Article Text

Abstract
Objective Sorafenib is the standard systemic therapy for advanced hepatocellular carcinoma (HCC). Survival benefits of resection/local ablation for early HCC are compromised by 70% 5-year recurrence rates. The phase 3 STORM trial comparing sorafenib with placebo as adjuvant treatment did not achieve its primary endpoint of improving recurrence-free survival (RFS). The biomarker companion study BIOSTORM aims to define (A) predictors of recurrence prevention with sorafenib and (B) prognostic factors with B level of evidence.
Design Tumour tissue from 188 patients randomised to receive sorafenib (83) or placebo (105) in the STORM trial was collected. Analyses included gene expression profiling, targeted exome sequencing (19 known oncodrivers), immunohistochemistry (pERK, pVEGFR2, Ki67), fluorescence in situ hybridisation (VEGFA) and immunome. A gene signature capturing improved RFS in sorafenib-treated patients was generated. All 70 RFS events were recurrences, thus time to recurrence equalled RFS. Predictive and prognostic value was assessed using Cox regression models and interaction test.
Results BIOSTORM recapitulates clinicopathological characteristics of STORM. None of the biomarkers tested (related to angiogenesis and proliferation) or previously proposed gene signatures, or mutations predicted sorafenib benefit or recurrence. A newly generated 146-gene signature identifying 30% of patients captured benefit to sorafenib in terms of RFS (p of interaction=0.04). These sorafenib RFS responders were significantly enriched in CD4+ T, B and cytolytic natural killer cells, and lacked activated adaptive immune components. Hepatocytic pERK (HR=2.41; p=0.012) and microvascular invasion (HR=2.09; p=0.017) were independent prognostic factors.
Conclusion In BIOSTORM, only hepatocytic pERK and microvascular invasion predicted poor RFS. No mutation, gene amplification or previously proposed gene signatures predicted sorafenib benefit. A newly generated multigene signature associated with improved RFS on sorafenib warrants further validation.
Trial registration number NCT00692770.
- hepatocellular carcinoma
- molecular oncology
- clinical trials
- tumour markers
- cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
There is no standard adjuvant treatment after resection/ablation for hepatocellular carcinoma and thus this is an unmet medical need. The phase 3 STORM study did not show benefits for sorafenib compared with placebo in terms of recurrence-free survival (RFS). In some solid tumours, however, cancer therapies based on predictive biomarkers assessed post hoc have entered clinical practice (eg, KRAS mutations status for cetuximab in colorectal cancer). We are herein reporting a level B evidence of a companion study of the phase 3 STORM trial exploring prognostic factors and predictors of response to sorafenib.
What are the new findings?
In terms of prediction of response to sorafenib, none of the previously reported gene signatures or biomarkers (amplification VEGFA, pERK) were predictive to sorafenib efficacy. We have generated a new gene signature that is independent of whatever clinical-pathological variable and is able to discriminate patients who respond to sorafenib in terms of RFS. In terms of prognosis, we identified pERK and microvascular invasion as prognostic biomarkers of poor RFS. However, none of the tested gene-signatures or mutations were predictive of recurrence.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The study confirms the lack of known biomarkers as predictors of response to sorafenib, and proposes a gene signature at level B evidence (Simon et al, 2009) that once validated might help at selecting patients able to benefit from sorafenib in the setting of adjuvancy.
Background
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy, with nearly 850 000 new cases/year worldwide.1 At early stages of the disease, surgical resection, liver transplantation and local ablation are considered the main treatments.2 However, up to 70% of patients treated with resection or local ablation present tumour recurrence at 5 years.1 Therefore, prevention of HCC recurrence—through the clearance of micrometastasis or de novo tumours by adjuvant treatments—is a major unmet medical need. In the past decade, a significant improvement in the management of advanced HCC has occurred. Sorafenib was first approved a decade ago3, and more recently other therapies have shown clinical benefits and/or US Food and Drug Administration approval as first (lenvatinib)4 or second-line treatments (regorafenib5, cabozantinib6, nivolumab7 and ramucirumab8). Sorafenib is an oral multikinase inhibitor targeting the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway, vascular endothelial growth factor receptors (VEGFR) and KIT, among other targets,9 that provides nearly 3-month median survival benefit and a 31% reduction of risk of death in patients with advanced HCC.3 Regorafenib displays more potent pharmacological activity than sorafenib and provided a 38% reduction of risk of death in patients progressing on sorafenib treatment.5 10
Several randomised studies testing adjuvant therapies such as interferon (IFN), chemotherapy, chemoembolisation, internal radiation, immune therapies and retinoids have failed to provide outcome benefits or led to inconclusive results and therefore are not recommended in clinical practice.2 11 In this context, the STORM trial was designed to evaluate the efficacy and safety of sorafenib as an adjuvant therapy in patients with HCC with a complete radiological response following surgical resection or local ablation.12 This phase 3, randomised, double-blind, placebo controlled trial included 1114 patients. The primary endpoint of recurrence-free survival (RFS) was not met (33.3 months in the sorafenib group vs 33.7 months in the placebo group; HR 0.94; 95% CI 0.78 to 1.13, p=0.26)12 and therefore further research is required to identify strategies to prevent HCC recurrence.
