Article Text

Abstract
Pancreatic ductal adenocarcinoma (PDA) is notoriously aggressive and hard to treat. The tumour microenvironment (TME) in PDA is highly dynamic and has been found to promote tumour progression, metastasis niche formation and therapeutic resistance. Intensive research of recent years has revealed an incredible heterogeneity and complexity of the different components of the TME, including cancer-associated fibroblasts, immune cells, extracellular matrix components, tumour vessels and nerves. It has been hypothesised that paracrine interactions between neoplastic epithelial cells and TME compartments may result in either tumour-promoting or tumour-restraining consequences. A better preclinical understanding of such complex and dynamic network systems is required to develop more powerful treatment strategies for patients. Scientific activity and the number of compelling findings has virtually exploded during recent years. Here, we provide an update of the most recent findings in this area and discuss their translational and clinical implications for basic scientists and clinicians alike.
- pancreatic fibrosis
- pancreatic cancer
- molecular carcinogenesis
- molecular oncology
- molecular mechanisms
Statistics from Altmetric.com
Introduction
Despite intense research efforts over the last decade, pancreatic ductal adenocarcinoma (PDA) is still considered one of the most aggressive and lethal solid tumours.1 Survival statistics have hardly changed, with most patients struggling to survive the first year after diagnosis, and a 5-year survival rate at 7%.1 Early diagnosis is still an unresolved problem as the vast majority (>80%) of patients are diagnosed with advanced or metastatic disease, and even for early stage tumours, relapse rate is high. Given the extraordinary progress in other tumour entities such as melanoma, colorectal, lung and breast cancer, PDA is projected to become the second leading cause of cancer-related death in the USA by 2030.2
After surgical resection of PDA, the nucleoside analogue gemcitabine was widely used as standard adjuvant treatment based on results from the European Study Group for Pancreatic Cancer-3 (ESPAC-3) trial.3 In 2017, results from the ESPAC-4 trial were published showing a significantly prolonged median overall survival for patients receiving gemcitabine plus capecitabine compared with gemcitabine alone (28.0 vs 25.5 months, p<0.04).4 Despite a higher rate of grade 3–4 adverse events in the combination treatment group (608 vs 481 events), gemcitabine plus capecitabine should nowadays be considered for adjuvant treatment for patients with PDA.
In the palliative setting, gemcitabine monotherapy has been the standard of care for many years. The regimen is relatively well tolerated, but the survival benefit is marginal at best.5 More recently, two combination chemotherapeutic regimens have been introduced for patients with PDA that provide oncologist with more effective treatment options in the palliative setting.6 7
Over the last few years, several areas of biomedical research have emerged that combine basic, translational and clinical research concepts to explore the complex biology of PDA with potential implications in patient care and prognosis. Classical molecular biology concepts are increasingly replaced by complex multidisciplinary approaches by multinational teams that address epithelial and stromal determinants and their interplay in PDA. The importance of the dialogues between both compartments has been recently shown in an elegant PDX mouse model where molecular profiling of both the tumour cells (human origin) and the stroma (mouse origin) has enabled to establish two stroma molecular subgroups that mirror those two described at the tumour cell level.8 Based on recent precision oncology concepts in other tumour entities, several whole-genome sequencing studies and transcriptional profiling analyses have been conducted aiming at the discovery of distinct molecular subtypes in PDA that could be exploited for therapeutic stratification. Besides known signature mutations such as KRAS, TP53, CDKN2A and SMAD4, numerous recurrently mutated or transcriptionally altered genes were discovered, that converge on core signalling pathways and cellular processes.9–11 Furthermore, genome-wide transposon screening and quantitative insertion site sequencing has become a powerful new tool for the discovery of cancer drivers that are difficult to uncover with other methods.12 13 Yet again, these studies showed that PDA is a highly heterogeneous and genetically diverse disease composed of numerous subgroups with different molecular make ups. However, subtype-driven therapy decisions are not yet implemented in clinical routine and are restricted to very few molecular alterations such as BRCA mutations, that occur in about 5% of patients with PDA and respond well to platinum-based chemotherapy and poly (ADP-ribose) polymerase inhibitors.14
Besides genetic and transcriptional alterations in tumour cells, the tumour microenvironment (TME) plays a pivotal role during pancreatic carcinogenesis and therapeutic resistance. This research topic has virtually exploded during recent years. In 2011, we first summarised important findings in this area in Gut,15 followed by a second comprehensive review in 2015.16 Since then, numerous novel and exiting preclinical and clinical studies have been conducted that will be discussed here. Furthermore, the concept of precision oncology and subtyping of PDA involves genetic and transcriptional alterations in epithelial cells and entails complex cellular alterations within the TME such as in immune cells, nerve cells, cancer-associated fibroblasts (CAFs) and multiple biochemical or epigenetic crosstalks within the pronounced tumour stroma in PDA. Although stromal subtyping is still in its infancy, it may turn out to be an important stratification tool for future therapies.
Targeting Sonic Hh as one of the central drivers of the stroma in pancreatic cancer
The Hedgehog (Hh) signalling system is central to the interplay between the tumour cell compartment and the surrounding stroma, thus it was judged to be a prime target for therapeutic approaches.17 Hedgehog ligands (mostly Sonic Hh (SHH), and to a lesser extent Indian Hh and Desert Hh) are expressed by pancreatic epithelial cells on inflammatory insults (eg, pancreatitis) or after experimental expression of mutant KRAS, implicating Hh signalling early in PDA development.18 19 In the setting of pancreatic cancer, the released Hh ligands do not seem to function in an autocrine manner but rather in a paracrine mode acting on surrounding mesenchymal cells.20–24 This fact is of utmost importance for the interpretation of results obtained using genetically engineered mouse models or the use of Hh inhibitors in mice and men.
Earlier in vitro and cell transplantation experiments had shown that tumour cell-derived Hh reprograms quiescent pancreatic fibroblasts and stellate cells into activated and contractile CAFs, resulting in the reciprocal promotion of cancer cell proliferation, survival, invasion and chemoresistance.25 26 Based on these observations and the finding that the abundant stroma can suppress tumour angiogenesis and impair therapeutic drug delivery,17 Hh inhibitors (targeting the essential signal transducing component Smoothened (SMO)) were included in clinical trials of metastatic PDA. Disappointingly however, clinical trials reported that the addition of SMO antagonists to gemcitabine was not superior when compared with gemcitabine alone (NCT01064622, NCT01195415).27 28 Even worse, one particular study using gemcitabine in combination with the SMO inhibitor Saridegib (IPI-926) had to be prematurely terminated as this group displayed a decreased survival compared with gemcitabine monotherapy (web page info from Infinity Pharmaceuticals). As a result, many clinical trials with Hh inhibitors were either withdrawn or came to a halt (a list of patient trials using Hh inhibitors can be found in online supplementary table 1).
