Article Text

Abstract
Objective Alcoholic liver disease (ALD) is a leading cause of death among chronic liver diseases. However, its pathogenesis has not been completely established. MicroRNAs (miRNAs) are key contributors to liver diseases progression. This study investigated hepatocyte-abundant miRNAs dysregulated by ALD, its impact on hepatocyte injury and the underlying basis.
Design Alcoholic hepatitis (AH) human and animal liver samples and hepatocytes were used to assess miR-148a levels. Pre-miR-148a was delivered specifically to hepatocytes in vivo using lentivirus. Immunoblottings, luciferase reporter assays, chromatin immunoprecipitation and immunofluorescence assays were carried out in cell models.
Results The miRNA profile and PCR analyses enabled us to find substantial decrease of miR-148a in the liver of patients with AH. In mice subjected to Lieber-DeCarli alcohol diet or binge alcohol drinking, miR-148a levels were also markedly reduced. In cultured hepatocytes and mouse livers, alcohol exposure inhibited forkhead box protein O1 (FoxO1) expression, which correlated with miR-148a levels and significantly decreased in human AH specimens. FoxO1 was identified as a transcription factor for MIR148A transactivation. MiR-148a directly inhibited thioredoxin-interacting protein (TXNIP) expression. Consequently, treatment of hepatocytes with ethanol resulted in TXNIP overexpression, activating NLRP3 inflammasome and caspase-1-mediated pyroptosis. These events were reversed by miR-148a mimic or TXNIP small-interfering RNA transfection. Hepatocyte-specific delivery of miR-148a to mice abrogated alcohol-induced TXNIP overexpression and inflammasome activation, attenuating liver injury.
Conclusion Alcohol decreases miR-148a expression in hepatocytes through FoxO1, facilitating TXNIP overexpression and NLRP3 inflammasome activation, which induces hepatocyte pyroptosis. Our findings provide information on novel targets for reducing incidence and progression of ALD.
- alcohol-induced injury
- inflammation
- cell death
- cell signalling
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Excessive alcohol consumption promotes hepatocyte dysfunction and death during the progression of liver diseases.
A set of microRNAs (miRNAs) including miR-182, miR-155 and miR-217 showed abnormal increases in alcoholic liver disease (ALD). However, the downregulated miRNAs have been scarcely explored.
MiR-148a belongs to miRNAs abundant in hepatocytes and regulates hepatocyte differentiation. MIR148A gene is regulated by the transcription factors, SREBP-1, MYB and p53.
Thioredoxin-interacting protein (TXNIP) in association with NLRP3 activates inflammasome pathway during the progression of non alcoholic fatty liver disease.
What are the new findings?
MiR-148a levels are dysregulated in the liver of patients with alcoholic hepatitis (AH) or ethanol-fed animals.
Forkhead box protein O1 is identified as an unrecognised transcriptional regulator of MIR148A gene, and its hepatic expression is diminished in patients with AH or animal models, lowering miR-148a levels.
TXNIP is discovered as a direct target of miR-148a and is overexpressed during the progression of ALD.
Alcohol treatment elicits caspase-1-mediated pyroptosis through TXNIP overexpression, and this event is reversed by miR-148a transfection. Consistently, hepatocyte-specific delivery of miR-148a alleviates alcoholic liver injury.
How might it impact on clinical practice in the foreseeable future?
Our findings demonstrate that miR-148a ameliorates alcohol-induced inflammasome activation and pyroptosis in hepatocytes through TXNIP inhibition, which shed light on novel targets and potential strategies for the treatment of ALD.
Introduction
Excessive alcohol drinking promotes alcoholic liver disease (ALD) such as steatosis, steatohepatitis, fibrosis to cirrhosis and finally to hepatocellular carcinoma, the leading causes of death among all chronic liver diseases. Alcoholic hepatitis (AH) with liver cirrhosis accounts for approximately 50% of the mortality in Western countries.1 Because of the rapid increase in alcohol consumption in other countries such as China and India, the prevalence of ALD has increased globally.2 However, successful therapeutic remedies are not available presumably because of inadequate patient characterisation, poor diagnostic as well as prognostic measures and lack of proper medication. An understanding of the pathogenesis of ALD and the molecular regulation may assist in the development of good diagnostic markers, along with preventive and/or therapeutic approaches.
The microRNAs (miRNAs) participate in a variety of pathophysiological processes. A set of miRNAs including miR-182, miR-155 and miR-217 showed an abnormal increase in ALD.3–5 More importantly, decrease of miRNA and the resultant overexpression of its target may also be crucial in understanding the process of hepatocyte death and disease progression. However, the significance of downregulated miRNAs has been scarcely explored in ALD (except the dysregulation of miR-122).6 This study aims at identifying a liver-specific miRNA, which is downregulated during the progression of ALD along with its target and investigates their roles in determining hepatocyte fate and the underlying molecular basis.
Alcohol consumption and detoxification processes have direct effects on hepatocytes, such as release of sterile danger signals, uric acid and extracellular ATP.7 The signals released from damaged hepatocytes activate multiple inflammatory pathways, promoting ALD.8 In addition, patients with AH showed increased levels of tumour necrosis alpha and interleukin 1 (IL-1),9 10 supporting the hypothesis that inflammatory response is the major driving force during ALD progression. Moreover, alcohol metabolism in hepatocytes facilitates production of reactive oxygen species (ROS) and mitochondrial dysfunction, sensitising hepatocytes to inflammatory cytokines.11 12 In addition to the immune cell-mediated inflammation, inflammasome-dependent IL-1β production has also been reported in hepatocytes.13 Nonetheless, whether alcohol consumption promotes inflammasome activation in hepatocytes, and if so, what the consequence is for hepatocyte fate had not been explored.
Here, we report that miR-148a is markedly decreased in patients with AH and experimental animal models and that dysregulation of miR-148a promotes thioredoxin-interacting protein (TXNIP) overexpression in hepatocytes facilitating pyroptosis, a caspase-1-dependent programmed cell death, in ALD. Moreover, we discovered forkhead box protein O1 (FoxO1) as a transcription factor for MIR148A gene expression and the ability of alcohol to inhibit FoxO1 expression. Two different alcohol-feeding animal models, cell-based assays and molecular approaches were used to assess the impact of decrease in miR-148a levels on TXNIP-mediated inflammasome activation in hepatocytes and consequent cell fate determination (ie, pyroptosis). Towards the end, we employed a hepatocyte-specific in vivo miRNA delivery system to confirm the identified pathogenesis of ALD.