The mechanism of action of sorafenib includes inhibition of both MAPK/ERK-mediated cell proliferation and angiogenesis driven by VEGF signalling.9 Moreover, it is known that sorafenib acts both on tumour cells as well as on endothelial cells.9 Despite multiple studies, robust predictive biomarkers of response to sorafenib (including sorafenib targets such as MAPK/ERK or VEGF) in patients with HCC have not yet been identified. While sorafenib provides benefits across all subgroups of patients, a recent meta-analysis of individual data from the phase 3 SHARP and Asia-Pacific HCC trials showed that patients with HCV aetiology and liver only disease have a significantly greater benefit with sorafenib.13 14 In terms of biomarkers, correlative analyses in the SHARP trial showed a non-significant trend towards higher survival benefit from sorafenib treatment in tumours with high c-Kit or low hepatocyte growth factor (HGF) plasma concentration.15 Nonetheless, no biomarker has been identified to clearly predict sorafenib efficacy. In this scenario, a thorough analysis of biomarker-defined subpopulations that might benefit from sorafenib in the adjuvant setting is required.
Herein we used a cohort derived from the phase 3 STORM trial population to conduct a companion biomarker analysis aimed at identifying biomarkers predicting sorafenib efficacy in preventing recurrence of HCC (hereafter, predictive biomarkers) and factors independently associated with prognosis (hereafter, prognostic biomarkers). Because our biomarker study was conducted on specimens collected in a randomised controlled trial (RCT) and were assayed after trial completion, our results hold a level B of evidence.16 This level of evidence goes beyond observational or retrospective biomarker studies (levels C and D) and is the closest to an RCT specifically designed to address tumour marker value as a predictor of outcome (level A).16
Materials and methods
Tumour samples
Formalin-fixed paraffin-embedded (FFPE) tissue blocks from patients with HCC resected between 2008 and 2010 and randomised into treatment in the STORM clinical trial (NCT00692770)12 were collected from the highest enrolling centres after ad hoc institutional review board clearance (online supplementary table 1). Out of the 202 collected samples, 188 were suitable for the study (hereinafter, BIOSTORM cohort) (figure 1). See also online supplementary materials and methods.
Supplemental material
Supplemental material

Flow chart of the BIOSTORM study. FISH, fluorescence in situ hybridisation; IHC, immunohistochemistry; VEGFA, vascular endothelial growth factor A.
Immunohistochemistry, fluorescence in situ hybridisation, whole transcriptome analysis and targeted exome sequencing
See online supplementary materials and methods.
Endpoints, clinical data and statistical analysis
RFS was defined as the time from randomisation to the first documented disease recurrence by independent radiological assessment or death by any cause, whichever happened first. The primary endpoint was to identify biomarkers predicting sorafenib efficacy in preventing HCC recurrence in terms of RFS. The secondary endpoint was to define prognostic biomarkers of RFS and/or validate those previously reported. In the BIOSTORM cohort, all events were recurrences (70 RFS events in 188 patients), and thus time to progression was equivalent to RFS. Biomarkers were considered predictive of sorafenib efficacy in preventing HCC recurrence when p of interaction was <0.05. Patients who had undergone less than one treatment cycle (4 weeks) were excluded from the predictive biomarker analysis (sorafenib: n=9 out of 83; placebo: n=6 out of 105). All statistical tests were two sided. Data were analysed with SPSS V.23.0 (SPSS) and the R statistical package. See also online supplementary materials and methods.
Results
Baseline and clinical characteristics of the BIOSTORM cohort
The BIOSTORM cohort consists of a subgroup of 188 patients representing 21% of the 900 STORM patients who had been treated with surgical resection. From those, 83 had been treated with sorafenib and 105 with placebo. All baseline patient demographics and disease characteristics were balanced in both BIOSTORM treatment groups (table 1). In addition, most BIOSTORM clinical-pathologic characteristics were similar to the STORM cohort, which only differed in age (p<0.001), prevalence of hepatitis C (p<0.001) and microvascular invasion (p=0.003).
Baseline patient demographics and disease characteristics by treatment group within BIOSTORM and compared with the STORM population undergoing resection
In BIOSTORM, as in STORM, median duration of treatment was shorter in sorafenib-treated patients compared with placebo (17 vs 30.6 months; p=0.007). Follow-up was shorter in sorafenib patients compared with placebo (30.9 vs 41.8 months; p<0.001), partially due to a higher rate of treatment discontinuation. In BIOSTORM, median RFS in placebo arm was 42.0 months and was not reached with sorafenib. Moreover, no significant benefit of sorafenib on RFS was observed (HR=0.82; 95% CI 0.50 to 1.35; p=0.430) (online supplementary figure 1). This was consistent with the STORM subgroup analysis of patients undergoing resection (HR=0.94; 95% CI 0.76 to 1.16).
Molecular factors predicting benefit to sorafenib
None of the baseline clinical variables (summarised in table 1) such as microvascular invasion (p=0.164) or presence of satellites (p=0.627) predicted response to sorafenib by p of interaction test. Thus, we explored molecular variables from MAPK pathway, VEGF pathway and previously reported molecular signatures and drivers to assess their predictive capacity.