Supplementary file 1
Additional preclinical studies employing a conditional Shh gene knock-out crossed into the LSL-Kras G12D/+; LSL-Trp53 R172H/+; Pdx-1-Cre (KPC) mouse model found a similarly reduced overall survival in animals lacking Shh expression. This was associated with a drastically reduced α-smooth muscle actin (α-SMA)-positive fibroblast stroma and an undifferentiated tumour histology.29 Comparable data were obtained when a clinically approved SMO inhibitor was applied as a monotherapy in KPC mice prior to tumour detection,29 or when CAFs were depleted in vivo using a SMA-TK allele.30 Intriguingly, tumours, which arose in animals with reduced Hh signalling and/or under conditions of stromal CAF ablation, were sensitive to angiogenesis or immune checkpoint inhibitors, respectively, suggesting novel possibilities for clinical combination therapies in the future.29 30
Taken collectively, these recent data imply that the Hh-activated stroma exerts both an anti-angiogenic function and impaired chemosensitivity but simultaneously a more dominant tumour-restraining activity in PDA. In line with this view, pharmacological stimulation of SMO has been shown to slow PDA progression, with fewer pancreatic intraepithelial neoplasia and less tumour cell proliferation in KPC mice.20 The concept of stromal Hh signalling showing tumour suppressive traits is not fully unprecedented as similar findings have been made in mouse models of bladder, prostate and colon cancer.31–33 In bladder cancer, for instance, Hh-activated stroma delivers pro-differentiating bone morphogenetic protein factors, thereby facilitating urothelial differentiation and interfering with carcinogenesis.31 It will be interesting to learn whether the tumour-suppressive functions of Hh signalling occur exclusively at certain tumour stages or whether they represent a general feature, applying to late as well as early stages. Furthermore, it is currently unclear whether Hh signalling impacts on the relative proportion of tumour-suppressive versus tumour-promoting CAF subpopulations within the stroma. Additional and currently unresolved issues in the field include the exact contribution of Hh ligands to the tumour immune infiltrate as well as non-canonical Hh effects such as the modulation of cellular metabolism.34
Biophysical properties of the tumour stroma: depletion or fine-tuning?
The notion whether and to which extent the pronounced tumour stroma contributes to disease progression and therapeutic resistance in PDA is widely debated in the field, and several mechanisms have been suggested. For instance, the stroma was shown to increase the stiffness of the tumour and to compress tumour vessels resulting in impaired tumour perfusion and drug accumulation. Besides biophysical properties of the stroma, stroma-specific subtypes have been proposed recently in analogy to the epithelial subtypes that may be prognostic for patients with PDA and represent novel therapeutic vantage points.10 35 36
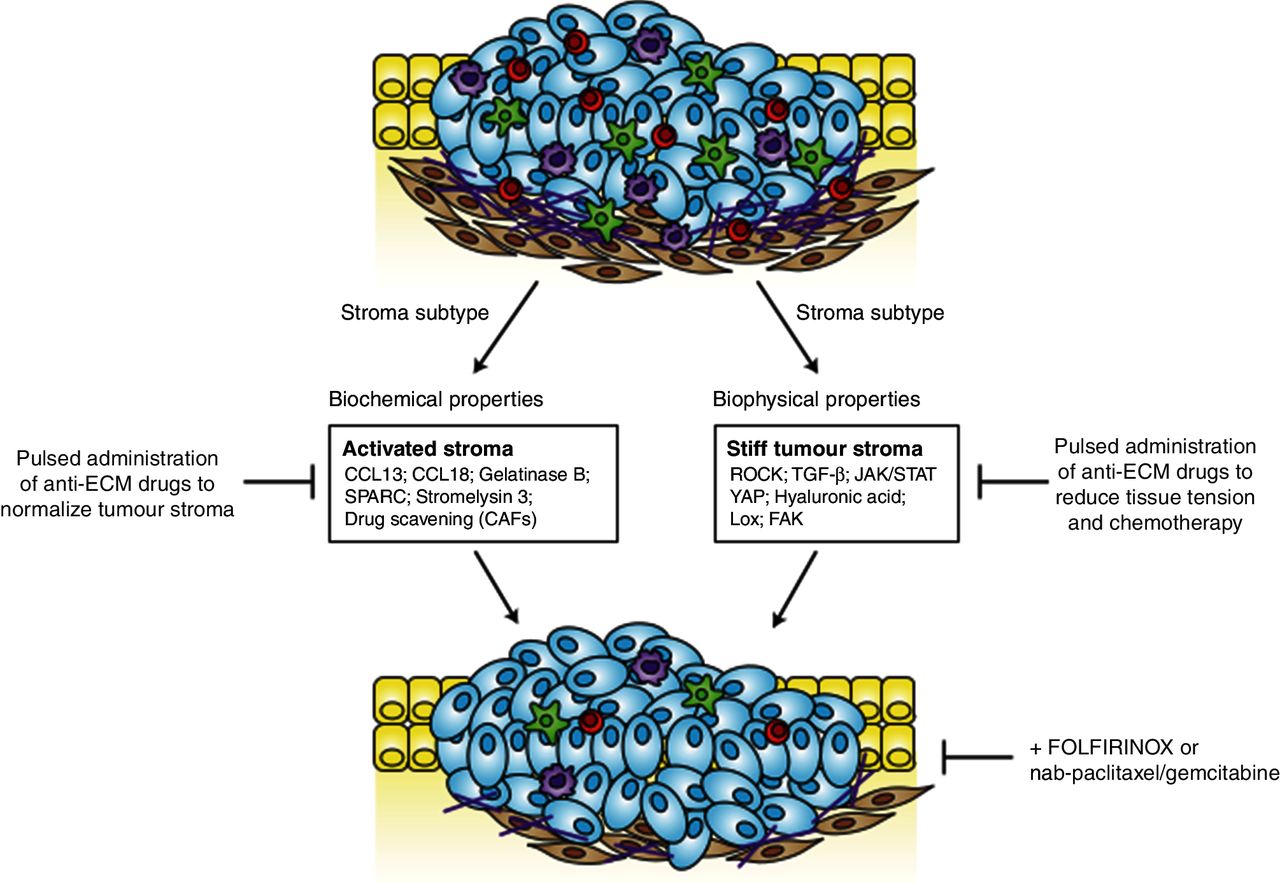
The ‘normal’ stroma subtype was found to highly express pancreatic stellate cell (PSC) markers such as α-smooth muscle actin, vimentin and desmin (median survival 24 months). In contrast, the activated pro-inflammatory and macrophage-rich phenotype was characterised by upregulation of a diverse set of genes such as chemokine ligands CCL13, CCL18, gelatinase B, stromelysin 3 and secreted protein acidic and rich in cysteine, thus promoting disease progression (median survival 15 months). These findings exemplify that the complexity of the stroma goes well beyond the bulk stromal phenotype. However, it is unknown whether and how different stroma subtypes (‘activated’ vs ‘normal’; ‘stiff’ vs ‘soft’), mediate therapeutic response in patients. An increasing body of evidence suggests that stromal subtypes regulate tumour cell tension and contractility. Using clinical specimen and experimental mouse models, Laklai et al identified a distinct, highly stiff, matricellular-stromal phenotype that was specifically associated with reduced epithelial transforming growth factor (TGF)-β signalling, elevated β-integrin mechano-signalling and Stat3 activation resulting in therapeutic resistance and significantly shortened survival in mice and humans.37 In line with these findings, ATRA, an active metabolite of vitamin A, was reported to reprogram mechanical quiescence in PSCs via a mechanism involving a retinoic acid receptor beta-dependent downregulation of actomyosin contractility.38 Furthermore, Rho kinase (ROCK) signalling, Yes-associated protein 1 and Janus kinase 2 and signal transducer and activator of transcription 3 signalling pathways have been associated with tissue tension and tumour cell contractility.39–41
Alternative stromal targets included the TGF-β pathway (LY364947),42 and the megadalton glycosaminoglycan hyaluronan (HA) using PEGPH20.43–45 Moreover, collagen maturation is mediated by lysyl oxidase (LOX), which is highly expressed in the hypoxic tumour microenvironment. Interestingly, the combination of gemcitabine and LOX inhibitors resulted in prolonged survival and reduced metastasis in the KPC mouse model.46 Furthermore, inhibition of focal adhesion kinase in mouse models of PDA reduces the collagen deposition in the extracellular matrix (ECM) and decreases the number of immune suppressive cells in the tumour, leading to prolonged survival and responsiveness to immunotherapy.47 Although the concept of increasing drug delivery seems appealing and feasible due to the pronounced desmoplasia, collapsed lymphatic drainage system and hypovascularity, the failure of numerous clinical studies has somewhat dampened the initial euphoria of this strategy. As discussed above, the disappointing clinical outcome of SHH inhibition in patients with PDA fuelled scepticism in the field whether stromal depletion would be the appropriate therapeutic strategy to overcome chemoresistance.27 However, the highly heterogenous tumour stroma may have to be assessed in much more detail to identify subgroups of patients that might benefit from specific, pharmacological stroma modifications. A first step towards personalised antistroma therapies might represent enzymatic depletion of HA by PEGPH20. Encouraging results from a phase I/II study (NCT01839487) combining PEGPH20 and gemcitabine showed that patients with high levels of HA had improved response rates.48 These results led to the initiation of a large phase III study that is currently enrolling patients (NCT02715804) combining gemcitabine with nab-paclitaxel and PEGPH20 or placebo. Figure 1 depicts an overview about key players mediating the stiff and activated tumour stroma subtype in PDA.

