Article Text

Abstract
Objective Liver injury impacts hepatic inflammation in part via Toll-like receptor (TLR) signalling. Triggering receptor expressed on myeloid cells 2 (TREM-2) modulates TLR4-mediated inflammation in bone marrow (BM)-derived macrophages but its function in liver injury is unknown. Here we hypothesised that the anti-inflammatory effects of TREM-2 on TLR signalling may limit hepatic injury.
Design TREM-2 expression was analysed in livers of humans with various forms of liver injury compared with control individuals. Acute and chronic liver injury models were performed in wild type and Trem-2-/- mice. Primary liver cells from both genotypes of mice were isolated for in vitro experiments.
Results TREM-2 was expressed on non-parenchymal hepatic cells and induced during liver injury in mice and man. Mice lacking TREM-2 exhibited heightened liver damage and inflammation during acute and repetitive carbon tetrachloride and acetaminophen (APAP) intoxication, the latter of which TREM-2 deficiency was remarkably associated with worsened survival. Liver damage in Trem-2-/- mice following chronic injury and APAP challenge was associated with elevated hepatic lipid peroxidation and macrophage content. BM transplantation experiments and cellular reactive oxygen species assays revealed effects of TREM-2 in the context of chronic injury depended on both immune and resident TREM-2 expression. Consistent with effects of TREM-2 on inflammation-associated injury, primary hepatic macrophages and hepatic stellate cells lacking TREM-2 exhibited augmented TLR4-driven proinflammatory responses.
Conclusion Our data indicate that by acting as a natural brake on inflammation during hepatocellular injury, TREM-2 is a critical regulator of diverse types of hepatotoxic injury.
- acute liver failure
- chronic liver disease
- hepatic stellate cell
- inflammation
- immune-mediated liver damage
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
- acute liver failure
- chronic liver disease
- hepatic stellate cell
- inflammation
- immune-mediated liver damage
Significance of this study
What is already known on this subject?
Increasing evidence suggests the gut-liver axis and innate immunity play a significant role in liver injury. Impaired intestinal permeability may promote liver inflammation due to the translocation of bacterial components into the liver.
The consequent hepatic inflammatory response is in part mediated by toll-like receptors (TLRs). Fine tuning of TLR-driven inflammation during liver injury is key. The liver requires mechanisms to control the intensity and duration of TLR-driven cytokine production, which can contribute to the pathogenesis of acute and chronic liver disease.
Triggering receptor expressed on myeloid cells 2 (TREM-2) has been proposed to attenuate TLR4-mediated inflammation and to be important in injury responses in colon and brain.
What are the new findings?
TREM-2 is expressed on non-parenchymal liver cells and is upregulated during diverse forms of liver injury in humans and mice.
Trem-2-/- mice display increased liver damage and inflammation during acute and chronic carbon tetrachloride treatment and acetaminophen (APAP) intoxication. Effects of TREM-2 in the context of chronic liver injury are dependent on both liver resident and infiltrating immune cells.
TREM-2 promotes survival during drug-induced APAP challenge.
Liver injury in Trem-2-/- mice is associated with elevated levels of reactive oxygen species and lipid peroxidation.
In line with TREM-2 effects on inflammation and injury, Kupffer cells and hepatic stellate cells isolated from Trem-2-/- mice exhibit increased proinflammatory cytokine and chemokine production upon TLR4 stimulation.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Strategies that aim to therapeutically activate this receptor may provide a novel route towards suppressing inflammation caused by toxin-induced hepatocellular injuries.