MAPK pathway
Given that sorafenib is a multikinase inhibitor that targets predominantly pathways involved in proliferation and neoangiogenesis, we sought to explore the predictive capability of different members of the MAPK pathway and VEGF signalling.
It is well known that the RAS/RAF/MAPK/ERK cascade plays a role in the regulation of cell proliferation, survival and differentiation,17 and phosphorylated extracellular signal-regulated kinase (pERK) is a commonly used surrogate of MAPK pathway activation. In BIOSTORM, nuclear and cytoplasmic pERK staining was observed in tumour hepatocytes in 11% of the patients (n=20/175), whereas it was identified in 86% of patients in tumour-associated endothelial cells (n=150/175) (figure 2A,B). These results were consistent with our previous data.18 Tumours with positive pERK hepatocyte staining significantly correlated with the proliferative marker Ki67 (p=0.048) (online supplementary figure 2). In terms of clinical-pathologic correlations, tumours displaying pERK hepatocyte staining were significantly associated with poorly differentiated tumours (p=0.002). In parallel, tumours with pERK-positive endothelial cells were smaller compared with pERK-negative cases (mean tumour size 41 vs 60 mm; p=0.033). Samples with negative pERK endothelial staining were positive for CD31 (online supplementary figure 2), suggesting that this negative staining was not due to absence of vascularisation.

Representative examples of (A) immunohistochemical pERK staining of tumour hepatocytes, (B) pERK staining of tumour endothelial cells, and (C, D) pVEGFR2 positive and negative staining of tumour hepatocytes. (E, F) Representative images of fluorescence in situ hybridisation (FISH) staining to determine vascular endothelial growth factor A (VEGFA) copy number. VEGFA was labelled in red; the control reference Cep6 in green; and DAPI-stained cell nuclei in blue.
To evaluate if pERK predicted RFS according to treatment allocation, we analysed the interaction between pERK and sorafenib effect. Sorafenib-treated patients with negative hepatocyte pERK staining (n=64/75, 85.3%) presented increased RFS compared with patients in the placebo group with negative hepatocyte pERK staining (median RFS not reached vs 42.1 months; HR=0.49; p=0.043), a difference that was not significantly better when compared with RFS of those patients with positive hepatocyte pERK staining (p of interaction p=0.11; online supplementary figure 3A). Neither endothelial pERK (p of interaction p=0.10; online supplementary figure 3B) nor Ki67 (p of interaction p=0.19) was predictive of sorafenib efficacy in terms of RFS. Also, the predictive value of additional previously reported candidate biomarkers,15 such as expression levels of HGF, c-Kit, vascular endothelial growth factor A (VEGFA) and basic fibroblast growth factor/fibroblast growth factor 2 (bFGF/FGF2) did not retain significant predictive value.
VEGF signalling
We had previously identified focal chromosomal amplifications in the locus of VEGFA—the gene encoding for the VEGFR2 activating ligand—in approximately 7% of patients with HCC, as the second most prevalent focal amplification in HCC.1 19 Retrospective studies have suggested that VEGFA gene copy number predicts response to sorafenib-treated patients in the adjuvant setting.20 Thus, we analysed whether pVEGFR2—a surrogate of the proliferative VEGF pathway—or VEGFA levels predicted RFS in sorafenib-treated patients.
Immunohistochemical analysis of pVEGFR2 revealed two different patterns of staining: nuclear pVEGFR2 staining, detected in tumour hepatocytes in 36% of the patients (n=54/152) (figure 2C,D), and cell membrane pVEGFR2 staining, detected only in 4% of the patients (n=6/152). Nuclear pVEGFR2 positively regulates the expression of the VEGFR2 gene itself by interacting with the promoter region of the gene, and promotes endothelial cell growth and proliferation.21 When tested for interaction with the treatment, absence of nuclear pVEGFR2 staining presented a non-significant trend for better RFS with sorafenib (p of interaction p=0.083) (online supplementary figure 3C). Membrane pVEGFR2 was not tested because of the limited number of samples displaying this staining.
On the other hand, analysis of VEGFA copy number demonstrated that high-level gains of this gene (≥4 copies) were present in 10% of the patients (n=17/162) (figure 2E,F), consistent with our previous observations.19 Nonetheless, high-level gains of VEGFA did not correlate with recurrence prevention in sorafenib-treated patients (p of interaction p=0.938) (online supplementary figure 3D). Tumours with satellites (p<0.001) and microvascular invasion (p=0.019) were significantly enriched with VEGFA focal amplifications.