Materials and methods
Liver samples of patients with AH
AH liver samples were obtained from the Liver Unit of the Hospital Clinic of Barcelona. The clinical characteristics of patients with AH and healthy controls have been described previously.3
Animal treatments
Male C57BL/6 mice at 6–8 weeks of age were fed on a Lieber-DeCarli diet (Dyets) with 5% (vol/vol) ethanol (36% ethanol-derived calories) ad libitum or an isocaloric liquid diet for 4–6 weeks. For binge alcohol model, ethanol (5 g/kg body weight) was administered two times daily by gastric intubation for 7 consecutive days, whereas control mice received water. For in vivo miRNA delivery experiment, mice were injected with lentiviruses expressing control or pre-miR-148a through tail vein.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) assay was performed according to EZ-ChIP assay kit protocol (Upstate Biotechnology, Lake Placid, New York, USA). The procedures for analysis are described in online supplementary materials and methods.
Supplementary file 1
Flow cytometric analysis
Pyroptosis was analysed using the FAM-FLICA in vitro Caspase-1 Detection Kit (ImmunoChemistry Technologie, Bloomington, Minnesota, USA). The procedures for analysis are described in online supplementary materials and methods.
Data analysis
Statistically significant differences were assessed by the Student’s t-test or one-way analysis of variance tests followed by Bonferroni’s method or Fisher’s Least Significant Difference for multiple comparisons when necessary. The data were expressed as the mean±SEM. Coefficients of correlation (r) were determined by the Pearson correlation method. The criterion for statistical significance was set at P<0.05 or P<0.01.
Additional detailed information is in online supplementary materials and methods.
Results
Dysregulation of miR-148a in patients with AH or ALD animal models
To determine the identity of the miRNAs dysregulated in patients with AH, we first analysed the profile of miRNA microarray accomplished using patients with AH and normal liver samples (GSE59492). In this analysis, miR-148a levels were significantly downregulated in patients with AH compared with the controls (figure 1A). Quantitative reverse transcription PCR (qRT-PCR) assays verified the marked dysregulation of miR-148a in human AH samples (figure 1B). Since miR-148b and miR-152 shared the same seed sequence, we additionally examined miR-148b and miR-152 levels and found no changes in the miRNAs levels (see online supplementary figure 1A). Ad libitum or intragastric ethanol-feeding animal models have significantly enhanced understanding on the pathogenesis of ALD despite some limitations in the production of the clinical features.14 15 We employed Lieber-DeCarli alcohol diet and binge alcohol drinking methods. In both models, miR-148a levels were notably diminished in the liver with alcohol feeding (figure 1C). Again, miR-148b and miR-152 levels were not changed (see online supplementary figure 1B). We confirmed hepatocyte injury and accompanying inflammation in mice subjected to Lieber-DeCarli alcohol diet (figure 1D upper). Hepatic miR-148a levels correlated with changes in the blood biochemical parameters (ie, serum alanine aminotransferase (ALT) activities and triglyceride (TG) contents) (figure 1D lower). MiR-148a dysregulation mediated by alcohol was corroborated with the results obtained using the primary hepatocytes or AML-12 cells (figure 1E). MiR-148a levels were lowered by ethanol in a dose-dependent manner (50–200 mM, 48 hours) (see online supplementary figure 2A). In subsequent experiments, we primarily chose the condition of 100 mM ethanol treatment (for 48 hours). Collectively, our results showed that miR-148a levels were substantially lowered in ALD.

Dysregulation of miR-148a in patients with AH or ALD animal models. (A) Expression heatmap of the significantly downregulated miRNAs in patients with AH (n=13) as compared with normal controls (n=6) (GSE59492) (fold change <0.5, P<0.01). (B) qRT-PCR assays for miR-148a in normal control or liver samples of patients with AH (n=6 or 12 each). Each point represents one sample, and the horizontal line indicates the mean value. (C) qRT-PCR assays for miR-148a in mouse liver samples. Mice were fed on either control diet or Lieber-DeCarli alcohol diet for 6 weeks (n=6 or 10 each) or received oral gavage of 5 g/kg body weight of alcohol two times daily for 7 days (n=7 each). Data were shown as box and whisker plot. Box, IQR; whiskers, 5–95 percentiles and horizontal line within box, median. (D) Liver histopathology (H&E, scale bar, 50 µm), blood biochemical parameters and correlations between miR-148a levels and serum ALT activities (or TG contents) using the same samples from the Lieber-DeCarli mouse model (n=6 or 10 each). (E) qRT-PCR assays for miR-148a in hepatocytes. Mouse primary hepatocytes or AML-12 cells were exposed to 100 mM ethanol for 48 hours (n=3 each). For (D) and (E), data represent the mean±SEM, **P<0.01. AH, alcoholic hepatitis; ALD, alcoholic liver disease; ALT, alanine aminotransferase; Con, control; EtOH, ethanol treatment; H & E, hematoxylin and eosin; qRT, quantitative reverse transcription; TG, triglyceride.