Molecular hallmark features of the activated tumour stroma and stiff tumour stroma in pancreatic ductal adenocarcinoma. CAF, cancer- associated fibroblast; ECM, extracellular matrix; FAK, focal adhesion kinase; JAK, Janus kinase 2; LoX, lysyl oxidase; STAT, signal transducer and activator of transcription 3; TGF, transforming growth factor; YAP, Yes-associated protein 1.
Tumour stroma, drug delivery and drug scavenging
In general, pancreatic tumours are hypovascular and densely packed with ECM components such as collagen and HA leading to high intratumoural tissue pressure, collapse of blood vessels and dramatically decreased tumour perfusion. Therefore, reducing tissue tension and intratumoural pressure by ablation or modification of ECM components may result in improved tumour perfusion and therapeutic response. Drug delivery is a complex process that depends on blood flow, particle size, charge, pore size of vessels and systemic and local half-life of the respective compound. Vessel pores are in the range of 50–60 nm in pancreatic cancer,49 and delivery of targeted or systemic therapies work in a size-dependent manner.50 Several preclinical investigations using different pharmacological or genetic approaches were aimed to physically deplete or remodel acellular components of the stroma (reduction of fibrillar collagen) to increase drug delivery and response to antineoplastic agents.40 51 For instance, co-administration of low-dose cilengitide (angiogenesis inhibitor) and verapamil (Ca2+ channel blocker) promoted tumour angiogenesis, gemcitabine delivery and improved therapeutic efficacy in mouse models.52 Using the angiotensin inhibitor losartan it was shown that stromal collagen and hyaluronan content was reduced resulting in increased perfusion, drug and oxygen delivery.53 A more recent study highlighted the potential importance of ROCK inhibition using the oral inhibitor fasudil for dual targeting of tumour tension and vasculature.54 To this end, fasudil reduced intratumoural fibrillar collagen and improved sensitivity towards gemcitabine/nab-paclitaxel by M-phase cell cycle arrest. Furthermore, fasudil priming was accompanied by reduced metastasis formation on gemcitabine/abraxane treatment. This effect may be explained by increased numbers of microvessels in the liver and impaired extravasation suggesting that fasudil may operate as a potential anti-adhesion drug.54
However, experimental data comparing well-vascularised C26 colon tumours and poorly vascularised BxPC3 pancreatic tumours revealed that only small 30 nm micelles (amphiphilic molecules that form aggregates) accumulated intratumourally, whereas large 70 nm micelles showed severely impaired tumour distribution.55 Notably, most systemic chemotherapy molecules are <10 nm of size, for example, gemcitabine measures <1 nm, questioning the need for improving gemcitabine delivery at all. However, the therapeutic efficacy of increasing drug delivery may differ between men and mice considering the differences in stromal compositions between genetically engineered mouse models (GEMMs) and humans and the lack of direct comparative studies. Importantly, increased intratumoural drug delivery may not result in higher levels of anticancer compound that is metabolically available and active against tumour cells. Especially for gemcitabine, rapid systemic and intratumoural inactivation may play a more important role than biophysical delivery.
Drug scavenging has become another emerging theory that may explain the failure of chemotherapies in PDA. The phenomenon that other cells than tumour cells, or even microbes inactivate or scavenge antineoplastic drugs is indeed compelling. A recent study has shown that intratumoural bacteria may significantly add to gemcitabine resistance in PDA.56 In this study, the authors compared the growth kinetics between tumours established from mycoplasma-positive and mycoplasma-negative tumour cells following gemcitabine therapy. In vitro studies using high-performance liquid chromatography-tandem mass spectrometry showed decreased levels of native gemcitabine and significantly elevated levels of the deaminated inactive metabolite 2',2'-difluorodeoxyuridine in conditioned medium from the mycoplasma-infected dermal fibroblasts. The authors hypothesised that expression of bacterial cytidine deaminase (CDD) might be the underlying mechanism of gemcitabine inactivation. The group consequently tested the role of the different isoforms in conferring gemcitabine resistance, and indeed demonstrated the 880-nucleotide long isoform (CDDL) to be the most crucial isoform in this context. Further pharmacological and genetic in vitro experiments confirmed the role of CDDL in mediating gemcitabine resistance by drug scavenging. Using a colon carcinoma mouse model, Straussmann and colleagues provided first evidence of synergistic effects of gemcitabine and antibiotics.56 Notably, using various genetic methods as well as bacterial lipopolysaccharides, the authors demonstrated the presence of bacteria in 86 of 113 examined human PDA samples, whereas bacterial DNA was only present in 3 out of 20 normal pancreas controls.56 The relevance of these findings awaits clinical confirmation in trials involving antibiotics.
By analogy, our group recently discovered that murine CAFs sequester large amounts of activated gemcitabine (2',2'-difluorodeoxycytidine-5'-triphosphate (dFdCTP)) intracellularly, preventing its availability to kill neoplastic PDA cells.57 Mechanistically, key metabolic enzymes involved in gemcitabine inactivation such as Nt5c1A were expressed at low levels in CAFs in vitro and in vivo, and ectopic expression of Nt5c1A resulted in decreased intracellular dFdCTP concentrations in vitro.57 Although this drug-scavenging hypothesis needs further experimental and clinical proof, the most recent findings exemplify the complexity of drug delivery and drug availability in PDA.