Reported molecular signatures and driver genes
To further investigate candidate predictive biomarkers, we analysed whole-genome expression. We obtained transcriptomic data for 140 HCC samples and 90 non-tumour adjacent tissues. The transcriptomic profile of each sample was used to classify each tumour according to its molecular class, and to assess enrichment of signalling pathways, progenitor features and clinical characteristics. Around 40% of the patients belong to the proliferation subclass19 (n=53/140) (online supplementary table 2), as observed in other HCC cohorts. This group of tumours was significantly enriched in S1 subclass (p<0.001),22 aberrant activation of tyrosine-protein kinase Met or hepatocyte growth factor receptor (MET)23 (p=0.038), late transforming growth factor beta (TGFβ)24 (p=0.009), insulin-like growth factor 1 receptor (IGF1R)25 (p<0.001), Notch26 (p<0.001), mechanistic target of rapamycin (mTOR)27 (p<0.001), and gene signatures associated with recurrence28 (p=0.015), vascular invasion29 (p<0.001) and sorafenib resistance30 (p=0.002). Of note, tumours displaying pERK-positive hepatocytes were enriched in proliferation-related signatures (IGF1R25 (p<0.001), Notch26 (p<0.001), Cytokeratin 19 (CK19)31 (p=0.015) and G232 (p=0.004)) (online supplementary table 3). On the other hand, patients in the non-proliferation class19 (45%, n=63/140) were significantly associated with S3 subclass22 (p<0.001).
In terms of prediction, we tested 22 previously reported tumour-derived molecular signatures, and survival prognostic signatures generated from adjacent non-tumour samples capturing the microenvironment signalling (online supplementary table 2).31 33 34 None of them showed predictive capacity in terms of RFS (equivalent to recurrence) in the interaction analysis. This applies to signatures designed for/or associated to recurrence24 28 32 or survival33 34 in retrospective analysis. Finally, the prognostic capacity of survival of gene signatures was not tested due to the marginal number of events (24/188).
Further, we explored the mutational landscape of the BIOSTORM cohort using targeted exome sequencing to identify mutations in 19 known HCC drivers1 (online supplementary table 4; online supplementary figure 4). The mutational profile was similar to what has been previously reported for HCC drivers,1 with TERT promoter mutations (53%), TP53 (46%), CTNNB1 (21%) and AXIN1 (16%) as the four most prevalent mutations in our cohort (online supplementary table 4). No significant association was found between mutational profile and outcome in terms of RFS and treatment.
Novel 146-gene expression signature predicts role of sorafenib in prevention of recurrence
Given the lack of interaction between tested clinical and biomarker candidates and treatment benefit, we developed a gene expression signature able to predict the role of sorafenib in recurrence prevention of HCC in resected patients.
Using standard procedures, we first identified genes associated with prognosis (Cox score p<0.05) in terms of RFS in the set of 67 sorafenib-treated patients and in the set of 73 placebo-treated patients (figure 1). To exclude genes that were mainly associated with poor prognosis independently of treatment, we filtered out those genes identified with the same prognosis value in both cohorts, keeping only those exclusively detected in the sorafenib group. We obtained a 146-gene set able to discriminate precisely patients benefiting from sorafenib in terms of extended RFS (from herein referred to as ‘sorafenib RFS responders’) from patients where sorafenib had no effect (‘non-responders’) (figure 3). This gene signature was composed of 87 poor prognosis genes and 59 good prognosis genes (figure 3A and online supplementary table 5). The signature identified 30% of the patients (n=42/140) as ‘sorafenib RFS responders’. These patients did not reach median RFS with sorafenib, while ‘non-responders’ presented a median RFS of 28 months. The predictive value of the signature was statistically significant (p of interaction p=0.040) in the BIOSTORM cohort (figure 3B).