Identification of FoxO1 as a transcriptional factor of MIR148A
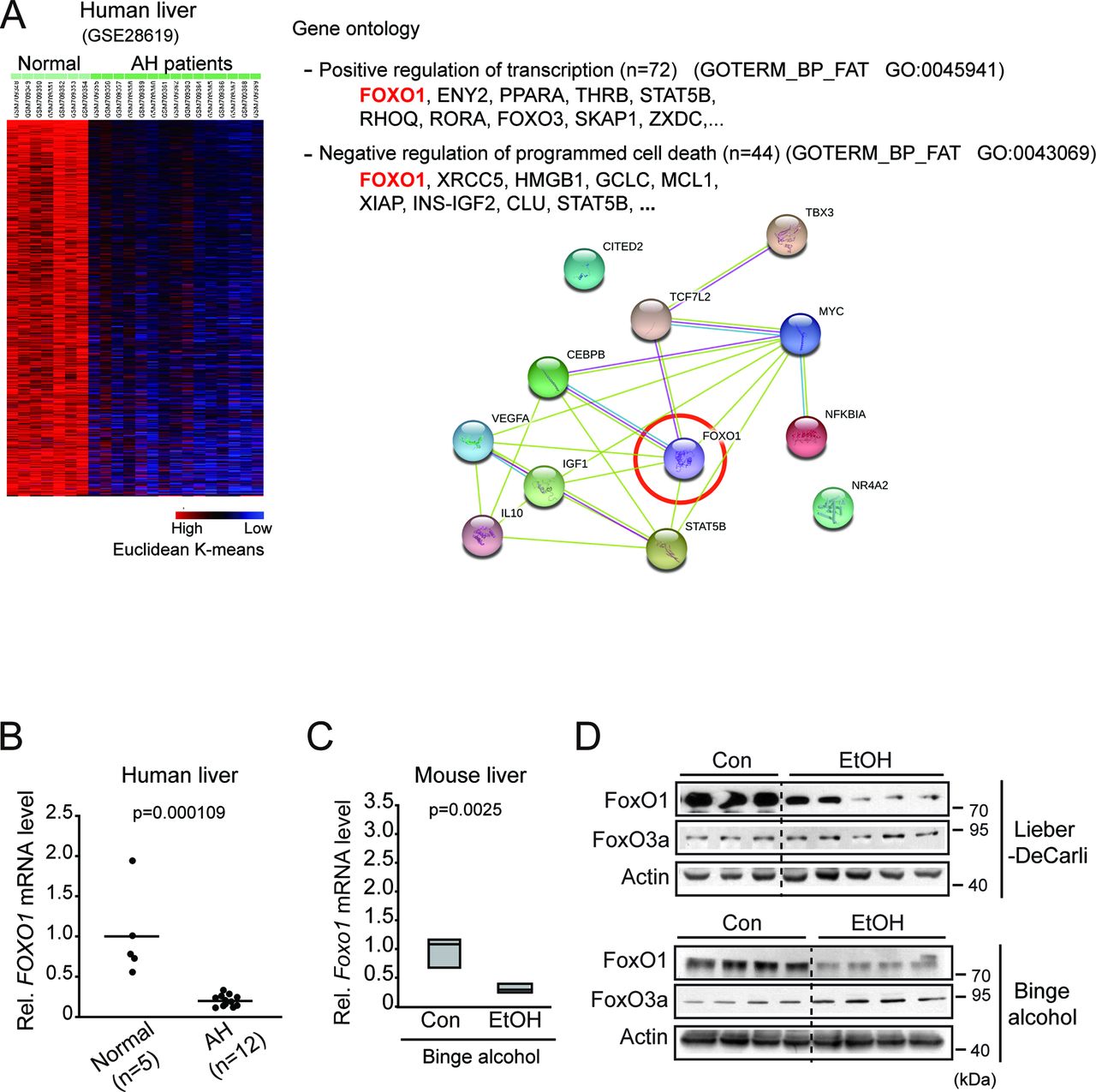
To explore the molecular mechanism by which alcohol decreases miR-148a expression, we next analysed Gene Expression Omnibus (GEO) dataset (GSE28619) using a K-mean clustering (K=4) and extracted clusters of genes downregulated in the liver of patients with AH. Of these, about 3310 downregulated genes were repressed more than the others (figure 2A left). Gene ontology analysis using DAVID database enabled us to find molecules involved in biological processes of ‘positive transcriptional regulation’ and ‘inhibitory regulation of programmed cell death’. Moreover, network analysis using STRING database yielded FoxO1 as the core molecule interacting with 12 other genes overlapping within the above two processes (figure 2A right). qRT-PCR assays verified dysregulation of FOXO1 in human AH liver samples (figure 2B). In the analysis of mice subjected to binge alcohol drinking, hepatic FoxO1 transcript levels were decreased (figure 2C). Immunoblottings confirmed the suppression of FoxO1 protein in both animal models (figure 2D). FoxO3a levels, assessed as a comparison, were not decreased.

Inhibition of FoxO1 in patients with AH or ALD animal models. (A) K-mean cluster and gene ontology analyses using the liver samples of patients with AH. FoxO1 was predicted as an inhibitor of alcohol-induced cell death among the most highly differentially expressed genes in GEO dataset (GSE28619). Blue, underexpression; red, overexpression (left). The genes were selected in the categories of two gene ontology pathways (P<0.05) associated with transcriptional regulation and programmed cell death (right). (B) qRT-PCR assay for FOXO1 in the liver of patients with AH (n=5 or 12 each). Each point represents one sample, and the horizontal line does the mean value. (C) qRT-PCR assay for FoxO1 in the liver of mice subjected to binge alcohol drinking (n=7 each). Data were shown as box and whisker plot. Box, IQR; whiskers, 5–95 percentiles and horizontal line within box, median. (D) Immunoblottings for FoxO1 and FoxO3a in the livers from the Lieber-DeCarli or binge alcohol animal models. AH, alcoholic hepatitis; ALD, alcoholic liver disease; Con, control; EtOH, ethanol treatment; FoxO1, forkhead box protein O1; GEO, Gene Expression Omnibus; mRNA, messenger RNA; qRT, quantitative reverse transcription.
In an effort to find the underlying mechanism of miR-148a dysregulation in conjunction with decrease in FoxO1 level, correlation analyses were performed. In human liver samples, FOXO1 levels were highly correlated with miR-148a levels (figure 3A). This positive correlation was also observed in animal models (figure 3B). We then used a cell model and found out that enforced expression of FoxO1 significantly enhanced the levels of the primary and the mature forms of miR-148a (figure 3C). A previous study has identified the MIR148A gene transcriptional start site.16 To validate the effect of FoxO1 on MIR148A transcription, we sought to evaluate FoxO1 binding to the MIR148A promoter. PROMO analysis enabled us to predict three putative FoxO1 binding sites within the −3 kb promoter region of the human MIR148A (figure 3D upper). In the ChIP assays, FoxO1 specifically bound to the proximal site (RE3) located at the region from −1546 bp to −1535 bp, but not to the others sites (RE1 and RE2). The specificity in the binding of FoxO1 to the DNA was supported by results of PCR assays using non-specific primers targeting an irrelevant region (figure 3D lower). The ability of FoxO1 to induce transactivation of MIR148A was reinforced by the outcomes of luciferase assays; knockdown of FoxO1 significantly inhibited luciferase activity (figure 3E), and this effect was completely abolished in the assays using the RE3 mutant construct. All of these results provide strong evidence that FoxO1 transcriptionally controls MIR148A expression.