Novel insight in CAF biology and heterogeneity
It is well established that CAFs are important in many steps during tumour development and progression.58 Traditionally, CAFs have been thought to represent a strong force driving tumour progression. This view is now gradually changing and an increasing body of evidence suggests that also CAFs, like immune cells, have variable, and sometimes opposing, functions in tumourigenesis.59
When discussing fibroblasts in cancer, it is important to keep in mind that ‘CAF’ is simply a collective term used to describe all cells within a tumour with morphological and functional features, similar to myofibroblasts that are active during wound healing processes. To date, there is no consensus on how to define CAFs on the molecular level. A plethora of markers has been suggested and used to identify CAFs, but there is no marker that is unique for CAFs or specific for all CAFs. When co-staining for the most frequently used CAF markers, such as α-SMA, vimentin, fibroblast-specific protein-1, platelet-derived growth factor receptor-β (PDGFR-β) and fibroblast activation protein-alpha (FAP-α), in different animal models of cancer and in human PDA tissue, an uneven distribution of these CAF markers is revealed.58 60 The variability in expression, and the different degree of overlap between the markers indicate that CAFs are in fact a very heterogeneous population of cells.
One obvious explanation to this heterogeneity is that there are multiple cellular sources for CAFs. In PDA, the major CAF source has been suggested to be activated PSCs, the cell type responsible for the production of most of the stromal collagens and a key modulator of the desmoplastic reaction.61 But recent studies also show that a subset of CAFs are derived from mesenchymal stem cells (MSCs) (figure 2). The MSCs associated with PDA show protumourigenic properties, partly because of their ability to induce tumour-promoting polarisation of macrophages,62 and by enhancing growth, invasion and the metastatic potential through secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF).63 In addition to PSCs and MSCs, there are other potential sources since CAFs have been shown to originate from activated resident fibroblasts, endothelial cells and fibrocytes in other tumour entities.59 64 In other terms, there are multiple cellular sources for CAFs, and this also makes it possible that CAFs originating from different progenitor cells have different functions and are involved in distinct tumour-related processes.
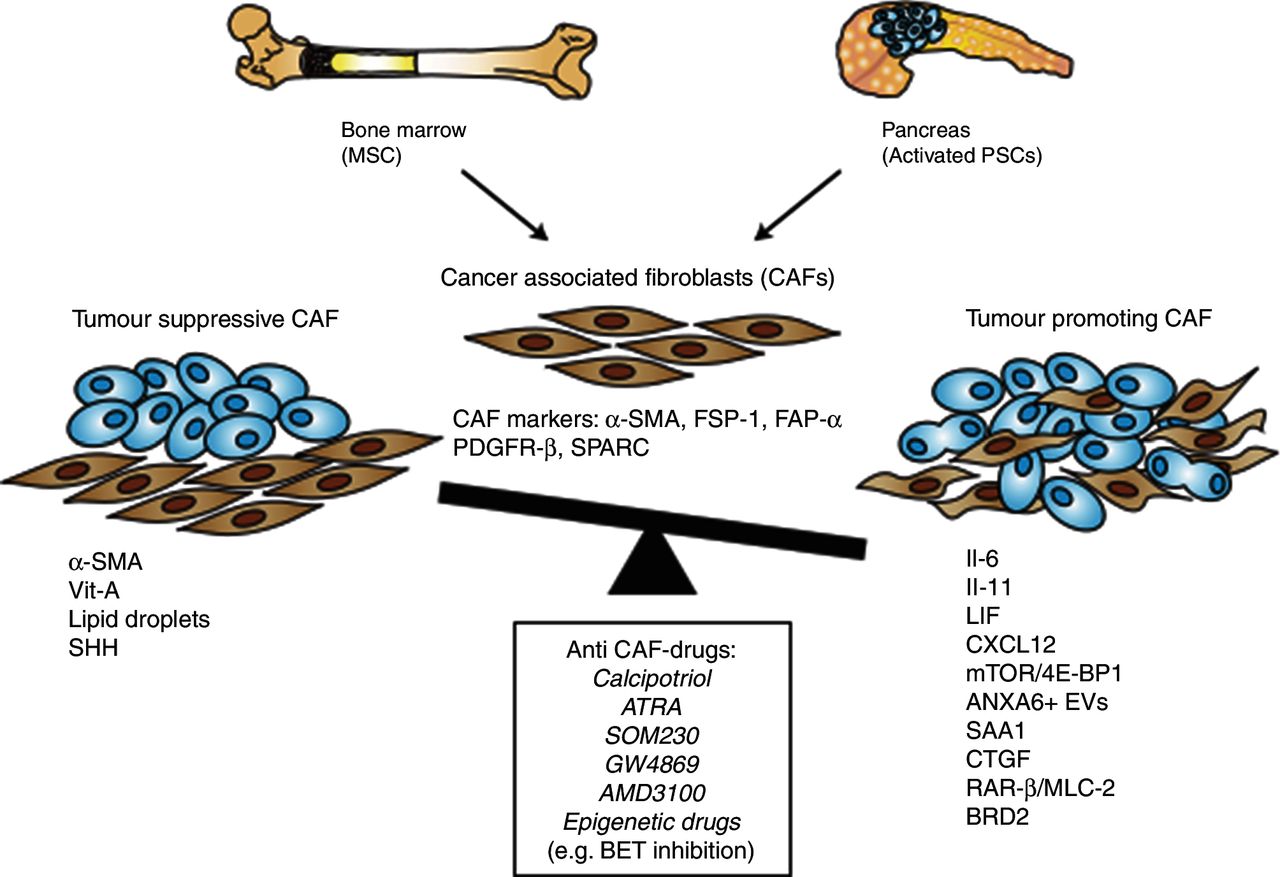
Evolution of cancer-associated fibroblasts (CAFs) from mesenchymal stem cells (MSCs) and pancreatic stellate cells (PSCs) with either tumour-promoting or tumour-restraining properties and potential anti-CAF drugs. FSP-1, fibroblast- specific protein-1; FAP-α, fibroblast activation protein-α; IL, interleukin; mTOR, mammalian target of rapamycin; PDGFR-β, platelet-derived growth factor receptor-β; RAR-β, retinoic acid receptor-β; SHH, Sonic Hh; SMA, smooth muscle actin; SPARC, secreted protein acidic and rich in cysteine.
An additional part of the explanation to the CAF heterogeneity is the fact that CAFs originating from the same cellular sources can transdifferentiate into different, functionally distinct subtypes depending on the juxtacrine and/or paracrine context these cells are found in. It is known that activated fibroblasts are highly plastic and can be driven by epigenetic alterations to transdifferentiate into many different cell types with different functions, such as chondrocytes, adipocytes, myocytes and endothelial cells.65 Traditionally, in PDA, CAFs have been considered to be a quite homogenous population derived mainly from PSCs, and that PSCs can be found in only two stages, either quiescent or activated into a ‘myofibroblastic’ state.66 67 Recent reports highlight that CAFs with myofibroblastic features, defined by high expression levels of α-SMA and referred to as myofibroblastic CAFs (myCAFs), are heterogeneously distributed in the PDA stroma, and located immediately adjacent to cancer glands. Another distinct subpopulation of CAFs is located more distantly from the cancer glands, lack elevated α-SMA expression and instead secrete interleukin-6 (IL-6) and additional inflammatory mediators, hence referred to as inflammatory CAFs (iCAFs).58 By using the pancreatic organoid system,68 and by co-culturing of cancer organoids with isolated PSCs, an organotypic model system to study stromal interactions was established. Interestingly, in this model cancer-naïve quiescent PSCs could transdifferentiate to either myCAFs or iCAFs depending on their spatial relation to the neoplastic cells.58 Furthermore, PSCs once induced to iCAFs could be reversed to myCAFs if the conditions were changed, implying a reversibility among these different subtypes. In addition to these subpopulations, another subpopulation of CAFs may be defined by the expression of CD10 and increased secretion of matrix metalloproteinase 3 in human PDA in vitro and in vivo.69
The complexity and function of CAFs is being investigated in several laboratories around the world, and it remains to be shown whether different CAF subtypes have different roles in tumourigenesis and response to treatment. We know that the expression levels of certain CAF markers and various ECM components correlate with worse prognosis.70 Also, the fibroblast activation grade is a predictive factor where patients with ‘immature’ fibroblasts are characterised by plump spindle-shaped morphology and correlate with a worse prognosis than patients with ‘mature’ fibroblasts with thin and wavy typical spindle-shaped morphology and symmetric/parallel orientation.71 Studies like these indicate that there are parts of the stroma that could potentially slow down tumour development, while others potentiate the progression.