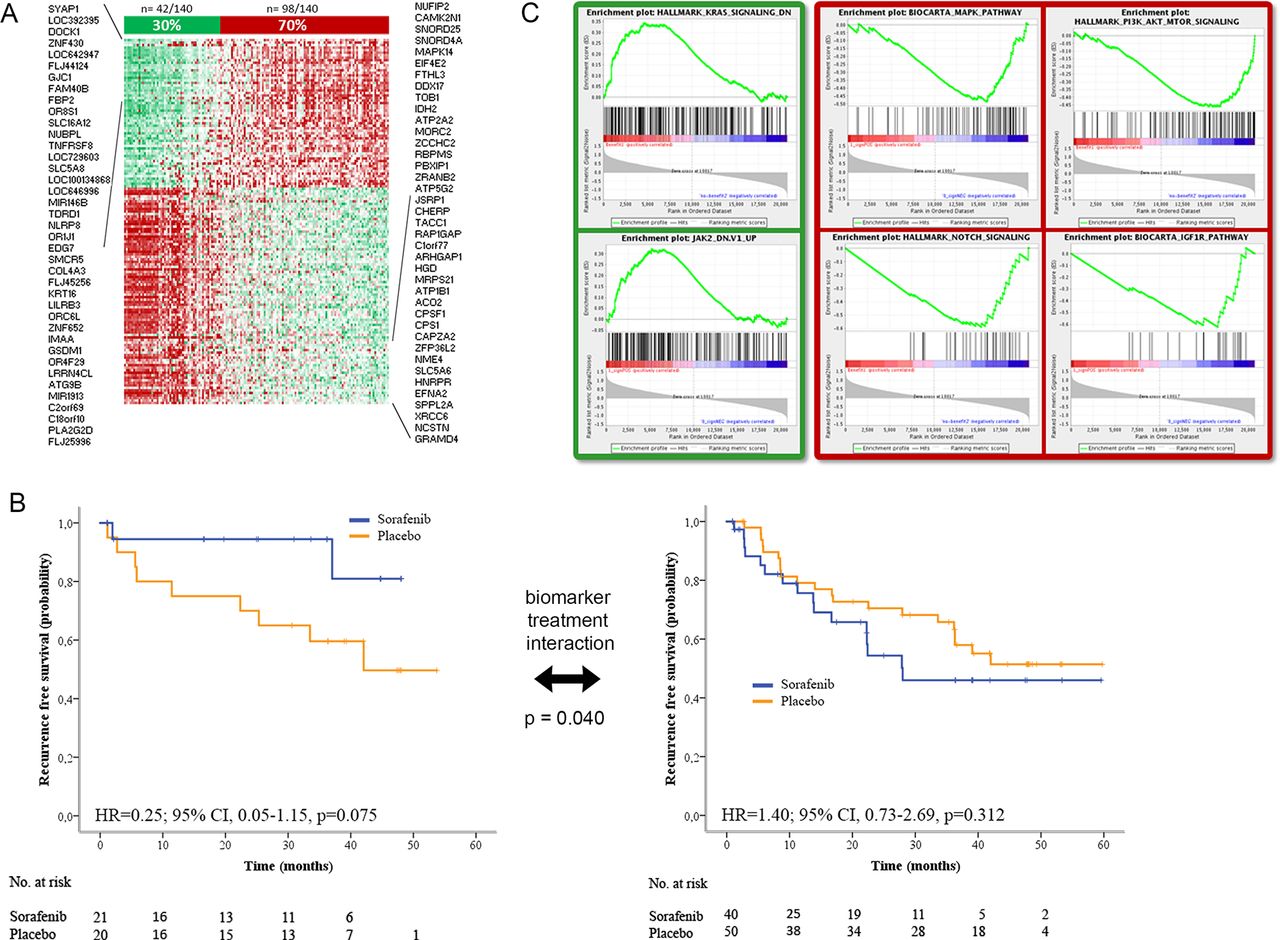
Novel 146-gene signature associated with better recurrence-free survival (RFS) in sorafenib-treated patients. (A) 146-gene signature identifying BIOSTORM patients with better RFS when treated with sorafenib. (B) Kaplan-Meier estimates measuring RFS probability over time of patients identified by the 146-gene signature capturing recurrence prevention in individuals treated with sorafenib (in blue) or placebo (in orange). P value of biomarker treatment interaction was 0.040. Left panel shows the Kaplan-Meier estimates for patients identified as ‘sorafenib RFS responders’ by the gene signature, and the right panel, the estimates for those identified as ‘non-responders’. (C) Patients identified by the 146-gene signature as ‘sorafenib RFS responders’ (in green) displayed downregulation of KRAS and JAK/STAT signalling. In contrast, patients recognised as ‘non-responders’ (in red) were enriched in MAPK, mTOR, IGF1R and Notch signalling.
We characterised ‘sorafenib RFS responders’ and ‘non-responders’ at the molecular level using enrichment analysis tools (figure 3C). In brief, ‘sorafenib RFS responders’ showed (A) downregulation of pathways indicative of poor prognosis such as KRAS, (B) activation of EIF2 signalling—previously reported to be inhibited after sorafenib treatment9—and oxidative stress responses, (C) association with immune-related processes, such as T helper cell differentiation and B cell development, and (D) upregulation of bile acid and lipid metabolism-related pathways (ie, liver X receptor signalling and related farnesoid X receptor/retinoid X receptor) (online supplementary tables 6–8). All these molecular traits were also present in the non-tumour adjacent tissue (online supplementary tables 7 and 8). Conversely, ‘non-responders’ (98/140; 70%) were characterised by (A) activation of signalling pathways of poor outcome (PI3K-AKT-mTOR, KRAS, MAPK, IGF1R, Notch); (B) higher mRNA alpha-fetoprotein levels (p=0.03); and (C) microvascular invasion (p=0.042) and HCV-related HCC (p<0.001) (figure 3C, online supplementary figure 5C and online supplementary tables 6–9). Of note, the mutational profile of known oncogenes in HCC was not significantly different in ‘sorafenib RFS responders’ when compared with ‘non-responders’.
Supplemental material
Immune cell profiles in ‘sorafenib RFS responders’
Recent preclinical studies have suggested that sorafenib may exert immune-modulating effects on the tumour microenvironment of HCC through upregulation of T cell activity, inhibition of macrophage-induced tumour growth and activation of natural killer (NK) cells.35–37 Therefore, it is important to understand the immune profile of ‘sorafenib RFS responders’ tumours. To this end, we characterised the immune infiltrates of the BIOSTORM tumours using published gene signatures38 and data deconvolution methods (ie, Cibersort),39 and correlated the results with the prediction of the 146-gene signature and our recently reported HCC Immune class.38 In this regard, we have defined that ~30% of HCCs present an ‘inflamed’ microenvironment (so-called Immune class) with high infiltration of immune cells, adaptive T cell response, active IFN-γ signalling and markers of cytolytic activity.38 The Immune class did not directly predict recurrence or response to sorafenib.