Transcriptional induction of MIR148A by FoxO1. (A) Correlations between FOXO1 transcript and miR-148a levels in patients with AH. (B) Correlations between FoxO1 protein and miR-148a levels in ALD animal models. (C) qRT-PCR assays for primary or mature forms of miR-148a in AML-12 cells transfected with Mock or FoxO1. (D) ChIP assays for FoxO1 binding to the promoter regions of MIR148A. HepG2 cells were transfected with Mock or FoxO1 overexpression vector. DNA–protein complexes precipitated with anti-FoxO1 antibody were subjected to PCR amplifications using the primers flanking the indicated FoxO1 response elements (FoxO1-REs). IgG immunoprecipitation represents negative control. The specificity of FoxO1 binding was verified using the primers targeting an irrelevant region in the promoter. One-tenth of cross-linked lysates served as the input control. (E) MIR148A promoter reporter assays using FoxO1-RE3 WT or Mut luciferase constructs in HepG2 cells transfected with si Con or si FoxO1. For (C)–(E), data represent the mean±SEM (n=3 each, *P<0.05, **P<0.01). AH, alcoholic hepatitis; ALD, alcoholic liver disease; ChIP, chromatin immunoprecipitation; Con, control; EtOH, ethanol treatment; FoxO1, forkhead box protein O1; miRNA, microRNA; Mut, mutant; n.s., non-significant; qRT, quantitative reverse transcription; WT, wild type.
TXNIP overexpression by ethanol as a result of miR-148a dysregulation
Having identified the inhibitory effect of ethanol on miR-148a and the role of FoxO1 in regulating its transcription, we next tried to find the target gene(s) regulated by miR-148a and its physiological role in alcohol-induced hepatocyte death. Since hepatic miR-148a levels correlated with changes in the serum ALT activities as well as the TG levels in the Lieber-DeCarli model, we selected candidate genes involved in processes of lipid/glucose metabolism and programmed cell death pathway using Targetscan and DAVID databases. Among the molecules involved in these two pathways, we focused on TXNIP because of its unique overlapping function in both biological processes mentioned above (figure 4A). Moreover, TXNIP was predicted as a putative target of miR-148a in the analyses of other bioinformatic databases (PicTar, miRanda and DIANA-mirPath). The levels of TXNIP appeared to increase in patients with AH, but the change was not statistically significant (figure 4B upper). A significant inverse correlation existed between miR-148a and TXNIP messenger RNA (mRNA) levels (figure 4B lower). To validate changes in TXNIP protein level in patient samples, we additionally used two different sets of liver specimens from patients with fibrosis or cirrhosis and found that TXNIP levels were higher in patients with alcohol-induced fibrosis than those with idiopathic fibrosis (see online supplementary figure 3A). In patients with alcoholic cirrhosis, TXNIP staining intensity seemed to be unchanged - compared to normal controls presumably due to large loss of functional hepatocytes (see online supplementary figure 3B). In the binge alcohol drinking model, Txnip mRNA levels were also found to be significantly enhanced (figure 4C). However, this effect was statistically non-significant in the Lieber-DeCarli model. More importantly, TXNIP protein levels in the liver were notably elevated in both models. Additionally, TXNIP was overexpressed in both primary hepatocytes and AML-12 cells treated with ethanol (figure 4D), supporting the direct effect of ethanol on the identified target in the hepatocytes.

TXNIP overexpression as a result of miR-148a dysregulation by ethanol. (A) Gene ontology analysis of putative target genes of miR-148a using two selected gene ontology categories (programmed cell death and lipid/glucose metabolism pathways). (B) qRT-PCR assays for TXNIP in liver samples of patients with AH (upper). Each point represents one sample, and the horizontal line does the mean value. Correlations between miR-148a and TXNIP levels were analysed (lower). (C) qRT-PCR or immunoblotting assays for TXNIP in the liver samples from control or Lieber-DeCarli (n=6 or 10 each) and control or binge alcohol (n=7 each) mouse models. Data were shown as box and whisker plot. Box, IQR; whiskers, 5–95 percentiles and horizontal line within box, median. (D) Immunoblottings for TXNIP in the lysates of mouse primary hepatocytes or AML-12 cells treated with 100 mM ethanol for 48 hours. Band intensities represent values relative to respective control (control, 1). (E) qRT-PCR or immunoblotting assays for TXNIP in the lysates of AML-12 cells transfected with miR-148a mimic for 48 hours (left), or miR-148a ASO for 72 hours (right). (F) TXNIP-3′UTR luciferase activity assays using HEK293 cells transfected with control mimic or miR-148a mimic or HepG2 cells transfected with Con ASO or miR-148a ASO for 72 hours. (G) qRT-PCR or immunoblotting assays for miR-148a-modulated primary hepatocytes isolated from the mice that had been treated with binge alcohol for 7 days. For (D)–(G), data represent the mean±SEM (n=3 or 4 each, *P<0.05). ASO, antisense oligonucleotide; Con, control; EtOH, ethanol treatment; qRT, quantitative reverse transcription; n.s., non-significant; TXNIP, thioredoxin-interacting protein.
To assess the effect of miR-148a on TXNIP, we transfected miR-148a in AML-12 cells and found that miR-148a transfection lowered TXNIP mRNA and protein levels, whereas its antisense oligonucleotide (ASO) transfection increased their levels (figure 4E). Given the nearly perfect pairing between the TXNIP-3′ untranslated region (UTR) and the miR-148a seed sequence (figure 4F left), we next defined the ability of miR-148a to inhibit TXNIP more precisely using in vitro assays. As expected, transfection of HEK293 cells with miR-148a prevented luciferase expression from pEZX-TXNIP-3′UTR luciferase construct, whereas that with miR-148a ASO exerted the opposite effect (figure 4F right). A mutant TXNIP 3′UTR reporter (5′ -TCGTGAG-3′ and 5′ -GGTGACA-3′) construct showed no change in luciferase expression. To verify the effect of miR-148a on the identified target on alcohol challenge, we performed further experiments using primary hepatocytes. Transfection of hepatocytes isolated from the ethanol-treated mice (1 week) with miR-148a significantly inhibited TXNIP mRNA and protein levels (figure 4G). Consistently, miR-148a ASO transfection enhanced TXNIP expression. Modulations of miR-148a levels were confirmed in AML-12 cells or primary hepatocytes after transfection with miR-148a mimic or ASO (see online supplementary figure 4A,B).
