with reductions of 30.9%, 21.8% and 20.0% in the 22.5 mg, 75 mg and 225 mg PF-00547659 groups, respectively.
Circulating β7+ central memory CD4+ T cells increased in a dose-dependent manner at week 8 in PF-00547659-treated patients; they also increased at week 12 but not dose-dependently. At week 8, the increases of β7 + cells as a percentage of overall CD4+ expressing cells were 1.21-fold, 1.22-fold and 1.34-fold in the 22.5 mg, 75 mg and 225 mg PF-00547659 groups, respectively, compared with placebo (figure 4A); corresponding values after 12 weeks of PF-00547659 were 1.20, 1.32 and 1.27. These changes for per cent β7+ central memory T cell estimates were statistically significant for all doses at weeks 8 and 12. At week 8, the relative ratios of molecules of equivalent soluble fluorochrome (MESF), the unit measure of β7+ protein expression on central memory Tcells, to placebo for PF-00547659 22.5 mg, 75 mg and 225 mg were 1.63, 1.73 and 1.73, respectively (figure 4C); corresponding values after 12 weeks were 1.68, 1.52 and 1.48. Increases in the absolute number of β7+ central memory T cells (figure 4B) and in MESF (figure 4C) were also statistically significant for all doses of PF-00547659 compared with placebo at weeks 8 and 12.
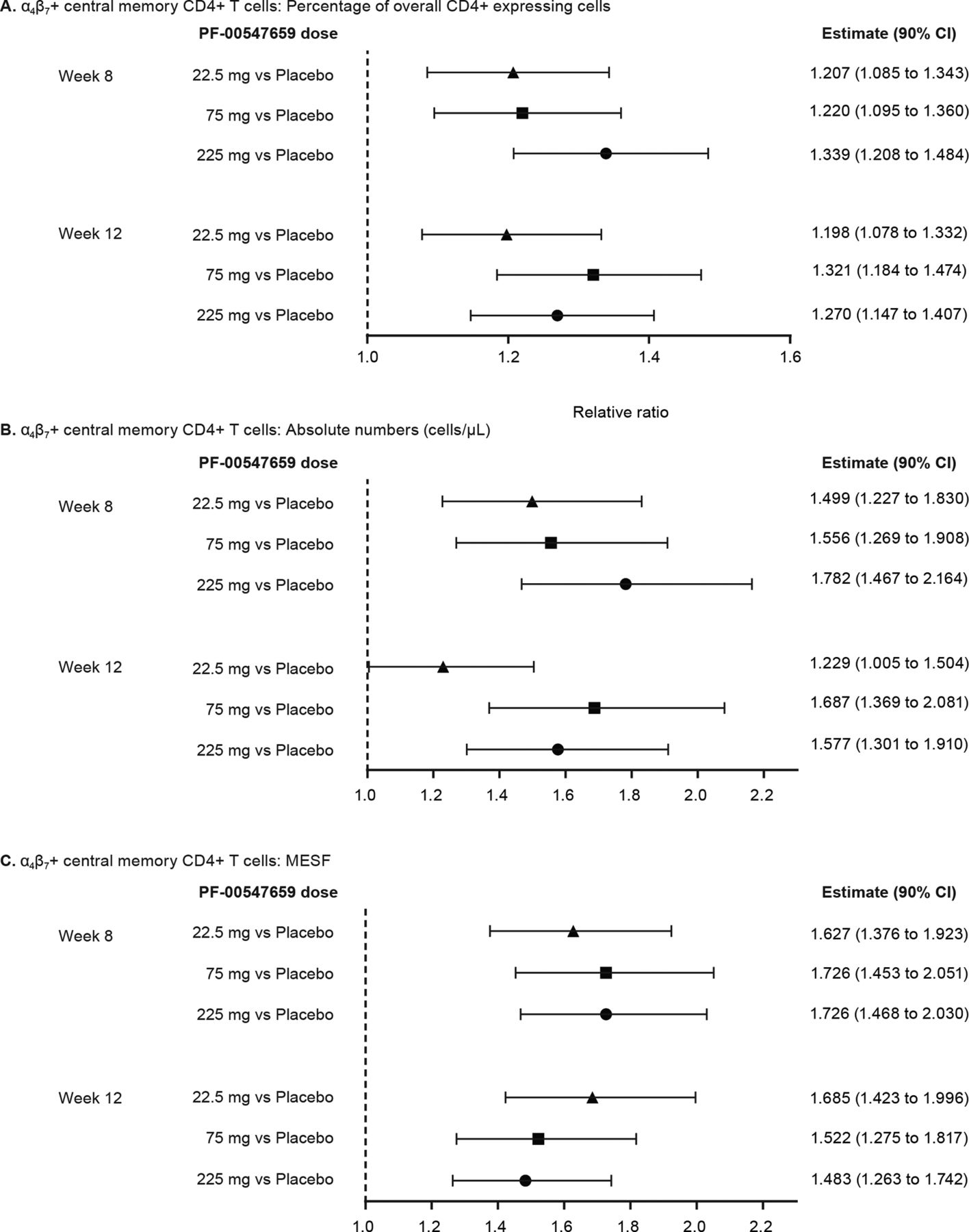