Here, our results indicate that the immune profile of ‘sorafenib RFS responder’ patients (n=42/140, 30%) was characterised by enrichment of signatures capturing the presence of B cells and CD4+ T cells and derivatives (type 1 and 2 T helper cells, and follicular helper T cells); and the exclusion of Immune class-associated features (ie, CD8+ T cells, effector memory T cells, and central memory T cells and tumour-associated tertiary lymphoid structures (TLS), generated during chronic immune stimulation to support infiltration and activation of the adaptive immune cells) (figure 4). Furthermore, the innate immune profile of ‘sorafenib RFS responders’ was enriched in activated Mast cells and cytolytic NK cells (NKCD56dim), and showed absence of activated macrophages, components from the major histocompatibility complex, and downregulation of signal transducer and activator of transcription proteins and focal adhesion kinase 1/protein tyrosine kinase 2 (FAK1/PTK2) (figure 4, online supplementary figures 5 and 6).39 In addition, ‘sorafenib RFS responders’ had significantly lower immunophenoscore—which has been associated with response to checkpoint blockade—and downregulation of IFN gene signatures predicting response to checkpoint inhibitors in other cancers (figure 4).39–41

Heatmap displaying the molecular class and immune characteristics of ‘sorafenib RFS responders’ and ‘non-responders’. Differences in RFS profiles in the sorafenib and placebo-treated patients identified by the signature as ‘sorafenib RFS responders’ suggest that placebo patients could have been candidates to respond to sorafenib. Immune profile of ‘sorafenib RFS responders’ highlights absence of CD8+ T cell lymphocytes but enrichment of other adaptive immune elements such as B cells and CD4+ T cells and derivatives. P values describe differences between ‘sorafenib RFS responders’ and ‘non-responders’. #P<0.01 between ‘sorafenib RFS responders’ and HCCs of ‘Immune class’. Signatures are referenced in online supplementary table 11. HCC, hepatocellular carcinoma; IFN, interferon; MHC, major histocompatibility complex; NK, natural killer cells; ns, non-significant differences; RFS, recurrence-free survival; TLS, tertiary lymphoid structures.
On the other hand, ‘non-responders’ (98/140; 70%) encompassed a subgroup of HCCs imprinted with Immune class traits (27/98; 28%), characterised by CD8+ T cells, TLS and programmed cell death protein 1 (PD1) signalling, along with another subgroup of HCCs characterised by Immune exclusion (CTNNB1 class: 27/98; 28%)38 (figure 4).
Prognostic biomarkers of HCC recurrence
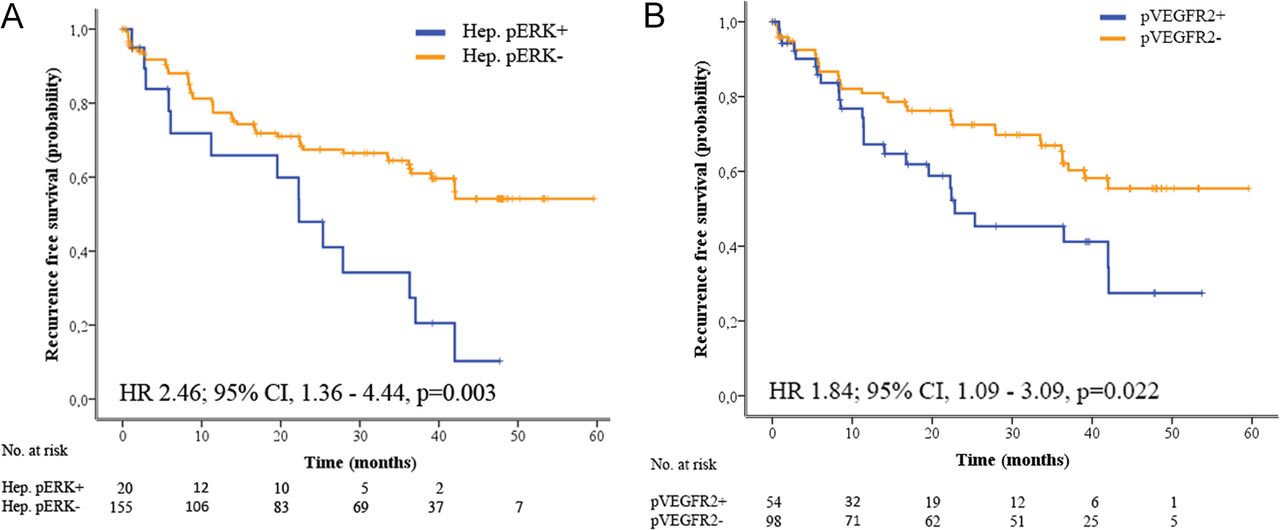
The second aim of the study was to assess the prognostic value in terms of RFS of all above described clinical and molecular features. In the univariate analyses, positive hepatocyte pERK (HR=2.46; p=0.003) and nuclear pVEGFR2 (HR=1.84; p=0.022) were significantly associated with poor RFS (figure 5). In addition, three gene signatures (G332 enriched in cell cycle control genes (HR=1.73; p=0.049); MET23 (HR=2.28; p=0.017); and IFN1α42 (HR=2.09; p=0.019)) were associated with poor RFS (table 2 and online supplementary table 10). Previously reported gene signatures from tumour and adjacent tissue designed to predict survival33 34 or recurrence24 28 32 did not predict RFS in the current study (online supplementary table 10). In the multivariate model, only hepatocyte pERK staining and microvascular invasion retained independent prognostic value for RFS (table 2). Both biomarkers were associated with poor prognosis (pERK HR=2.41; 95% CI 1.21 to 4.80; vascular invasion HR=2.09; 95% CI 1.14 to 3.83).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prognostic value of hepatocyte pERK and pVEGFR2. Tumours with hepatocyte pERK staining (A) or pVEGFR2 staining (B) have significantly poorer outcome compared with pERK negative or pVEGFR2 tumours.