Introduction
Toll-like receptors (TLRs) are pattern recognition receptors that recognise conserved microbial molecules. A variety of TLRs are expressed on different liver cell types, including hepatocytes, hepatic stellate cells (HSCs) and resident Kupffer cells (KCs), with the latter cell type being located in the hepatic sinusoids and the first cells to encounter gut derived danger signals, such as lipopolysaccharide (LPS).1–3 The crucial role of TLR signalling within the liver is highlighted by studies showing that Tlr4 -/- mice are protected from various forms of liver injury.4 5 Illustrating the critical role of the fine tuning of TLR4-driven inflammation during liver injury, interleukin 1 receptor-associated kinase-M (IRAK-M) and A20, prototypical negative regulators of TLR4 signalling, protect from alcohol-induced and ischaemia/reperfusion liver injury, respectively.6 7
The triggering receptor expressed on myeloid cells 2 (TREM-2) is a relatively recent described regulator of TLR-mediated inflammatory responses. Similar to IRAK-M and A20, TREM-2 acts to dampen TLR-driven cytokine responses within bone marrow derived macrophages (BMDM).8 This is in contrast to TREM-1 which augments TLR-induced inflammation.9 TREM-1 and TREM-2 both signal via the ITAM motif of the adaptor protein DNAX-activation protein 12, which becomes tyrosine phosphorylated leading to increases in intracellular calcium and extracellular regulated kinase (ERK1/2) phosphorylation.9–11 TREM-1 is expressed on KCs and HSCs and is important in promoting inflammation-driven hepatocellular carcinoma and diethylnitrosamine liver injury.12 TREM-2 has been shown to be expressed on KCs where it serves to control liver stage malarial infection.13 14 Given that TREM-2 has been proposed to attenuate TLR4-mediated inflammation and has previously been shown to be important in injury responses in the colon and brain, we undertook this study to examine if it could play a role in liver injury and inflammation.11 15–17
Materials and methods
Patients
To perform gene expression analysis at mRNA level, liver tissue was obtained from control individuals (n=21) and patients with liver cirrhosis of different aetiology (n=23). Patient characteristics and liver injury-related serology are indicated in online supplementary table 1. Tissue samples of normal liver used as controls were obtained from background non-neoplastic tissue of liver resection specimens for colorectal metastasis. The selected sections were located furthest away from the tumour mass lesion. Samples and data from patients included in this study were provided by the Basque Biobank (http://www.biobancovasco.org) and were processed following standard operation procedures with appropriate approval by the Clinical Research Ethics Committees of the Basque Country and Donostia Hospital. An informed consent was obtained from all subjects. Human hepatic myofibroblasts were isolated from livers of adult male patients after surgical resection as approved by the Newcastle and North Tyneside Local Research Ethics Committee, subject to patient consent (10/H0906/41).
Supplementary file 1
Experimental models of liver injury
Wild type (WT) and Trem-2 knockout (Trem-2-/- ) male mice (25–30 g) were intraperitoneally injected one time (acute) or two times a week for 8 weeks (chronic) with carbon tetrachloride (CCl4) at 2 µl/g body weight (CCl4: olive oil at 1:1 (vol:vol) (acute) and 1:3 (vol:vol) (chronic)). For the acetaminophen (APAP) model WT and Trem-2-/- male mice (25–30 g) were overnight-starved and intraperitoneally injected once with APAP (Sigma-Aldrich) at a concentration of 300 mg/kg or 500 mg/kg dissolved in sterile warm saline. For survival studies, mice were injected with a lethal dose of APAP (750 mg/kg) and survival was monitored every hour for 30 hours, after which it was monitored every day. Animal care and procedures were approved by the Animal Care and Use Committee of the Medical University of Vienna and the Austrian Ministry of Sciences as well as by the Animal Experimentation Ethics Committee (CEEA) of Biodonostia Research Institute and the Newcastle Ethical Review Committee under a UK Home Office project license. Trem-2 -/- mice backcrossed onto a >98% C57BL/6 background were obtained from Marco Colonna (Washington University) and generated as previously described.8 For all in vivo experiments, we used age-matched (8–10 week old) male Trem-2-/- mice and WT controls bred at the Medical University of Vienna or Biodonostia Research Institute. For bone marrow (BM) transplantation, Trem-2 -/- mice were crossed with mice that ubiquitously express green-fluorescent protein (GFP) on a C57BL/6 background (WT-UbGFP+) to generate (Trem-2 -/-/Ub-GFP+) mice.
Isolation of mouse hepatocytes, KCs, HSCs, BMDMs and treatments
Hepatocytes, KCs and HSCs were isolated from mice and rats as previously described.18 19
Additional Materials and Methods are included as online supplementary files.