In the analysis of GEO dataset (GSE67456), mice fed on Lieber-Decarli diet showed an increase in Txnip transcript level in the liver (see online supplementary figure 5A). A significant inverse correlation existed between Foxo1 and Txnip transcripts. FoxO1 inhibition of TXNIP and the antagonising effect of miR-148a ASO were confirmed in cell-based assays. FoxO1 overexpression prevented alcohol-induced TXNIP expression, which was resumed by miR-148a ASO transfection (see online supplementary figure 5B). Flow cytometric analyses corroborated the effects of FoxO1 and miR-148a ASO transfection on TXNIP in cells treated with ethanol (see online supplementary figure 5C). All of these results indicate that exposure of hepatocytes to alcohol induces TXNIP overexpression through dysregulation of the FoxO1-mediated miR-148a expression.
Activation of NLRP3 inflammasome in hepatocytes by ethanol
Inflammasome activation is responsible for promoting the progression of liver diseases including ALD.17 Since TXNIP facilitates NLRP3 inflammasome activation through binding to NLRP3,18 we focused on the role of TXNIP-NLRP3 inflammasome in alcohol-induced hepatocyte injury. In the analysis of GEO dataset (GSE97234), Nlrp3, ASC (Pycard) and Il1b mRNA levels were all upregulated in the liver of AH mice compared with controls (figure 5A). Ethanol treatment increased NLRP3 expression in primary hepatocytes or AML-12 cells (figure 5B and C, see online supplementary figure 6A). The active caspase-1 and ASC levels were also upregulated (figure 5C). In patients with AH, NLPR3 transcript levels tended to be diminished (see online supplementary figure 6B). However, NLRP3 protein expression was increased in alcohol-fed mice (see online supplementary figure 6C). The caspase-1 (CASP1) and ASC (PYCARD) transcript levels also increased and inversely correlated with FOXO1 mRNA levels in the dataset of patients with AH (GSE28619) (see online supplementary figure 7). To assess the localisation of NLRP3 and TXNIP, immunocytochemical assays were done. As expected, TXNIP and NLRP3 were colocalised after ethanol treatment, and their staining intensities were enhanced (figure 5D). Moreover, ethanol treatment increased the intensity of TXNIP and NLRP3 binding, which was prevented by miR-148a mimic transfection (figure 5E upper). Knockdown of TXNIP resulted in a decrease in the level of active caspase-1 with no change in NLRP3 level (figure 5E lower). Consistently, miR-148a transfection attenuated colocalisation of TXNIP and NLRP3 (figure 5F). Since caspase-1 facilitates pyroptosis,19 we wondered whether alcohol was capable of eliciting hepatocytes pyroptosis (200 mM ethanol was used in this assay as reported previously20). In the flow cytometric analysis using the biomarkers for pyroptosis (ie, active caspase-1 and propidium iodide (PI), it was observed that ethanol treatment augmented the population of caspase-1/PI double positivity in AML-12 cells, and this effect was diminished by miR-148a transfection (figure 5G). Similar results were obtained by TXNIP knockdown. In addition, miR-148a ASO transfection increased the population of caspase-1/PI-double positive cells. FoxO1 overexpression led to a decrease in the caspase-1/PI double-positive cell populations (see online supplementary figure 8).

Pyroptosis of hepatocytes as mediated by TXNIP-dependent inflammasome activation. (A) Expression heatmap of Nlrp3, Pycard (ASC) and Il1b in the RNA-seq database (GSE59492) from control and AH mice (n=3 each) (P<0.01). (B, C) qRT-PCR or immunoblotting assays for NLRP3 or other inflammasome markers in mouse primary hepatocytes or AML-12 cells treated with 100 mM ethanol for 48 hours. Data represent the mean±SEM (n=4 each, *P<0.05, **P<0.01). Band intensities represent values relative to respective control (control, 1). (D) Confocal images of TXNIP and NLRP3 in AML-12 cells treated with 100 mM ethanol for 48 hours. (E) Immunoprecipitation or immunoblottings for TXNIP and NLRP3 inflammasome markers in AML-12 cells exposed to 100 mM ethanol for 48 hours immediately after transfection with miR-148a mimic (or Con mimic) (upper) or TXNIP siRNA (or Con siRNA) for 48 hours (lower). (F) Confocal images of TXNIP and NLRP3 in AML-12 cells treated with 100 mM ethanol for 48 hours after miR-148a mimic (or Con mimic) transfection. (G) Flow cytometric analyses using propidium iodide and active caspase-1 markers. AML-12 cells were similarly transfected, and then treated with 200 mM ethanol for 48 hours. miR-148a mimic or miR-148a ASO transfection and TXNIP knockdown experiments were separately done. AH, alcoholic hepatitis; ASO, antisense oligonucleotide; Con, control; EtOH, ethanol treatment; qRT, quantitative reverse transcription; siRNA, small-interfering RNA; TXNIP, thioredoxin- interacting protein.