These findings prompted the hypothesis that CAFs possess tumour-restraining properties. In fact, not all fibroblast features are tumour promoting,72 and it is well known that normal fibroblasts have the ability to inhibit the proliferation of different cancer cell lines,73 and that ECM from normal fibroblasts restrict proliferation of pancreatic cancer cell lines.71 It is therefore apparent that at least some components of the stroma can mediate tumour-restraining properties as part of an innate host defence to protect from tumour formation. In other words, some stromal elements are good and others are bad, and this might differ during different stages of tumourigenesis and progression.
The role of CAFs in the process of epithelial to mesenchymal transition (EMT) and metastasis formation has recently attracted increasing attention. Microscopically, liver metastasis from patients with PDAC resemble the primary tumour including the dense desmoplastic reaction. Several studies have emerged that investigate different cellular and acellular stromal components indicating that stromal components are similar between the primary tumour and liver metastasis with distinct differences occurring during metastasis evolution and depending on metastasis size.57 74 75 The partly conflicting data might be explained by differences in mice and humans, and whether paired primary tumour and metastasis samples were used. Furthermore, elegant studies using fluorescently labelled cells in genetically engineered mice suggest that the process of liver metastasis occurs very early before invasive PDAC develops and is driven by EMT.76 Microenvironmental cues from early recruited, granulin-secreting macrophages induce activation of resident hepatic stellate cells that promote a fibrotic microenvironment and metastatic tumour growth.77 These data support the notion of a systemic tumour disease and has triggered widespread discussion about a broader use of neoadjuvant treatment strategies in operable patients with PDAC.
Therapeutic targeting of CAFs in PDA
Blindly depleting the stroma would decrease tissue tension and in turn reduce tumour aggressivity,78 but would also risk to remove components of the ECM and subtypes of CAFs with tumour-inhibiting properties. Results from this approach could therefore give effects opposite to the intended. As discussed above, genetic and/or pharmacological approaches to eliminate certain components of the stroma by either depletion of α-SMA-positive proliferating cells or inhibition of the SHH pathway revealed a more aggressive disease and shorter survival in animal models of PDA.29 30 These findings suggest that general depletion of the heterogeneous stroma is not a feasible approach. Thus, approaches targeting key pathways in pro-tumourigenic CAFs appear to be a more promising strategy. Preclinical studies have now started to emerge showing that this is possible and that targeted therapeutic strategies directed towards specific components of the stroma or pathways of CAFs can be fruitful.79–83 Furthermore, tumour cell metabolism can also be driven by PSCs,84 and autophagy is required for the activation of PSCs, tumour progression and growth.83 Importantly, CAFs can be therapeutically targeted to enhance the response to standard chemotherapy. For instance, CAFs have been shown to crosstalk with tumour cells through extracellular vesicles (EV). In this context, gemcitabine has been identified to promote release of EVs from CAFs thus inducing expression of Snail in pancreatic cancer cells and promote proliferation and drug resistance.85 Furthermore, EVs carrying annexin A6/LDL receptor-related protein 1/thrombospondin 1 (ANXA6/LRP1/TSP1) complex were discovered to be crucial for the CAF—tumour cell crosstalk promoting PDA aggressiveness.86 Another important pathway for tumour-stromal interactions is the hepatocyte growth factor/tyrosine-protein kinase Met (HGF/c-MET) pathway. HGF is secreted by activated PSCs that in turn stimulate PDAC cells that express c-MET. Disruption of this crosstalk has been shown to enhance therapeutic efficacy in preclinical studies.87 88 In addition, FAP-α-positive CAFs were identified as major source of CXCL12, and blockade via AMD3100, a CXCL12 receptor chemokine (C-X-C motif) receptor 4 inhibitor, or genetic ablation of FAP-α-positive CAFs induced rapid T-cell accumulation and subsequent sensitisation to T-cell checkpoint antagonists.89 Recently, Guerra and colleagues identified murine Saa3, a member of the serum amyloid A (SAA) apolipoprotein family, as important pro-tumourigenic factor of PDGFR-α-positive CAFs.81 The human orthologue SAA1 was overexpressed in CAFs of patients with PDA and correlated with worse prognosis indicating a potential novel therapeutic target.81 Another interesting study from Bousquet and colleagues in Toulouse recently showed that selective pharmacological activation of somatostatin receptors in CAFs blocks the mTOR/4E-BP1 pathway and subsequent synthesis of several CAF-derived secreted proteins,82 including IL-6 that resulted in sensitisation to gemcitabine and inhibition of cancer cell metastasis.90
In addition, recent data suggest that CAFs induce widespread increases in histone acetylation at transcriptionally enhanced genes in tumour cells, suggesting the PDA epigenome as a potential therapeutic target of stromal cues.91 In particular, inhibition of the bromodomain and extraterminal family of epigenetic readers may be a promising therapeutic strategy in subgroups of patients with PDA.91 Figure 2 depicts the various sources and markers of CAFs, as well as potential anti-CAF drugs that have been used experimentally and are described above in detail.
In summary, the complexity in CAF biology is only beginning to emerge. The next years will provide more compelling studies to fully appreciate the heterogeneous nature of CAFs in PDA to fully exploit its potential for novel treatment strategies.
Biological implications of immune cells in PDA: lymphoid cells
Interplay between innate and adaptive immune cells within tumours induces and regulates cell-based immunity against malignancies. Tumour infiltration with cytotoxic CD8+ T cells is associated with better prognosis of patients with pancreatic cancer.92–94 Suppression of CD8+ T-cell infiltration into tumours results in increased tumour growth and represents an effective tumour escape strategy. Reduced infiltration of CD8+ T cells into the juxtatumoural stroma compartment as compared with the panstromal compartment has been described for the KPC model.95 Exclusion of CD8+ T cells from the tumour microenvironment was demonstrated to be mediated by extratumoural macrophages.96 By using recently developed multiplex staining protocols, proximity of intratumoural T cells to PDAC cancer cells was correlated with increased patient survival.97
Importantly, the number of infiltrating CD8+ T cells alone is not a sufficient predictor of a successful antitumour response. Intratumoural T cells transform into a dysfunctional state, resulting in loss of cytotoxic effector function, such as release of interferon-γ, and characterised by metabolic alterations.98 Intratumoural T-cell dysfunction is characterised by upregulation of inhibitory receptors, termed immune checkpoints, such as T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene-3 and programmed death receptor-1 (PD-1). Recently, it was described that the majority of the intratumoural T cells of patients with pancreatic cancer expressed the co-inhibitory checkpoint receptor PD-1.99 As a result, successful antitumour T-cell responses are compromised.