Univariate and multivariate analysis of clinical and molecular prognostic factors in terms of RFS
Discussion
The success of sorafenib to inhibit tumour cell proliferation and angiogenesis43 and its proven efficacy in advanced HCC3 provided the rationale for testing this compound as adjuvant therapy to prevent recurrence after resection or local ablation. The phase 3 trial did not achieve the primary endpoint of superiority in RFS comparing sorafenib vs placebo. In the companion BIOSTORM study presented here, we explored biomarkers predicting the efficacy of sorafenib in preventing HCC recurrence, along with prognostic markers. Neither the proliferation or angiogenic biomarkers tested, nor the reported prognostic gene signatures or mutations identified in driver genes were associated with improved RFS in the sorafenib arm. The fact that none of the tested biomarkers predicted sorafenib RFS underscores the complexity of the mechanisms of action related to this drug. We generated a 146-gene signature able to identify 30% of patients who might benefit from sorafenib in terms of recurrence prevention. In addition, we further confirmed the role of pERK and microvascular invasion as two independent predictors of recurrence in early HCC. Previously reported gene signatures from tumour and adjacent tissue designed to predict survival33 34 or recurrence24 28 32 did not predict RFS in the current study. Our results hold a B level of evidence, which is based on subgroup or retrospective analysis of prospective phase 3 investigations.16
Thus far, more than 20 predictive biomarkers have entered clinical practice (eg, Her-2/neu expression as biomarker of response to trastuzumab; KRAS mutations for resistance to cetuximab or panitumumab).44–46 For sorafenib, validated biomarkers of response have not yet been identified. A companion study of the SHARP trial described that plasma concentrations of soluble c-Kit and HGF showed a non-significant trend towards predicting response to sorafenib.15 Several studies have proposed pERK, proxy of activation for the RAS/MAPK pathway inhibited by sorafenib in vitro and in solid tumours,9 as a candidate biomarker associated to outcome after sorafenib treatment, though with contradictory results.47–49 The absence of a validated scoring system for immunostaining for pERK, and the differences in detection methods, cohorts and endpoints could account for these inconsistencies. On the other hand, VEGF-related biomarkers have been proposed as potential predictors of response to sorafenib, considering that this drug inhibits pVEGFR2.43 In this sense, VEGFA amplification was reported as a predictive factor of recurrence prevention in patients treated with sorafenib after liver resection.20 However, all the above studies claiming the predictive value of pERK or VEGF-related molecules have been tested in the setting of retrospective non-randomised studies.
In BIOSTORM, neither pERK, nor pVEGFR2 or VEGFA copy number status was significantly associated with recurrence prevention of sorafenib. Only sorafenib-treated patients tended towards better outcome if their tumours were hepatocyte-pERK negative and/or endothelial-pERK positive. Similarly, previously reported tumour-derived signatures associated with poor outcome in terms of recurrence or survival did not show predictive capacity for RFS on interaction analysis.
Our generated signature identifying patients benefiting from sorafenib adjuvancy in terms of recurrence prevention classified 30% of cases as ‘sorafenib RFS responders’. These displayed downregulation of poor prognosis oncogenic pathways and deregulation of bile acid/lipid metabolism-related signalling, were enriched in B cells and CD4+ T cells (and derivatives) and presented exclusion immune features related to the HCC Immune class,38 and were associated to signatures of poor response to immune checkpoint inhibitors.39 41 On the other hand, ‘non-responders’ tumours presented traits of cancer aggressiveness, with more vascular invasion and activation of poor prognosis signalling cascades, among others. The fact that HCV was more prevalent in non-responders does not align well with our previous report where HCV-related HCC had a better response to sorafenib in advanced cases.14 Thus, clarification of this association would require further research.
In terms of prognostic biomarkers, here we report pERK staining and microvascular invasion as two independent predictors of RFS in early HCC. While microvascular invasion is a well-established predictor of recurrence, the role of pERK is less documented. In our hands, patients displaying tumour pERK hepatocyte staining were characterised by poor differentiation degree and enhanced proliferation traits. pERK is a key element in angiogenesis and apoptosis. Nuclear pERK has been involved in proliferation processes, and the cytoplasmic form in cell differentiation. In the setting of early HCC, only one previous retrospective study reported a correlation between ERK activation and poor survival.50 In terms of prognostic value of gene signatures, the univariate analysis revealed that three gene signatures (G3,32 MET,23 IFN1α42) predicted poor RFS, whereas previously reported gene signatures from tumour and adjacent tissue designed to predict survival33 34 or recurrence24 28 32 were not significantly associated with RFS. None of the above described signatures retained independent prognostic value in the multivariate analysis.