Supplementary file 2
Results
TREM-2 is upregulated during liver injury in mice and humans
We first examined TREM-2 transcript levels in a cohort of control (n=21) and diseased liver (n=23) as described in online supplementary table 1. By grouping all the cirrhotic livers despite of different disease aetiology (HCV or HBV infection, HCV/HIV coinfection and alcoholic or other aetiology) and comparing these with control liver tissues, we could observe significantly elevated TREM-2 transcript levels in cirrhotic versus control liver (figure 1A). Correlating these data with levels of markers of hepatocyte injury, fibrosis and inflammation revealed that increases in TREM-2 expression correlated with collagen levels and with alanine aminotransferase, aspartate aminotransferase and the inflammation markers IL-8 and IL-6 (figure 1B,C, online supplementary figure 1), suggesting that increases in TREM-2 may serve to dampen hepatic inflammation and injury. Evaluating TREM-2 expression using immunohistochemistry in an independent cohort of human control liver and cirrhotic liver of variable aetiology (online supplementary table 2), verified that TREM-2 expression was more extensive in diseased compared with control liver (online supplementary figure 2). Mild cytoplasmic TREM-2 immunostaining was observed in monocytes and KCs in control liver, while there were no TREM-2 positive endothelial cells or lymphocytes. TREM-2 immunohistochemical expression in cirrhotic liver was observed in neutrophils, monocytes and macrophages in the fibrous septa, within sinusoids and in parenchymal inflammatory foci. Collagen deposition in fibrous septa was confirmed in cirrhotic versus control liver by Sirius red staining (online supplementary figure 2). Further, Trem-2 was upregulated during both acute and repetitive CCl4 induced injury in mice compared with olive oil controls and augmented Trem-2 levels occurred during bile duct ligation compared with sham operated mice (figure 1D–F). In the chronic CCl4 model, we noted that on cessation of liver damage levels of Trem-2 transcript declined over a 10-day period of recovery, but remained slightly higher than in controls (figure 1E). We conclude liver injury in mice and man is associated with a substantial induction in the expression of TREM-2 indicating a role for the receptor in hepatic wound repair.

Expression of TREM-2 during human and mouse liver disease. (A) qRT-PCR analysis of TREM-2 expression in normal human liver (controls) and cirrhotic samples. (B,C) Correlation between TREM-2 and COL1A1 levels (B) or ALT (C). n=21 controls and 23 cirrhotic livers. (D) qRT-PCR of Trem-2 expression in livers of mice after the indicated acute CCl4 time points. Olive oil treated mice were used as controls. n=3 mice per condition and time point. (E,F) Trem-2 mRNA expression in the liver of mice treated with CCl4 for 12 weeks and sacrificed 1, 3, 7 or 10 days after the last CCl4 injection (E) and 14 or 21 days after BDL (F). n=3–5 (E) and 4–5 mice per time point (F). Statistical analysis used was Mann Whitney test (A) and non-parametric Spearman’s correlation test (B,C). Data represent mean±SEM and **,*** denote a P value of ˂0.01 and ˂0.001, respectively versus olive oil (D,E) or sham (F) determined using one-way analysis of variance, followed by Tukey’s posthoc test. ALT, alanine aminotransferase; BDL, bile duct ligation; CCl4, carbon tetrachloride; TREM, triggering receptor expressed on myeloid cells.