High mutational burden results in neoepitopes that can be recognised by intratumoural T cells. Immunoediting allows tumours to adapt to this immunological pressure and escape T-cell-mediated killing. It is debated to what extent neoantigen formation is relevant for the immune control of pancreatic cancer. In a K-Ras-driven mouse model of pancreatic cancer, the mutational load was low, resulting in minimal neoepitope burden and a lack of immunoediting phenomena.100 However, using genomic profiles of 221 PDAC cases extracted from public databases, Bailey et al found that nearly all PDAC samples expressed neoantigens. Tumour samples demonstrated a clear T-cell signature, indicating that T cells do in fact infiltrate pancreatic carcinoma tissue. However, active suppression mechanisms mitigate these T-cell responses, mediated by the presence of regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs), inhibitory cytokines like TGF-β and IL-10 or nitric oxide synthase (iNOS).101
Formation of tertiary lymphoid aggregates indicates a successful adaptive immune response against tumour antigens and is associated with improved prognosis,93 as evidenced by microarray analysis of microdissected aggregates.102 T-cell receptor (TCR) deep-sequencing confirmed that clonal T-cell expansion takes place in tertiary lymphoid structures that are found in tumour biopsies of patients with PDAC.103 These lines of evidence indicate that the common paradigm of immunologically ‘cold’ pancreatic carcinoma has to be modified. In fact, antitumoural immune reactions are found in the vast majority of patients with PDAC,103 and have been associated with improved prognosis.104
However, PDAC tumours exert mechanisms by which they escape immune detection. Metabolic alterations, such as upregulation of indoleamine 2,3-dioxygenase (IDO), an enzyme that starves T lymphocytes of tryptophan, result in an increased number of Treg cells.105 The tumour-promoting role of γδ T cells in pancreatic carcinoma has been highlighted by a recent report that deletion of intrapancreatic γδ T cells resulted in an influx of immunogenic Th1 cells and CD8+ T cells to the TME. Interestingly, pancreas-infiltrating γδ T cells expressed high levels of T-cell exhaustion ligands, such as programmed death-ligand 1 (PD-L1) and galectin-9.106 As a result of these adaptive immune suppression mechanisms, cytotoxicity is low despite presence of a pool of T cells that recognise tumour antigens, resulting in a reduced activation signature of T cells within the TME.101
Biological implications of immune cells in PDA: myeloid/monocytic cells
The contribution of myeloid cells for the activation versus suppression of the intratumoural lymphoid compartment has found intense interest in pancreatic cancer research recently.107
Tumour-infiltrating cells of the myeloid lineage, including TAM, tumour-associated neutrophils (TAN), monocytes and MDSCs, have been recognised as important mediators of immune evasion.108 These myeloid cells are recruited from the bone marrow to the site of the tumour by distinct chemokine pathways co-opted by the tumour cells to facilitate myeloid cell attraction.
TAMs may acquire different functional states during tumour initiation, progression and therapeutic intervention. This functional plasticity has been categorised in two different opposing polarisation states, called M1 and M2. In general, M1 macrophages are known to secrete pro-inflammatory cytokines with predominantly antineoplastic effects while M2 macrophages produce anti-inflammatory signals, which may facilitate tumour progression.109 110 However, it is increasingly being recognised that this simplified classification does not fully represent the complexity of TAM action.
Several studies have demonstrated an inverse correlation between patient prognosis and TAM infiltration in various tumour entities including pancreatic cancer.111 By releasing miscellaneous cytokines, proteases and growth factors such as vascular endothelial growth factor, TAMs mediate angiogenesis and tumour progression.112 In addition, TAMs are able to impair the efficacy of chemotherapy. In pancreatic cancer, it has been shown that TAMs influence the activity of Cda, a key metaboliser of gemcitabine, thereby driving resistance to gemcitabine-based chemotherapy.113 Moreover, TAMs impair the efficacy of therapeutic irradiation and targeted anti-angiogenic treatment by promoting angiogenic escape mechanisms.114
Based on these profound tumour-promoting effects of TAMs, various approaches are currently being tested to target TAMs in pancreatic cancer. These include antibodies and small molecules against colony-stimulating-factor-1 receptor (CSF1R),115–117 small molecule inhibitors against the chemokine CCR2,118 the macrophage-targeting chemotherapeutic agent trabectedin119 and bisphosphonates such as zoledronate that have been shown to induce apoptosis of phagocytic myeloid cells.112 These approaches have shown promising effects in preclinical models and are currently tested in a variety of cancers including pancreatic cancer. Combination therapies of substances, which did not show sufficient efficacy as single agents, are explored for synergistic efficacy. Combining a CSF1R inhibitor and a CXCR2 antagonist results in improved efficacy, however, when a checkpoint inhibitor is added, dramatic therapy effects have been reported.117 Mace et al reported that PD-L1 immunotherapy can be boosted in a CD8+ T-cell-dependent manner by blockade of IL-6, which is produced by pancreatic stellate cells.120
In addition to TAM, the role of TAN has recently attracted increasing interest after numerous reports have identified elevated numbers of neutrophils in the circulation and in the tumour tissue (defined as TAN) as poor prognostic markers.121 Nywening et al recently showed that simultaneous targeting of both CXCR2+ neutrophils (TAN) and CCR2+ macrophages (TAM) by small molecule inhibitors augmented antitumour immunity and improved response to chemotherapy in a preclinical model, suggesting that dual targeting of CCR2+ TAM and CXCR2+ TAN is superior to either strategy alone.118
MDSCs, which represent a mixture of immature stages of macrophages, granulocytes and dendritic cells (DCs), are strongly increased in numbers both in circulation as well as the microenvironment of human PDA, and MDSC levels correlate with clinical cancer stage.122 123 Results from GEMMs suggest that GM-CSF produced by tumour cells from early cancer stages is a major factor stimulating MDSC differentiation.124 125 In the tumour, MDSCs contribute to suppressing CD4+ and CD8+ T-cell function, and stimulate expansion of immunosuppressive Tregs. Consequently, targeted depletion of MDSCs in GEMMs by different experimental strategies was demonstrated to activate an effective endogenous antitumour T-cell response in established tumours, and to significantly impair KrasG12D-driven pancreatic cancer initiation when induced at early stages.107 126 Zhang et al described myeloid cells as mandatory for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Authors used CD11b-diphteria toxin receptor (DTR) mice to deplete myeloid cells, finding that CD11b+ myeloid cells regulate CD8+ T-cell exhaustion by upregulation of PD-L1 on tumour cells. This effect was mediated by activation of epidermal growth factor receptor (EGFR) signalling in tumour cells. It was concluded that myeloid cells are mandatory for sustained mitogen-activated protein kinase (MAPK) signalling in pancreatic epithelial cells. Accordingly, treatment with MAPK inhibitors lowered intratumoural expression of PD-L1 and rendered pancreatic cancer susceptible to PD-1 blockade. Within this paradigm, CD11b+ cells employ EGFR/MAPK-dependent regulation of PD-L1 expression on tumour cells to induce a state of dysfunction/exhaustion in CD8+ T cells. Moreover, MDSCs have the potential to subvert innate antitumour immunity through a variety of different avenues. Among others, they have been shown to inhibit natural killer cell cytotoxicity through cell contact-dependent mechanisms and promote the conversion of macrophages towards an M2 phenotype in model systems of lung and breast cancer,127–129 thus underscoring their role as central inhibitors of antitumour immunity and their attractiveness as targets for novel therapeutic concepts.