Relative ratios of α4β7+ central memory CD4+T cells as (A) percentage of overall CD4+expressing cells, (B) absolute number (cells/µL) and (C) MESF for PF-00547659 22.5 mg, 75 mg and 225 mg versus placebo. MESF, molecules of equivalent soluble fluorochrome.
Soluble MAdCAM concentrations in PF-00547659- but not placebo-treated patients decreased significantly, in a dose-related manner, at week 2 compared with baseline and remained low during the study. After 12 weeks, median suppression of soluble MAdCAM of 88.7%, 96.5% and 97.8% was observed following monthly dosing of 22.5 mg, 75 mg and 225 mg, respectively, whereas a median increase of 6.7% was seen with placebo.
Pharmacokinetics and ADA
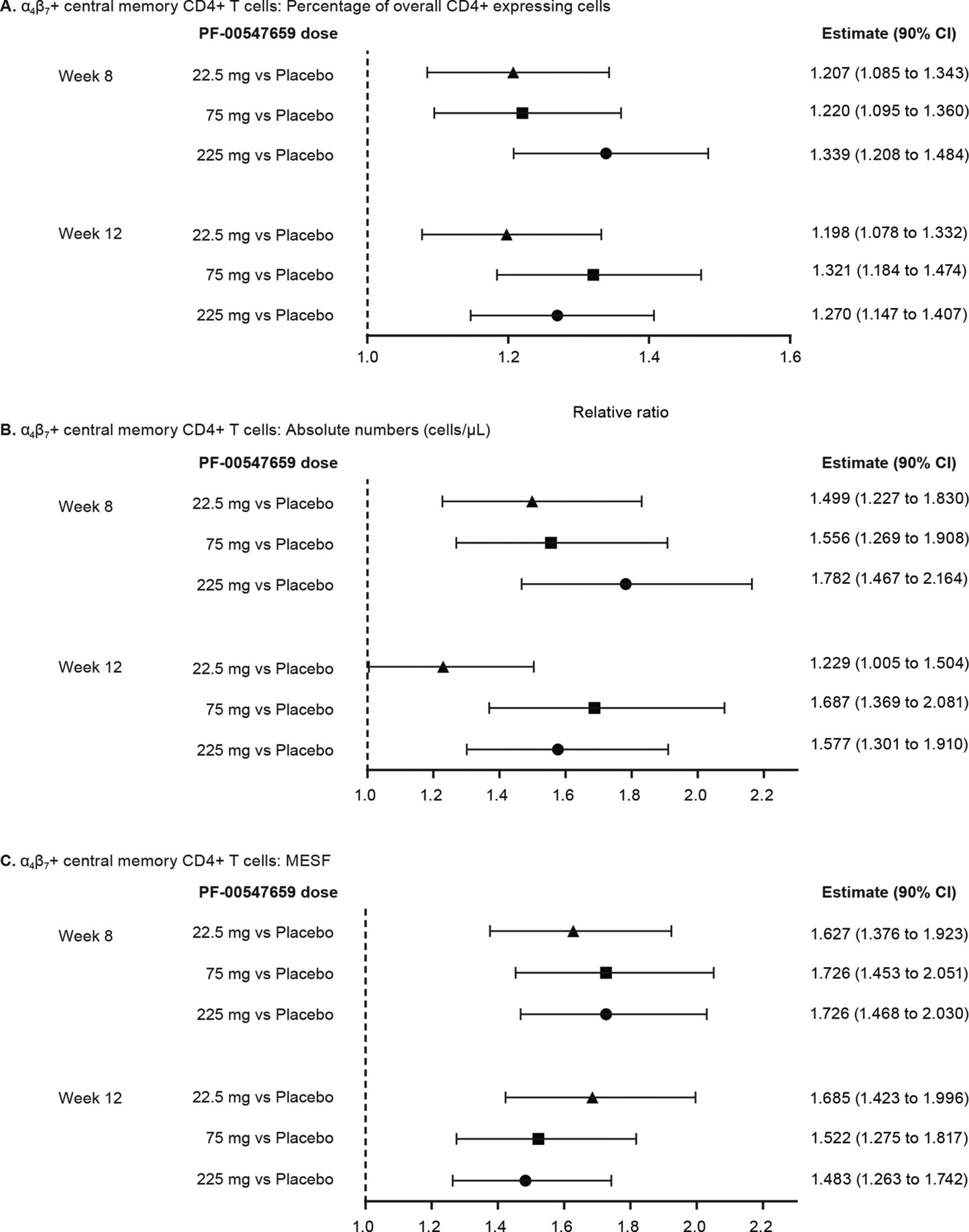
Findings from intensive pharmacokinetic sampling in 30 patients are presented in table 3. Further analyses showed no concentration-effect relationship for the central memory T cells, as the increase in β7+ cells in the blood was similar across all pharmacokinetic quartiles (figure 5). No relationship was found between hsCRP or faecal calprotectin and trough concentration of PF-00547659 (figure 6A and B). CDAI-70 and CDAI-100 response and remission were poorly correlated with PF-00547659 trough concentrations (figure 7A and B).
Pharmacokinetic parameters (n=30)

Median percentage of changes from baseline in MESF of β7+ cells after PD-00547659 treatment across pharmacokinetic quartiles. MESF, molecules of equivalent soluble fluorochrome.

Systemic exposure–response relationship between PD-00547659 at week 12 (by dose) and inflammation measured by (A) hsCRP or (B) faecal calprotectin. Data for two patients with a percentage change from baseline in hsCRP >2000 are not shown in (A) to allow clearer presentation of the other points; data for one patient with a percentage change from baseline in faecal calprotectin >1000 are also not shown in (B) for this reason. In addition, data are not included for patients in the placebo group and patients in the active treatment groups with no pharmacokinetic information. hsCRP, high-sensitivity C reactive protein.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportions of patients who achieved (A) CDAI-70 and (B) CDAI-100 responses by quartile of exposure at week 12. CDAI-70/CDAI-100, Crohn’s Disease Activity Index 70/100-point decrease from baseline.
A total of 588 of 618 (95%) samples from 190 patients on active treatment were reported negative for ADA. Overall, 5% of patients were confirmed to be ADA positive, with antibody titres generally low and close to the cut-off (4.64; none were higher than 9.3). In patients with postbaseline confirmatory ADA (n=15), ADA positivity did not have an obvious effect on exposure, CDAI scores or the safety profile. (Additional details of ADA analysis are provided in the online supplementary text.)