Lastly, we acknowledge the strengths and limitations of the study. A major strength is that the study was conducted with a subgroup of patients randomised in the setting of the phase 3 STORM trial.12 Therefore, BIOSTORM patients were prospectively followed, and management of adverse events, drug prescriptions and patients’ outcomes were properly scrutinised. In addition, the primary endpoint RFS (which in BIOSTORM study overlaps with time to recurrence) was assessed by central radiological reviewers. Finally, we used FFPE samples (standardised method of tissue processing) to ensure the highest preanalytical validity and followed standardised scoring criteria or, when absent, published criteria to ensure analytical validity. All these positive factors contrast with the conventional retrospective nature of most biomarker studies.51 Moreover, our study follows the Reporting Recommendations for Tumor Marker guidelines,52 and together with the SHARP biomarker investigation15 is one of the few HCC studies for biomarker discovery with level B of evidence16 (meaning retrospective study conducted in the setting of a randomised phase 3 trial). These types of studies have changed the standard of care in cases where the primary endpoint was negative, but the biomarker companion investigation yields positive results. This is the case of cetuximab for patients with colorectal cancer with wild-type RAS.45 In terms of limitations, we acknowledge that despite a huge collecting effort, the number of patients recruited (n=188) is not ideal for defining bona fide predictive biomarkers.16 This fact reinforces the need of budget allocation for ensuring the collection of tissue samples in the setting of controlled investigations in HCC.
Overall, our biomarker BIOSTORM study (A) established lack of predictive value of sorafenib response for previously reported molecular biomarkers, (B) generated a predictive 146-gene signature to discriminate patients where sorafenib would prevent recurrence after resection, (C) established pERK and microvascular invasion as independent prognostic tools to identify recurrences in patients resected from early HCC tumours, and (D) did not validate previously reported signatures associated with recurrence.24 28 32 For any of these biomarkers to be used as surrogates of efficacy of sorafenib recurrence prevention, a validation study using an independent cohort and conducted by independent investigators would be required.
Supplemental material
Supplemental material
![[gutjnl-2018-316408-SP4.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
Supplemental material
![[gutjnl-2018-316408-SP5.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
Supplemental material
![[gutjnl-2018-316408-SP6.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
Supplemental material
![[gutjnl-2018-316408-SP7.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
Supplemental material
![[gutjnl-2018-316408-SP9.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC9/embed/inline-supplementary-material-9.jpg?download=true){kind=link}
Supplemental material
![[gutjnl-2018-316408-SP10.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/10/01/gutjnl-2018-316408/DC10/embed/inline-supplementary-material-10.jpg?download=true){kind=link}
Acknowledgments
We thank Marta Boteller, Judit Peix and Laia Cabellos for technical assistance and Juan José Lozano for suggestions in the transcriptomic analysis. This study has been in part developed at the building Centre Esther Koplowitz from IDIBAPS/CERCA Programme/Generalitat de Catalunya.
References
Footnotes
Contributors RP, RM, LB, AV, ST, DS, AM, LRC, JC, MS, SNT and ZZ were involved in study concept and design; data acquisition, analysis and interpretation; manuscript drafting. TT, GYC, VM, SR, NK, HCL, EG, CV, AEC, UC, MM, LL, SS, JWP, CP and GM provided specimens for genomic analysis or clinical data, and critically revised the manuscript. JML and JB were involved in study concept and design; interpretation of data; critical revision of the manuscript; study supervision; and obtained funding.
Funding This study was supported by Bayer HealthCare Pharmaceuticals and Onyx, a wholly owned subsidiary of Amgen. JML is supported by the European Commission (EC)/Horizon 2020 Program (HEPCAR, Ref 667273-2), US Department of Defense (CA150272P3), Asociación Española Contra el Cáncer (AECC), National Cancer Institute (P30-CA196521), Samuel Waxman Cancer Research Foundation, Spanish National Health Institute (SAF2016-76390) and the Generalitat de Catalunya/AGAUR (SGR-1162 and SGR-1358). JB has received grant support from Instituto de Salud Carlos III (ISCIII) (PI14/00962), AECC, AGAUR (SGR-605), WCR (AICR) 16-0026, and Spanish Health Ministry (Plan Estrategico Nacional contra la Hepatitis C). Centro de Investigacion Biomedica en Red de Enfermedades Hepaticas y Digestivas is funded by ISCIII. RP and AM are funded by AECC and EC. ST, RM, LB and JC are supported by MINECO (BES-2014-068300), Río Hortega (ISCIII-SEOM), Beatriu de Pinós (AGAUR) and Miguel Servet (ISCIII-CP13/00160) grants, respectively. AV is supported by the US Department of Defense (CA150272P3), the Tisch Cancer Institute, and the American Association for the Study of Liver Diseases Foundation (AASLDF) Alan Hofmann Clinical and Translational Award.
Competing interests JML, JB, VM and AEC received research support and consultancy fees from Bayer. AV and SS received consultancy fees from Bayer. CP and GM are employees of Bayer HealthCare Pharmaceuticals.
Patient consent Not required.
Ethics approval Hospital Clinic, Barcelona, Spain. Each participating centre obtained their own required institutional review board approval.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. The abstract and significance of this study box has been updated.