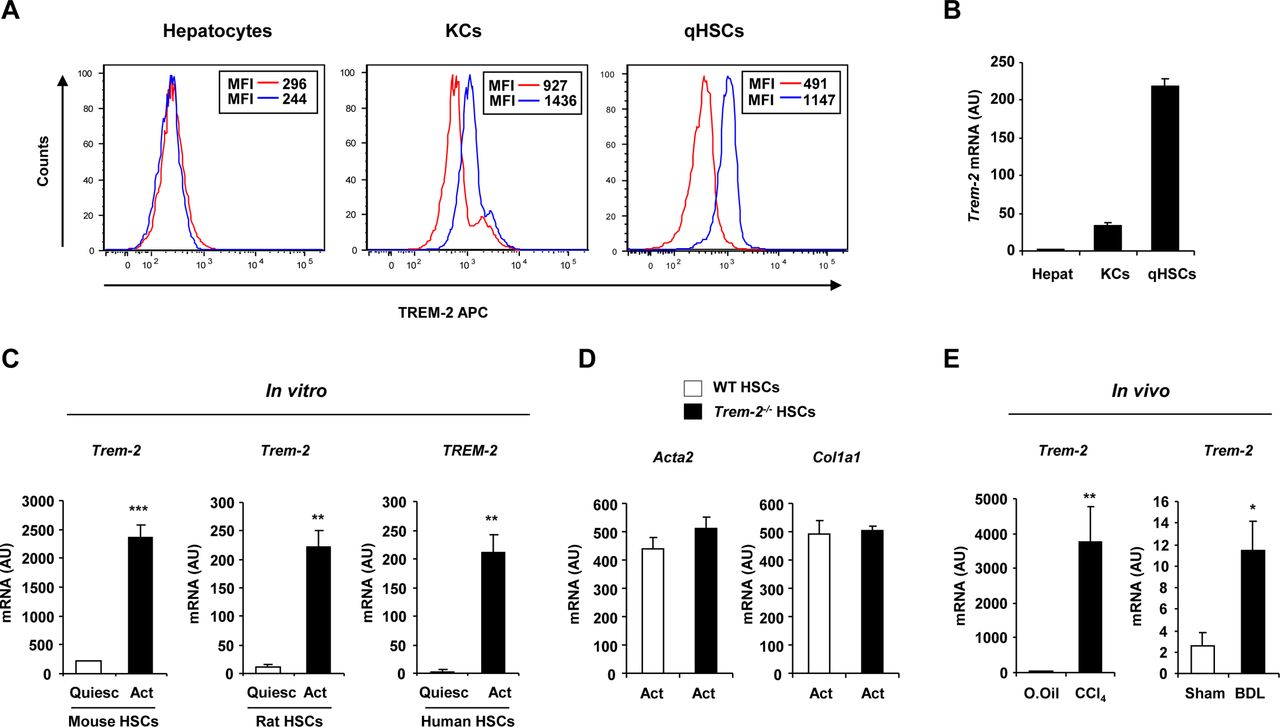
TREM-2 expression in non-parenchymal liver cells and activated HSCs during liver injury. (A) TREM-2 expression in primary mouse hepatocytes, KCs or qHSCs detected by flow cytometry. Red and blue lines depict the isotype and TREM-2 antibodies respectively. (B) qRT-PCR analysis of Trem-2 expression in various liver cell types. n=10 (hepatocytes), 4 (KCs) and 5 (qHSCs). (C) TREM-2 expression in mouse, rat and human HSCs during trans-differentiation in vitro at day 1 (quiescent) and day 7 (activated) after the isolation. n=3. (D) Transcript levels of Acta2 and Col1a1 in WT and Trem-2-/- activated mouse HSCs. n=3. (E) Trem-2 expression in rat HSCs activated in vivo from CCl4 and BDL treated rats. n=4–5 per condition. Data represent mean±SEM and *, **, *** denote a P value of ˂0.05, ˂0.01 and ˂0.001, respectively versus quiescent (C) or olive oil or sham (E) and statistical analysis used was unpaired Student’s t-test. Data in (C,E) are representative of two and in (D) is representative of three independent experiments BDL, bile duct ligation; CCl4, carbon tetrachloride; HSCs, hepatic stellate cells; KCs, Kupffer cells; qHSC, quiescent HSC; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
TREM-2 is expressed on non-parenchymal liver cells and is upregulated during HSC activation
Flow cytometry and qRT-PCR for TREM-2 in murine hepatic cells demonstrated expression on KCs and quiescent HSCs, but not on hepatocytes demonstrating that TREM-2 was expressed by non-parenchymal liver cells (figure 2A,B). We next investigated the expression of Trem-2 during HSC transdifferentiation, which was induced in vitro by culturing freshly isolated murine quiescent HSCs on plastic for 7 days. By doing so, we could observe that Trem-2 mRNA expression was strongly induced during mouse HSC transdifferentiation in vitro. This response was conserved as TREM-2 expression during activation in vitro was similarly increased in rat and human HSCs (figure 2C). Examining the activation status of both genotypes of HSCs revealed that activated Trem-2-/- HSCs displayed similar α-smooth muscle actin (encoded by Acta2) and α1(I) procollagen (encoded by Col1a1) expression to WT (figure 2D). To address if Trem-2 was induced during HSC activation in vivo, we isolated HSCs from bile duct ligated or chronic CCl4 injured rats. Trem-2 was significantly induced in HSCs in vivo during liver injury (figure 2E). Together, these data suggested that conserved increases in TREM-2 expression during HSC activation might play a role during chronic liver injury, a process where HSCs are crucial fibrogenic effector cells and are important mediators of innate immunity that cross-talk with other liver resident and infiltrating cells during hepatocellular damage.20 21
TREM-2 attenuates chronic hepatic inflammation and injury