Safety
Overall, the incidence of AEs, serious AEs (SAEs) and discontinuations was similar across treatment groups (table 4 and online supplementary table S1). No evidence was seen of a safety signal or an increased incidence of AEs at higher doses of PF-00547659.
Incidence of AEs
SAEs were reported more frequently in patients receiving PF-00547659 versus placebo, with no evidence of a dose response (table 3). Most SAEs were due to or related to CD (n=5, n=6, n=7 and n=8 in the placebo vs 22.5 mg, 75 mg and 225 mg treatment groups, respectively). Reported SAEs were considered unlikely to be due to the study drug. Injection site reactions—reported as bruising, erythema, pain and swelling—were uncommon and occurred at similar rates in the placebo and PF-00547659 groups.
During the 12-week study period, among the 35 patients who discontinued treatment, 24 withdrew due to AEs. More discontinuations were observed in the 22.5 mg (n=13) and 75 mg (n=12) PF-00547659 treatment groups compared with the placebo and 225 mg PF-00547659 groups (n=5 each) (figure 1). Withdrawals due to AEs were also higher in the 22.5 mg and 75 mg PF-00547659 groups (n=9 and n=8, respectively) than in the placebo and 225 mg PF-00547659 groups (n=3 and n=4, respectively). Most AE withdrawals were due to CD or its complications. No evidence was seen of a PF-00547659 drug or dose relationship.
Discussion
The differences in the rates of CDAI-70 response, CDAI-100 response and CDAI remission between PF-00547659 and placebo were not statistically significant at weeks 8 or 12 in patients with moderately-to-severely active CD, 95% of whom had previously failed or were intolerant to anti-TNF therapy. However, subgroup analyses for the more rigorous endpoint of CDAI remission did show a significant treatment effect for PF-00547659 in patients with a baseline hsCRP concentration >5 mg/L or >18.8 mg/L (median) and in those with baseline SES-CD >17.
A placebo effect in response and remission that rises quickly to a high level has been described previously in patients with CD.18 Factors that are associated with lower placebo-effect response and remission rates are objective evidence of inflammation at baseline (ie, elevated hsCRP and/or faecal calprotectin and/or presence of ulcers at colonoscopy)6 19–22 and prior anti-TNF therapy.6 7 23 Placebo remission rates in the latter patient population have been reported to range from 5% to 10%. In this study, patients were required to have a baseline hsCRP concentration >3 mg/L at screening, and ulcers present at a colonoscopy performed and interpreted at the local site within 8 weeks of screening or during screening. Although the study included patients who had failed immunosuppressive therapy, they comprised only 5% of the study population, with the remaining 95% of patients having failed or been intolerant to anti-TNF therapy. Thus, the high placebo-treatment response and remission rates observed in our study are highly unexpected and not easily explained.
Phase II studies of a similar size to the current study were conducted to evaluate two other anti-integrin agents, natalizumab24 and vedolizumab.25 Although these studies are not directly comparable to the current study of PF-00547659 because of differences in populations studied and timing of endpoints, like the current study, they reported both CDAI-70 and remission endpoints. The outcome at week 8 was not the primary endpoint in any of the studies, but it is the only time point reported in all three studies. At 8 weeks, similar proportions of patients receiving the lowest evaluated dose of the three agents achieved a CDAI-70 response (PF-00547659 22.5 mg, 53%; natalizumab 3 mg/kg, 56%; and vedolizumab 0.5 mg/kg, 49%), but the proportion of patients who received placebo and achieved this endpoint in the present study was considerably higher (48%) than that in the natalizumab (35%) and vedolizumab (41%) studies. In contrast, the proportions of patients in the placebo group who achieved remission at week 8 were similar in the three studies (17%, 16% and 21%, respectively), but the proportion of patients receiving the highest dose of PF-00547659 who achieved remission (27%) was paradoxically lower than that achieving remission with the highest dose of natalizumab (43%) and vedolizumab (37%).