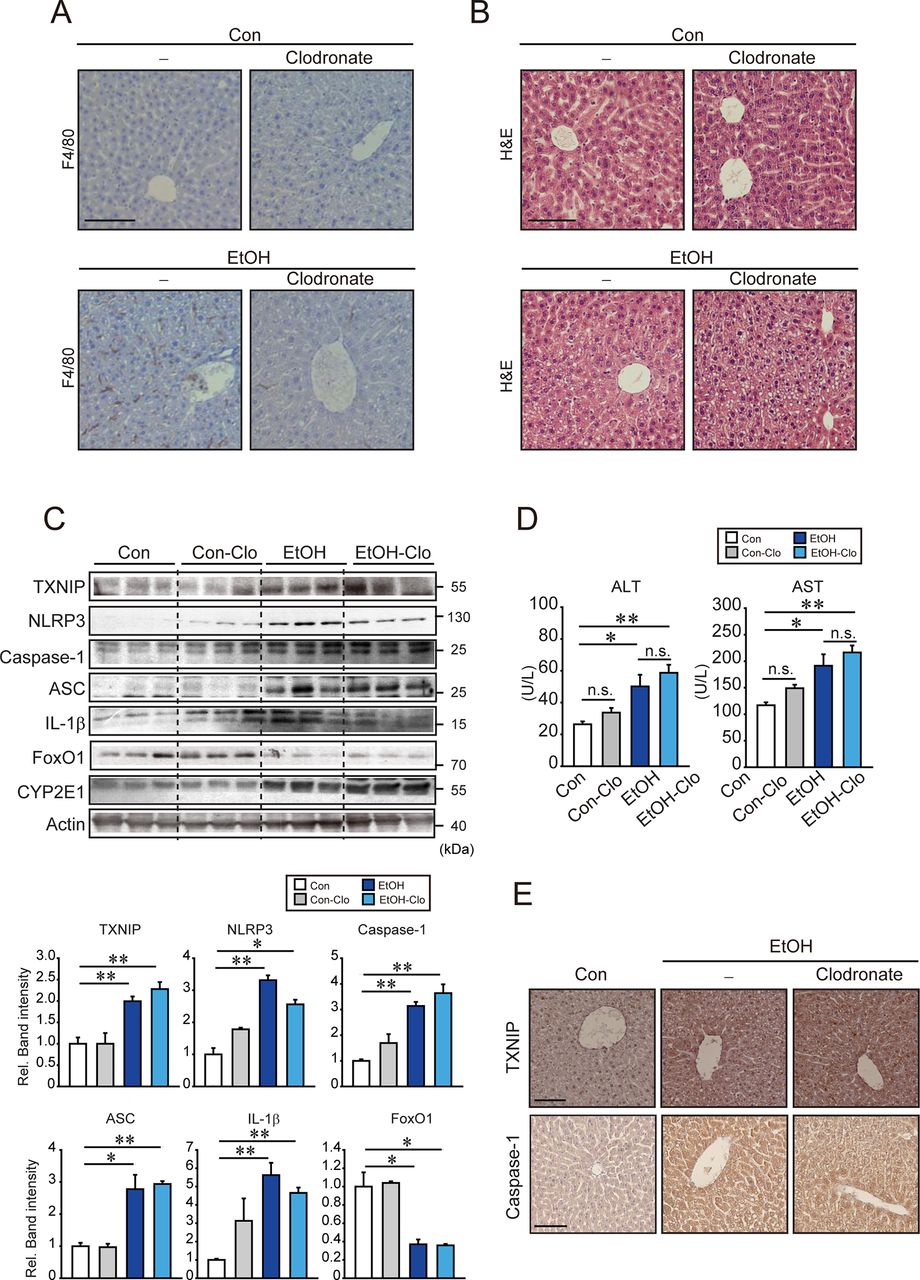
To understand the role of the inflammasome in hepatocytes during alcohol-induced liver injury, we next examined the effect of alcohol on inflammasome activation in the liver using an in vivo macrophage depletion model. In immunostaining assays, it was observed that the treatment with clodronate liposomes diminished the F4/80 staining intensity in the liver of mice fed Lieber-DeCarli diet (figure 6A). Clodronate neither induced any histopathological change in the liver nor enhanced ethanol-induced liver injury (figure 6B). TXNIP, active caspase-1 and ASC levels were all elevated in both ethanol and ethanol plus clodronate treatment groups (figure 6C). The levels of NLRP3 and IL-1β were partly diminished in livers of ethanol plus clodronate treated group. FoxO1 expression was decreased in ethanol fed groups regardless of clodronate treatment. Clodronate did not significantly change ALT or AST activities (figure 6D). In immunohistochemistry (IHC) staining, TXNIP and caspase-1 levels were increased after feeding the ethanol with or without clodronate treatment (figure 6E). This effect was confirmed by the result of terminal deoxynucleotidyl transferase nick end labelling (TUNEL) (see online supplementary figure 9). Collectively, our results implicate that alcohol induces hepatocytes pyroptosis via TXNIP-mediated NLRP3 inflammasome activation as a consequence of miR-148a dysregulation.

Ethanol-induced inflammasome activation in hepatocytes from mice treated with clodronate. (A, B) Representative IHC for F4/80 (scale bar, 100 µm), and H&E staining (scale bar, 50 µm). C57BL/6 mice were first intravenously injected with 10 mL/kg body weight of clodronate (or vehicle), and after a week, they were subjected to Lieber-DeCarli diet (or control diet) with additional injections of clodronate (5 mL/kg body weight, intraperitoneal, every 4 days) for 5 weeks. (C) Immunoblottings for TXNIP, inflammasome or other biomarkers. Densitometric band intensities represent values relative to respective control (control, 1). (n=3 each). (D) Serum ALT and AST activities. (n=3–6/group). (E) IHC for TXNIP or caspase-1. Representative liver sections were shown (n=3 each). Scale bar, 100 µm. For (C)and (D), data represent the mean±SEM (statistical comparisons among groups by one-way ANOVA with Fisher’s least significant difference (C) or Bonferroni correction (D) for multiple comparisons, *P<0.05, **P<0.01). AH, alcoholic hepatitis; ALT, alanine aminotransferase; ANOVA, analysis of variance; ASO, antisense oligonucleotide; AST, aspartate transaminase; Con, control; EtOH, ethanol treatment; IHC, immunohistochemostry; IL-1β, interleukin 1β; n.s., non-significant; qRT, quantitative reverse transcription; siRNA, small-interfering RNA; TXNIP, thioredoxin-interacting protein.