To formally investigate a function for TREM-2 in chronic liver disease, we employed a genetic approach comparing damage-induced responses of Trem2-/- mice with WT controls. We thus performed chronic CCl4 injury and first determined differences in hepatic levels of the inflammatory mediator monocyte chemoattractant protein 1 (Mcp1). We observed significantly elevated Mcp1 mRNA in WT at peak injury (day 1 after the final administration of CCl4) which declined close to control levels with recovery (day 5 postinjury) (figure 3A). Similarly, as expected, levels of the matrix metalloproteinase-13 (Mmp13) transcript were induced with peak injury in WT liver and declined to control levels with recovery (figure 3A). Both Mcp1 and Mmp13 were induced at significantly elevated levels in peak injured Trem-2-/- livers compared with WT, although with recovery no differences were observed between genotypes. Intriguingly we did not observe differences for damage-induced expression of interleukin-6 (Il6), interleukin-1 beta (Il1b), tumour necrosis factor-alpha (Tnf) or transforming growth factor beta-1 (Tgfb1) (online supplementary figure 3). Although at peak injury there was a tendency for higher Acta2 and Col1a1 liver transcript levels in Trem-2-/- mice, Sirius red staining verified that TREM-2 exerted only minor effects on fibrosis (online supplementary figure 4). Augmented inflammation during peak injury in Trem-2-/- livers was however associated with significantly enhanced transaminase release, suggesting that increased death and damage of hepatocytes occurs in the absence of TREM-2 (figure 3B). In agreement, histology disclosed significantly more necrosis and apoptosis in Trem-2-/- livers but unremarkable effects of TREM-2 on fibrosis (figure 3C,D). Monitoring transcription of genes associated with cellular stress (Heme oxygenase 1 (Hmox-1), cytochrome b-245 heavy chain (Cybb), nitric oxide synthase 2 (Nos2), Heat-shock family member 70 (Hspa1b)) and apoptosis (B-cell CLL Lymphoma 2 (Bcl-2), BCL2-like 1 (Bcl2l1), BCL-2 associated-X protein (Bax)) revealed that these pathways were both operational at peak injury and that levels of the stress-associated marker Hspa1b and the antiapoptotic gene Bcl2l1 were significantly upregulated in Trem-2-/- compared with WT livers (figure 3E). We conclude that an important function of TREM-2 is to protect against excessive liver injury in the context of iterative hepatotoxic damage.

TREM-2 impacts chronic CCl4-induced liver injury. (A–E) WT and Trem-2-/- mice were treated with CCl4 for 8 weeks, sacrificed 1 or 5 days after the last CCl4 injection and (A) liver Mcp1, Mmp13 (B) ALT and AST levels were determined. (C) Necrosis, apoptosis and fibrosis histology score from WT and Trem-2-/- mice that were treated with CCl4 for 8 weeks and sacrificed 1 day after the last CCl4 injection. (D) Representative H&E (Magnification is 10x) from day 1 are depicted. (E) Transcript levels of the indicated genes associated with stress and apoptosis were determined. n=3 mice per genotype (olive oil) and 4–8 mice per genotype (CCl4 both time points). Data represent mean±SEM and *, ** denote a P value of ˂0.05 and ˂0.01, respectively versus WT at the same time point (Mann Whitney test). ALT, alanine aminotransferase; AST, aspartate aminotransferase; CCl4, carbon tetrachloride; TREM, triggering receptor expressed on myeloid cells; WT, wild type.