One design element of this trial that differs from that of previous studies is that the required washout period for previous anti-TNF therapy was 6 weeks, rather than 8 weeks, prior to baseline. Theoretically, the prior anti-TNF antibody may still have been present in the systemic circulation following recent anti-TNF treatment, creating a carryover effect; a similar observation was made previously in patients with CD treated with adalimumab who had previously failed infliximab but still had infliximab in the circulation.6 Duration of the study period may also have been a factor in the high placebo effect. In a meta-analysis of 23 randomised controlled trials in active CD, the most important factor contributing to the CDAI remission rate among placebo-treated patients appeared to be length of the study; estimated remission rates were 12% for studies <2 months, 19% for studies ≥2 to 4 months and 32% for studies ≥4 months in duration.18 The higher rate observed at 12 weeks compared with 8 weeks in the current study seems to be consistent with these findings. This effect also appeared to be pronounced in the subgroup analyses assessing remission rates at different baseline hsCRP levels. Another possible contributing factor may have been that the baseline colonoscopy examinations were not centrally read by a blinded central reader, a clinical trial design feature that has been associated with higher placebo response and remission rates in patients with ulcerative colitis.26 The relative overrepresentation of patients who had failed anti-TNF therapy (73% vs 27% who had not failed, including only 8% who were anti-TNF–naïve) should also be noted. In contrast, 48% of patients with CD treated with vedolizumab had failed anti-TNF therapy, and among them the difference in remission rates between active and placebo groups was 6%, whereas the difference in CDAI-100 response rates was 1%.6 Finally, the use of CDAI as an entry criterion and as an endpoint, while common to studies of CD, raises the possibility of regression to the mean as a confounder.
The unexpectedly high rates of response and remission in patients receiving placebo ultimately render this trial a technical failure. We are unable to determine with certainty whether the trial failed because the high placebo rates impeded detection of a treatment difference or the drug simply was not effective. Findings of subgroup analyses suggest potential clinical benefits of treatment with PF-00547659, as significant differences between placebo and active treatment were seen in patients with higher levels of inflammation, measured by baseline hsCRP, or more active disease, measured by SES-CD. These findings need to be corroborated in a prospective study, but they suggest that PF-00547659 may be an effective treatment for CD, and that the failure of the trial was due to unknown technical factors that resulted in an unexpectedly high response rate in patients who received placebo, rather than lack of efficacy.
Although the primary endpoint was not met, PF-00547659 was pharmacologically active, as shown by a dose-related increase in circulating β7+ central memory T cells and a sustained dose-related decrease in soluble MAdCAM. The increase in circulating lymphocytes in PF-00547659-treated patients observed in this and earlier studies27 28 may represent a reduction of MAdCAM/integrin-mediated migration of lymphocytes to the gut, resulting in increased numbers of central memory T cells in the blood circulation. In future trials, these biomarkers should be further evaluated as potential markers for drug exposure that may be directly correlated with clinical efficacy. Due to the elevated hsCRP entry criterion (>3 mg/L), mean values in all groups were elevated at baseline. The geometric mean values for patients in the treatment arms remained below baseline for all postbaseline time points, but by week 12 the geometric mean for placebo-treated patients had increased above the baseline.
The three dose levels of PF-00547659 all appeared to be safe and well tolerated in this population. Most common AEs were related to the underlying disease, with no evidence of a dose response in any AE group. This safety profile needs to be interpreted with some caution, as a larger number of patients and a longer duration of therapy will be required to fully assess the safety of PF-00547659 in patients with CD.
In conclusion, although prospective efficacy endpoints were not met in this study, signals of robust pharmacological activity based on circulating soluble MAdCAM and increased circulating β7+ central memory T cells were observed. The placebo clinical response and remission rates were higher than expected, making assessment of efficacy difficult. Nonetheless, posthoc analyses showed evidence of efficacy in patients with greater endoscopic or biomarker signals of active inflammation.