Amelioration of hepatic injury by hepatocyte-specific delivery of miR-148a
To validate the physiological impact of miR-148a in hepatocytes on alcohol-induced liver injury, we finally performed in vivo gene delivery using lentivirus. A lentiviral vector comprising gene transcribing the miR-148a, cloned downstream from the albumin promoter, was constructed and the packaged viruses were injected to mice (the albumin promoter was used for hepatocyte-specific gene expression). Hepatocyte-specific delivery of miR-148a ameliorated the pathogenesis of ALD in mice-fed Lieber-DeCarli diet, as evidenced by an improvement in the histopathological features such as reduction in the hepatocyte ballooning and abnormal lipid accumulation (figure 7A upper). qRT-PCR assays confirmed the delivery of miR-148a (figure 7A lower). Immunoblottings for TXNIP, active caspase-1, ASC and IL-1β in liver homogenates verified inhibition of inflammasome activation (figure 7B). FoxO1 level was restored, whereas NLRP3 or CYP2E1 levels remained unchanged. In IHC, TXNIP and caspase-1 staining intensities in the liver tissue were lowered after lentiviral delivery of miR-148a compared with the controls (figure 7C). The ability of miR-148a to inhibit inflammasome activation was verified by a decrease in serum IL-1β level (see online supplementary figure 10A). Inhibition of hepatocyte death by miR-148a delivery was confirmed by TUNEL staining for the liver sections (see online supplementary figure 10B) and immunoblotting for PARP-1 cleavage (see online supplementary figure 11). Consistently, hepatocyte-specific delivery of miR-148a significantly repressed ethanol-induced increases in the liver-to-body weight ratio, ALT activity and serum and hepatic TG contents (figure 7D). Overall, our results demonstrate that miR-148a protects hepatocytes from alcohol-induced inflammasome activation and hepatocyte pyroptosis by inhibiting TXNIP (figure 7E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of inflammasome and liver injury after hepatocyte-specific lentiviral delivery of miR-148a. (A) H&E staining (scale bar, 50 µm) and qRT-PCR assays for miR-148a in the liver. Mice were intravenously injected with a single dose of albumin promoter-pre-miR-148a lentiviruses (or Lv-control), and after a week, they were fed on a Lieber-DeCarli diet for 4 weeks as depicted in the scheme (n=4–6/group). (B) Immunoblotting assays for TXNIP, inflammasome or other biomarkers in liver homogenates. Densitometric band intensities represent values relative to respective control (control, 1) (n=3 each). (C) IHC for TXNIP or caspase-1. Representative liver sections were shown (n=3 each). Scale bar, 100 µm. (D) Liver-to-body weight ratio and representative blood biochemical parameters. (E) A scheme illustrating the mechanism by which ethanol induces pyroptosis of hepatocytes. For (A), (B) and (D), data represent the mean±SEM. Significantly different compared with each control, *P<0.05, **P<0.01. For (A), statistical significance of differences between two groups indicated (unpaired t-test). For (B) and (D), statistical comparisons among multiple groups by one-way ANOVA with Fisher’s least significant difference (B) or Bonferroni correction (D). ALD, alcoholic liver disease; ALT, alanine aminotransferase; ANOVA, analysis of variance; AST, aspartate transaminase; Con, control; EtOH, ethanol treatment; FoxO1, forkhead box protein O1; IL-1β, interleukin 1β; qRT, quantitative reverse transcription; TG, triglyceride; TXNIP, thioredoxin-interacting protein.
Discussion
MiRNAs regulate diverse biological functions in the liver. Alteration in the levels of specific miRNAs promotes the incidence and progression of liver diseases (see online supplementary table 1).21 Since hepatocytes are primarily afflicted by ethanol toxicity, miRNAs abundant in hepatocytes may be more sensitive to alcohol exposure. In our GEO data (GSE59492) analysis of liver specimens of patients with AH, only 2% of the miRNAs were found to be significantly downregulated. Among them, miR-148a was profoundly and most significantly suppressed. Since miR-148a belongs to the most abundant miRNAs in the liver22 and had not been investigated before, we focused on miR-148a dysregulation in the progression of ALD. Here, we revealed marked and specific decrease in miR-148a level in patients with AH, and this decrease was further confirmed by the outcomes from two different ALD animal models. Moreover, our data obtained from the primary hepatocytes or cell lines corroborated the effect of alcohol on miR-148a dysregulation.
FoxO1 belongs to key transcription factors related to stress response pathway.23 In particular, FoxO1 serves as a metabolic regulator and tumour suppressor in the liver.24 25 In the present study, FoxO1 was predicted a central molecule interacting with other genes for ‘positive transcriptional regulation’ and ‘inhibitory regulation of programmed cell death’ processes. Moreover, we found marked suppression of FoxO1 in the liver of patients with AH and ALD animal models, which confirmed the inhibitory effect of alcohol on FoxO1. This is in line with our finding of negative correlation between the miR-148a levels and the serum ALT activities (or TG contents) or with the known effect of alcohol on NAD+/NADH ratio and lipid and glucose metabolism.26–28 Decrease in the level of FoxO1 facilitates ROS and proinflammatory cytokine production in 3T3L1 adipocytes.29 Similarly, hepatocytes exposed to ethanol and unsaturated fatty acids showed decrease of nuclear FoxO1.30 In another study, Foxo1 levels were rather enhanced in the Lieber-Decarli rat model,31 possibly reflecting species variation in the metabolic capacity of alcohol. Overall, our results strengthen the concept that a decrease in the level of FoxO1 promotes the progression of ALD in association with dysregulation of miR-148a. Here, we demonstrated that FoxO1 is a transcriptional factor for MIR148A. Although several transcription factors such as SREBP-1, MYB and p53 may regulate the miR-148a levels,16 32 33 they did not show any correlation with miR-148a in ALD. Our data revealed strong correlation between miR-148a and FoxO1, along with identifying the presence of a functionally active FoxO1 binding site in the human MIR148A as corroborated by the results of the mutation assay.
Ethanol consumption may cause epigenetic changes, which may also contribute to miR-148a dysregulation in the progression of ALD. Previously, it has been shown that hypermethylation of miR-148a gene promoter was associated with silencing of miR-148a in liver cancer34 and IL-6 enhanced DNMT1 activity and methylation-dependent tumour suppressor genes by decreasing miR-148a/152 in malignant cholangiocytes.35 In our study, however, ethanol treatment unchanged the methylation of miR-148a promoter region in hepatocytes (see online supplementary figure 12).