Therapeutic approaches targeting the immune system in PDA
Intratumoural T-cell exhaustion is not terminal. The molecular signature of T-cell dysfunction can be reversed by blocking receptors that are overexpressed during exhaustion, such as PD-1 and cytotoxic T lymphocyte antigen 4 (CTLA-4).130 131 Suppressive mechanisms of T-cellular transmembrane coinhibitory receptors are deeply linked to antigen-dependent TCR signalling.132 PD-1 is the most prominent molecule associated with T-cell exhaustion. Mechanistically, binding of PD-L1 or PD-L2 results in ectodomain competition leaving activating ligands without their respective costimulatory receptors, modification of intracellular signalling pathways and induction of inhibitory genes.133 PD-1 activation reduces T-cell motility,134antagonises TCR signalling through the phosphoinositid-3 kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) and Ras pathways,135 and suppresses effector gene transcription.136 For PDA, expression levels of PD-1 and its ligand PD-L1 inversely correlate with survival.137 TIM-3 is another interesting candidate for therapeutic checkpoint inhibition, with TIM-3+PD-1+ double positive CD8+ T cells representing the most severely exhausted phenotype of intratumoural T cells. CTLA-4, in turn, represses the activity of the T-cell costimulatory receptor CD28. T-cellular surface expression of CTLA-4 suppresses T-cell activation by outcompeting CD28 in binding B7 molecules. CTLA-4 polymorphism has been linked to increased susceptibility to pancreatic cancer.138
However, most clinical trials on checkpoint inhibition in patients with pancreatic carcinoma had disappointing outcomes.139 A study on CTLA-4 antibody ipilimumab in 27 patients with metastatic or locally advanced pancreatic carcinoma found no statistically significant beneficial efficacy.140 Most clinical trials on combination of anti-PD-1 monoclonal antibodies (mAbs) nivolumab and pembrolizumab, with chemotherapies such as gemcitabine, nab-paclitaxel or irinotecan have not yet been published.139 Results on anti-PD-L1 treatment in pancreatic cancer have been reported141; however, no conclusive beneficial evidence has been reported yet for the patient populations investigated in those trials (current trials are listed in online supplementary table 2).
Supplementary file 2
A major advance in the field was the identification of subgroups of tumour entities that respond better to immune-based therapies than others. Referred to as personalised therapy, it has become clear that molecular signatures are more important than histological subtypes to define chances of treatment response, particularly when it comes to immune-based therapies. In general, somatic mutations encode non-self, immunogenic neoantigens. It has been hypothesised that tumours with a large number of somatic mutations are more susceptible to immune-mediated therapies, for example, checkpoint blockade.142 Mismatch-repair (MMR) defects are well-known for increasing the tumour mutational load, resulting in a higher number of neoantigens expressed by those tumours.
The hypothesis that MMR-deficient tumours stimulate the immune system has been suggested previously,143 and dense infiltration with immune cells and high levels of Th1 cytokines were reported in MMR-deficient tumours decades ago.144 145 With broad application of novel methods such as whole-exome next-generation sequencing and bioinformatics analysis, these findings could be put into a wider context. It was demonstrated that formation of neoantigens is the causal explanation for the association between somatic hypermutation and increased immune phenomena in MMR-deficient tumours.146
In 2015, a seminal paper by Le et al reported in a phase II study that MMR status predicts clinical benefit of immune checkpoint blockade with the anti-PD-L1 drug pembrolizumab. Pembrolizumab had beneficial efficacy on response rate, progression-free survival and overall survival for a diverse group of MMR-deficient, but not MMR-stable malignant neoplasms.142 Whole-exome sequencing described—in mean—1782 somatic mutations per tumour in MMR-deficient samples, compared with 73 in MMR-proficient tumours, a highly significant difference. High somatic mutational load was associated with prolonged progression-free survival. Non-colorectal carcinomas were included into the study, but no cases of pancreatic carcinoma were reported back then. However, very recently, the authors expanded their analysis to a panel of 12 different tumour types, including pancreatic carcinoma (ClinicalTrials.gov number, NCT01876511).147 Six patients with pancreatic carcinoma were evaluable, and all six of those responded to the treatment. In May 2017, the US Food and Drug Administration (FDA) granted accelerated approval to anti-PD-L1 drug pembrolizumab as a second-line agent for patients with unresectable or metastatic microsatellite instability-high and MMR-deficient solid tumours regardless of histotype. Figure 3 depicts the complex interaction between immune cells and pancreatic cancer cells with potential experimental and clinical compounds that block mechanisms of immune evasion. Since it is estimated that only 1%–2% of all pancreatic carcinoma are MMR deficient,148 the number of eligible patients with pancreatic cancer will be small. In contrast, MMR deficiency was reported to be more frequent in acinar cell carcinoma,149 and in intraductal papillary mucinous neoplasms (IPMN)-related tumours as compared with non-IPMN-related tumours.150

{kind=link}
{kind=link}
{kind=link}
Immune cell-cancer cell crosstalk mediating immune evasion and potential preclinical and clinical drugs that re-activate the tumour immune system. APC, antigen-presenting cell; LAG, lymphocyte-activation gene-3; MHC, major histocompatibility complex; PaCa, pancreatic cancer cells; PD-1, programmed death receptor-1; PD-L1, programmed death-ligand 1; TCR, T-cell receptor; Tregs, T-regulatory cells; TIM-3, T-cell immunoglobulin and mucin-domain containing-3.
New biomarkers might expand the number of patients eligible for checkpoint inhibition therapy. Indeed, a recent study by Balachandran et al found that long-term pancreatic cancer survivors following pancreatic resection were more prone to harbour neoantigens with similarity to known microbial peptides.104 This group established a neoantigen quality fitness model, which could guide the development of new vaccine and treatment approaches for the majority of patients with pancreatic cancer.