Acknowledgments
The authors wish to thank the patients who participated in the study and the investigators and medical staff of all participating study centres. They would also like to acknowledge Gail M Comer, MD, for her contributions to the conception and design of the study as well as the members of the Data Monitoring Committee: Dr Colm O’Morain (Chair), Dr Paola Cinque, Dr David Clifford, Dr Lauren Krupp, Dr Glen Cooke, and Dr Dan Anbar. Medical writing support was provided by John Bilbruck and Donna McGuire of Engage Scientific Solutions and was funded by Pfizer.
References
Footnotes
Contributors WJS, AB, LSB, FC, KJG, MHZ and GRD’H made substantial contributions to the conception or design of the study. All authors contributed to the acquisition or interpretation of the data; reviewed and revised the manuscript for important intellectual content and approved the final version of the manuscript prior to its submission. As guarantors, AB and KJG were responsible for the overall content of the manuscript.
Funding The study was sponsored by Pfizer.
Competing interests WJS has received grant support, personal fees and non-financial support from Pfizer during the conduct of the study; grant support from Pfizer, Exact Sciences, Amgen, the American College of Gastroenterology and the Broad Foundation; grant support and personal fees from Prometheus Laboratories, AbbVie, Boehringer Ingelheim, Takeda, Atlantic Pharmaceuticals, Janssen, Bristol-Myers Squibb, Genentech and Nutrition Science Partners and personal fees from Kyowa Hakko Kirin, Millennium Pharmaceuticals, Celgene Cellular Therapeutics, Santarus, Salix Pharmaceuticals, Catabasis Pharmaceuticals, Vertex Pharmaceuticals, Warner Chilcott, Gilead Sciences, Cosmo Pharmaceuticals, Ferring Pharmaceuticals, Sigmoid Biotechnologies, Tillotts Pharma, Am Pharma BV, Dr August Wolff, Avaxia Biologics, Zyngenia, Ironwood Pharmaceuticals, Index Pharmaceuticals, Nestle, Lexicon Pharmaceuticals, UCB Pharma, Orexigen, Luitpold Pharmaceuticals, Baxter Healthcare, Ferring Research Institute, Amgen, Novo Nordisk, Mesoblast, Shire, Ardelyx, Actavis, Seattle Genetics, MedImmune (AstraZeneca), Actogenix NV, Lipid Therapeutics Gmbh, Eisai, Qu Biologics, Toray Industries, Teva Pharmaceuticals, Eli Lilly, Chiasma, TiGenix, Adherion Therapeutics, Immune Pharmaceuticals, Celgene, Arena Pharmaceuticals, Ambrx, Akros Pharma, Vascular Biogenics, Theradiag, Forward Pharma, Regeneron, Galapagos, Seres Health, Ritter Pharmaceuticals, Theravance, Palatin, Biogen and the University of Western Ontario (owner of Robarts Clinical Trials) outside the submitted work. In addition, WJS reports patents related to the use of topical azathioprine to treat inflammatory bowel disorders (US 5,691,343), topical formulations of azathioprine to treat inflammatory bowel disorders (US 5,905,081), colonic delivery of nicotine to treat inflammatory bowel disease (South African patent 97/1020; US 5,846,983, 5,889,028 and 6,166,044; Mexico patent 209636; Europe patents 0954337 and 893998; Hong Kong patent HK1019043; China patent ZL97192177; Czech patent 293616; Canada patent 2,246,235), the use of azathioprine to treat Crohn’s disease (US 5,733,915), azathioprine compositions for colonic administration (New Zealand patent 306062; Singapore patent 45647; Australia patent 707168; Czech patent 290428), intestinal absorption of nicotine to treat nicotine responsive conditions (Australia patent 718052; US 6,238,689), the use of topical azathioprine and thioguanine to