Among the miRNAs abundant in hepatocytes, miR-148a plays important roles such as regulation of hepatocyte differentiation, inducing adult liver phenotype.34 However, the roles and targets of miR-148a had been mostly studied in tumour biology field. To our knowledge, miR-148a regulation of TXNIP is a novel event occurring in the progression of liver disease. MiR-148a transfection prevented alcohol-induced TXNIP expression in cell-based assay, indicating the ability of miR-148a to regulate ALD through TXNIP. This idea is in line with the reports that TXNIP rendered cells susceptible to oxidative stress and injury.18 36 37 Minor or moderate increases in TXNIP mRNA levels in patients with AH or Lieber-DeCarli diet mice may reflect the feature of miRNA-regulating protein translation. Increase of TXNIP protein in alcoholic fibrosis was also validated using human samples. Together, our results showing FoxO1 regulation of miR-148a, changes in TXNIP levels, reciprocal relationship between FoxO1 and TXNIP in animal experimentation and database analysis, and the ability of FoxO1 to restore miR-148a and inhibit TXNIP levels strongly support the role of FoxO1 in the regulation of TXNIP by miR-148a.
Another intriguing finding of our study is discovery of TXNIP overexpression induced by alcohol for the promotion of NLRP3 inflammasome activation in hepatocytes. The NLRP3 inflammasome pathway undergoes two steps for full functional activation. The first priming signal is transcriptional increases in inflammasome component genes, whereas the second step is formation of oligomer complex (NLRP3/ASC/caspase-1) and activation, which is enhanced by TXNIP. Full activation of NLRP3 inflammasome through TXNIP binding then induced active caspase-1 and IL-1β release as a hallmark. Hence, knockdown of TXNIP lowered active caspase-1 level but unaffected NLRP3 and ASC levels in the present study. These results are in line with previous reports that inhibition of TXNIP did not attenuate NLRP3 and ASC protein levels but diminished activation of inflammasome (ie, decrease in caspase-1 levels and IL-1β secretion).38 39
Host-derived danger signals, such as certain metabolites and ROS, activate the NLRP3 inflammasome pathway.40 NLRP3 may also be activated in hepatocytes in response to endotoxin challenge, a condition facilitated by alcohol intake.41 In NLRP3-deficient mice, alcohol-induced liver steatosis and injury were ameliorated.42 Thus, an increase in NLRP3 level would promote hepatocyte injury. Increase in NLRP3 protein level in our alcohol-fed mice is consistent with the report showing NLRP3 transcript upregulation after alcohol feeding.43 Our data showing a slight decrease in NLRP3 mRNA in patients with AH would be associated with compensatory responses. Additionally, the inverse relationship between the levels of FoxO1 and inflammasome markers in patients with AH strengthened the conclusion that decrease of FoxO1 by alcohol contributes to TXNIP-mediated NLRP3 inflammasome activation in ALD.
Multiple mechanisms facilitate hepatocyte death, resulting in the subsequent progression of ALD. Excessive and uncontrolled activation of inflammasome has a damaging effect on the host, inducing uncontrolled inflammatory responses and the consequent cell death.44 However, inhibition of apoptotic cell death is not enough to abrogate alcohol-induced liver injury,45 suggesting that other types of hepatocyte death mechanisms may also exist. An interesting aspect of our finding is that alcohol-induced activation of inflammasome pathway in hepatocytes promoted pyroptosis, which relied on TXNIP overexpression. This observation was consistent with the finding that caspase-1 was activated in hepatocytes of patients with AH.46 Consistently, TXNIP deficiency resulted in the enhancement of cell proliferation and an increased resistance to cell death.47 Moreover, miR-148a transfection ameliorated alcohol-induced pyroptotic cell death, supportive of the notion that miR-148a and the associated molecules may represent new targets for ALD.
Inflammasome activation was extensively studied in macrophages. Recently, attention was paid to hepatocytes per se in this aspect.48 49 However, the specific contribution of NLRP3 inflammasome activation in hepatic parenchymal cells to alcoholic liver injury and the associated molecular mechanisms had been unclear. Our result that alcohol intakes increased all of the hepatic inflammasome components examined in mice pretreated with clodronate fortifies the effect of alcohol on inflammasome activation in hepatocytes. In these animals, IL-1β secretion was diminished as compared with the control (data not shown) presumably because of the inhibition of inflammasome activation in macrophages. Other types of cells (eg, immune cells and hepatic stellate cells) may have some effects. Nevertheless, our data showing that the serum ALT activities remained elevated after macrophage depletion further supports the direct effect of alcohol on NLRP3 inflammasome activation in hepatocytes. More importantly, this hypothesis is strengthened by the outcomes of an in vivo experiment using hepatocyte-specific lentiviral delivery of miR-148a. Of note, miR-148a delivery caused recovery of hepatic FoxO1 expression in ethanol-fed mice, which may reverberate an improvement in liver function. This event may be associated with alterations in AKT activity, a kinase that degrades FoxO1 through phosphorylation, since acute ethanol treatment has been shown to activate PI3K/AKT pathway.50
Collectively, our findings demonstrate that alcohol induces pyroptosis of hepatocytes by activating the NLRP3 inflammasome mediated by miR-148a-dependent TXNIP overexpression. The present identification of miR-148a as a regulator of pyroptosis, along with the transactivating effect of FoxO1 on the gene expression, sheds light on novel targets and potential strategies for the treatment of ALD.
References
Footnotes
Contributors MJH: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; statistical analysis. THK: study concept and design; acquisition of data; analysis and interpretation of data. JSY study concept and design; acquisition of data; analysis and interpretation of data; administrative, technical or material support. DB: acquisition of data; analysis and interpretation of data; statistical analysis. PS-B: study concept and design; human sample analysis and interpretation of data; administrative, technical or material support. SGK: study concept and design; analysis and interpretation of data; writing of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; obtained funding; administrative, technical or material support and study supervision.
Funding This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIP) (NRF-2015R1A2A1A10052663) and MJH was supported by Basic Science Research Program of the Ministry of Education (NRF-2018R1A6A3A01011724). Sancho-Bru P was supported by Instituto de Salud Carlos III, Miguel Servet CP11/00071 and FIS PI14/00320 and cofinanced by Fondo Europeo de Desarrollo Europeo (FEDER), Unión Europea, ’Una manera de hacer Europa'.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics Committees of Seoul National University and the Hospital Clinic of Barcelona.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. The funding statement has been updated.
Presented at A part of the data included in this manuscript was presented at the 2017 Spring International Convention of the Pharmaceutical Society of Korea in Korea (2017).