Unfortunately, the majority of patients with PDA will not benefit from checkpoint inhibitor therapy unless combined with another form of therapy that is able to boost antitumour efficacy. Pancreatic cancer, at least the MMR-sufficient forms, have a low mutational load,151 as compared with lung cancer and malignant melanoma, in which cigarette smoke and ultraviolet light result in high numbers of somatic mutations.152 153 In this context, it has recently been shown in a PDAC mouse-model that combined treatment with agonistic anti-CD40 mAb is able to overcome resistance to PD-1 and CTLA-4 blockade.154
Besides anti-exhaustive checkpoint inhibition, the armoury of immunotherapy contains a multitude of immunostimulatory agents and methods, which partly aim at inducing de novo immune responses against tumour cells. Current reviews inform on combinatorial therapies of checkpoint inhibitor and T-cell-stimulatory therapies in PDA, such as adoptive cell transfer (ACT), vaccination or metabolic alteration by small molecule IDO inhibitor indoximod.155 156
ACT therapy expands and/or modifies autologous or allogeneic immune cells ex vivo before reinfusion,157 avoiding inhibitory signals from suppressive bystander cells as found in vivo. Recently, it was shown in an elegant study that T cells engineered to express affinity-enhanced TCR can overcome intratumoural immune suppression and induce pancreatic cancer cell death.158 A similar approach is the adoptive transfer of T cells genetically modified to express a chimeric antigen receptor (CAR). The extracellular CAR domain is a Fab fragment of an antibody that recognises a tumour antigen, whereas the intracellular domain contains the TCR signal transduction sequence. As antibodies bind to extracellular molecular structures, this strategy abrogates the requirement for MHC restriction. With promising preclinical data,159–161 efficacy of autologous T cells transiently expressing a mesothelin-specific CAR is currently tested in clinical trials in patients with pancreatic cancer. However, drawbacks include neurotoxicity and a cytokine release syndrome, high cost of therapy as well as poor efficacy in solid tumours in comparison to haematological malignancies. These difficulties could be overcome by careful use of gene editing tools such as Zinc finger nucleases, transcription activator-like effector nucleases and CRISPR-Cas9 systems. Current reviews inform on techniques to manufacture next-generation CAR-T cells.162
Tumour vaccines are based on the idea to boost a patient’s antitumour T-cell responses, for example, by adoptive transfer of DCs, pulsed with peptides, proteins, whole tumour cells, DNA or RNA. Clinical trials on DC vaccination in PDA demonstrated feasibility and promising therapeutic results.163 164 An alternative strategy is in vivo activation of DCs with vaccine adjuvants, such as ISOMATRIX, which showed efficacy in a murine pancreatic cancer model.165 166 Vaccination with allogeneic pancreatic cancer cell lines is a feasible strategy to circumvent the limitation of insufficient tumour material derived from patients with cancer. GVAX, a pancreatic cancer cell line transfected with GM-CSF, recruits immune cells to the tumour site, inducing an antitumour immune response.167 The same group that recently demonstrated efficacy of checkpoint inhibition in MMR-deficient tumours,147 reported earlier on efficacy of checkpoint inhibitor ipilimumab combined with the GVAX vaccine in a small clinical study in patients with pancreatic cancer.168 Similarly, oncolytic virotherapy has been combined with checkpoint inhibitor pembrolizumab in a phase II study on metastatic pancreatic carcinoma, demonstrating feasibility and antitumour activity, as evidenced by immune infiltration and reovirus infection in tumour biopsies.169
Conclusions
In our recent review on stromal biology and therapy in PDA in this journal, we had concluded that although targeting of various stromal components and pathways was considered a promising therapeutic strategy, none of the efforts had yet led to efficacious and approved therapies in patients.16 In addition, at that time recent data had changed the paradigm that tumour stroma is solely tumour promoting, and had highlighted the need to critically revisit the complexity and complicity of the tumour-stroma with translational implications for future therapy and clinical trial design. A tremendous amount of outstanding data have been generated in the past 3 years that has advanced the field and has been summarised in this update. Most prominently, the demonstration of the efficacy of checkpoint-inhibitor therapy in a small subgroup of patients with PDA with MMR deficiency by Le et al,147 has finally introduced the first FDA-approved treatment targeting a cellular stromal component. Furthermore, recent clinical trials of combination therapy of cytotoxic chemotherapy and targeting of the stromal component HA has shown efficacy in patients with PDA expressing high levels of this ECM protein. These studies indicate that indeed stromal targeting may now be on the verge of becoming a firm component of the therapeutic armamentarium for patients with PDA. However, both examples highlight that due to the complexity of tumour-stroma interactions in PDA, stromal targeting needs to be individualised and no single approach or compound will work for all patients with PDA. Thus, it can be expected that future therapeutic subtyping of patients with PDA will be restricted to genetic and epigenetic subgroups targeting intertumoural heterogeneity, and among others will have to account for stromal heterogeneity and mechanisms interfering with drug metabolism such as drug scavenging by stromal or bacterial cells in the tumour.
Key messages
Key points 1: basic science
A pronounced desmoplastic microenvironment is a histological hallmark feature in pancreatic cancer consisting of cancer- associated fibroblasts (CAFs), immune cells, soluble growth factors, matricellular proteins and extracellular matrix components such as collagen and hyaluronan (HA).
CAFs are a highly heterogenous cell population, and recent evidence has revealed several protumourigenic pathways such as serum amyloid A 1, mammalian target of rapamycin/4E-BP1, CXCL12 and extracellular vesicles as potential therapeutic targets.
Pancreatic tumours are characterised by T-cell exhaustion resulting in loss of cytotoxic effector function, and infiltration of tumour-promoting tumou r-associated macrophages, tumour-associated neutrophils and myeloid-derived suppressor cells mediating immune evasion and tumour progression.
Key points 2: clinical science
- Epithelial and stromal subtyping will pave the way for precision oncology concepts in pancreatic cancer and guide individualised antistromal therapies.
- A small group of patients with mismatch-repair-deficient tumours may respond to checkpoint inhibitors due to their pronounced immunogenic environment.
Enzymatic HA depletion in combination with gemcitabine/nab-paclitaxel is promising and currently tested in a HA-high subgroup of patients with PDA in a phase III trial.
This treatment may become the first stroma-targeted therapy in an HA-high subgroup of patients with PDA.
Future research directions and challenges
Identification of predictive stromal signatures and subgroups that will allow tailored therapy approaches.
Detailed characterisation of pro-tumorigenic and tumour-suppressive properties of CAFs and acellular stromal components.
Clarification whether tumour stroma acts as biophysical barrier for drug delivery.
Clinical trials that test combinatorial strategies of induction phase chemotherapy and consolidation phase immunotherapy.
Translational clinical trials that compare preclinical approaches in patient-derived xenografts and organoids with patients’ response to personalise therapies.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
Footnotes
AN and CAB are shared first authors.
Contributors AN, CAB and TMG conceptualised the review, designed figures and tables and wrote several paragraphs of the manuscript, DÖ, ML, PM, MB, DAT wrote paragraphs of the manuscript, all authors critically revised the manuscript.
Funding This work was supported by the State of Hesse (LOEWE-Schwerpunkt ’Medical RNomics') and EU FP7 grant no. 602783 (large-scale integrated project ’CAM-PaC') and by funds from the DFG (KFO325). This paper reflects only the authors' views and the European Union is not liable for any use that may be made of the information contained therein. DAT is a distinguished scholar of the Lustgarten Foundation, Director of the Lustgarten Foundation-designated Laboratory of Pancreatic Cancer Research, and supported by the Cold Spring Harbor Laboratory Association, the V Foundation and the National Institutes of Health (NIH 5P30CA45508-29, 5P50CA101955-07, P20CA192996-03, 1U10CA180944-04, 1 U01CA224013-01, U01CA210240-01A1, 1R01CA188134-01, 1R01CA190092-04 and 5U10CA180944-05). DÖ received funding from the Swedish Research Council (2017-01531), and the Swedish Society of Medicine (SLS-691681 and SLS-786661), federal funds through the county council of Västerbotten (VLL-643451), the Cancer Research Foundation in Northern Sweden (AMP 17-877), The Swedish Foundation for International Cooperation in Research and Higher Education (PT2015-6432), the Knut and Alice Wallenberg Foundation and the Swedish Cancer Society (CAN 2017/332 and CAN 2017/827). PM received funding by the DFG (MI710/8-1; Mi710/10-1). AN received funding from the Deutsche Krebshilfe, Max Eder Research Group (110972 and 70113213).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.