treat colorectal adenomas (US 6,166,024), enema and enterically coated oral dosage forms of azathioprine (US 6,432,967), a pharmaceutical composition for the treatment of inflammatory bowel disease (US 7,341,741), intestinal absorption of nicotine to treat nicotine responsive conditions (Canada patent 2,260,909) and obesity treatment and device (US 7,803,195 B2). SDL has received financial support for research from UCB, Janssen, Abbvie, Genentech, Amgen, Takeda and Pfizer; and consulting fees from UCB, Janssen and Takeda. DT reported no disclosures. EL has received educational grants from MSD, Abbvie; speaker fees from Abbvie, Ferring, MSD, Chiesi, Mitsubishi Pharma, Hospira, Janssen and Takeda and has served on advisory boards for Abbvie, Ferring, MSD, Takeda, Mitsubishi Pharma, Celltrion and Prometheus. MK has received speaker fees from Abbvie, Alvogen, Ferring and Takeda and fees for travel/accommodations/meeting expenses from Ferring, Alvogen and Abbvie. JK reported no disclosures. WR has received financial support for research from Abbott Laboratories, Abbvie, Aesca, Centocor, Falk Pharma GmbH, Immundiagnsotik and MSD; has served as a consultant for Abbott Laboratories, Abbvie, Aesca, Amgen, AM Pharma, Astellas, AstraZeneca, Avaxia, Bioclinica, Biogen IDEC, Boehringer-Ingelheim, Bristol-Myers Squibb, Cellerix, Chemocentryx, Celgene, Centocor, Celltrion, Covance, Danone Austria, Elan, Falk Pharma GmbH, Ferring, Galapagos, Genentech, Gilead, Grünenthal, ICON, Index Pharma, Inova, Janssen, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, MedImmune, Millenium, Mitsubishi Tanabe Pharma, MSD, Nestle, Novartis, Ocera, Otsuka, PDL, Pharmacosmos, Pfizer, Procter & Gamble, Prometheus, Robarts Clinical Trial, Schering-Plough, Second Genome, Setpointmedical, Takeda, Therakos, Tigenix, UCB, Vifor, Zyngenia and 4S and as a speaker for Abbott Laboratories, Abbvie, Aesca, Aptalis, Centocor, Celltrion, Danone Austria, Elan, Falk Pharma GmbH, Ferring, Immundiagnostik, Mitsubishi Tanabe Pharma, MSD, Otsuka, PDL, Pharmacosmos, Schering-Plough, Shire, Takeda, Therakos, Vifor and Yakult. XH has served on advisory boards for Abbvie, Fresenius Kabi, Janssens and Takeda and has participated in educational activities for Abbvie, Arard, Ferring, Fresenius Kabi, Mayoly Spindler, MSD, Nestlé, Norgine, Nutricia and Takeda. DIP reported no disclosures. SS has received consulting/speaker fees from AbbVie, Biogen, BMS, Boehringer, Celltrion, Ferring, Hospira/Pfizer, Jansen, Merck, Novartis, Takeda and UCB. SN, AB, LSB and MHZ are employees of Pfizer. AA, FC,KJG, JBC and RC were employees of Pfizer during the OPERA study. GRD’H has served as a consultant for Abbvie, Ablynx, Amakem, AM Pharma, Avaxia, Biogen, Bristol Myers Squibb, Boerhinger Ingelheim, Celgene, Celltrion, Cosmo, Covidien, Ferring, Dr FALK Pharma, Engene, Ferring, Galapagos, Gilead, GlaxoSmithKline, Hospira, Johnson and Johnson, Medimetrics, Millenium/Takeda, Mitsubishi Pharma, Merck Sharp Dome, Mundipharma, Novonordisk, Pfizer, Prometheus laboratories/Nestle, Receptos, Robarts Clinical Trials, Salix, Sandoz, Setpoint, Shire, Teva, Tigenix, Tillotts, Topivert, Versant and Vifor and has received speaker fees from Abbvie, Ferring, Johnson and Johnson, Merck Sharp Dome, Mundipharma, Norgine, Pfizer, Shire, Millenium/Takeda, Tillotts and Vifor.
Ethics approval The protocol was approved by the institutional review board or ethics committee at each centre.
Provenance and peer review Not commissioned; externally peer reviewed.