TREM-2 expression within liver resident and infiltrating immune cells is required for dampening of chronic CCl4 induced liver injury. (A–D) WT mice reconstituted with WT-GFP+bone marrow (WT-WT), Trem-2-/- mice reconstituted with Trem-2-/- -GFP+bone marrow (Trem-2-/--Trem-2-/- ) or chimeric mice (WT-Trem-2-/- ) and (Trem-2-/- -WT) were treated with CCl4 for 8 weeks and sacrificed 1 day after the last CCl4 injection and (A) serum ALT and AST levels, (B) H&E stain of livers (C) total number of macrophages (defined as CD45+CD11b+Ly6C+Ly6G-F4/80+GFP+ cells, (online supplementary figure 5) normalised to liver weight and (D) liver Mcp1 transcript levels were determined. Data represent mean±SEM and *, **, ***, **** denote a P value of ˂0.05, ˂0.01, ˂0.001 and <0.0001 respectively versus the indicated genotype (one-way analysis of variance, followed by Tukey’s posthoc). n=3 per genotype (olive oil) and 3–5 per genotype (CCl4). ALT, alanine aminotransferase; AST, aspartate aminotransferase; CCl4, carbon tetrachloride; GFP, green fluorescent protein; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
Combined action of liver resident and infiltrating cells are required for TREM-2 to dampen chronic injury
To understand the contribution of TREM-2 expression on inflammatory cells versus HSCs to liver damage, we next reconstituted lethally irradiated WT or Trem-2-/- mice with WT or Trem-2-/- BM that ubiquitously expresses GFP within both genotypes, to generate four groups of mice. Nearly 100% of blood leucocytes were GFP+ within all groups, proving successful transplantation (online supplementary figure 5). Corroborating observations that TREM-2 attenuated chronic liver injury (figure 3B), elevated transaminases were observed in Trem-2-/- mice reconstituted with Trem-2-/- BM compared with WT (figure 4A). Although Trem-2-/- mice reconstituted with WT BM displayed a tendency for more liver damage compared with WT mice transplanted with WT BM, significantly elevated damage was only observed when TREM-2 was deficient within immune and resident cells (such as HSCs), demonstrating TREM-2 protects from immune-mediated hepatocellular damage via its combined functions in these cell types. These observations were confirmed by histology (figure 4B). Monitoring hepatic immune cell influx revealed equal numbers of recruited neutrophils between the groups of mice during chronic injury, while recruited monocyte derived macrophages were significantly elevated in Trem-2-/- mice reconstituted with Trem-2-/- BM, which correlated with elevated levels of Mcp1 (figure 4C,D, online supplementary figures 5 and 6). Together, these observations further support the concept that in the context of chronic CCl4 injury, TREM-2 dampens liver injury and indicates that these effects are dependent on both resident and infiltrating immune cells.

Cytokine responses and mitogen activated protein kinase (MAPK) signalling in WT and Trem-2-/- non-parenchymal hepatic cells after TLR4 stimulation. (A,B) WT and Trem-2-/- KCs were treated with LPS (100 ng/mL) (A) or (B) heat-killed E. coli (2×107 CFU/mL) for the indicated time points (n=4–5 per condition and time point) and levels of Cxcl1, Tnf, Il6 and Il1b were determined by qRT-PCR. (C, D) Cxcl1 (C) and MCP-1 (D) levels in WT and Trem-2-/- activated mouse HSCs treated with 2×107 CFU/mL heat-killed E. coli or 100 ng/mL LPS. n=3 (E. coli) and 4 (LPS). (E) Human HSC LX-2 cells were transfected with a control or TREM-2 overexpressing plasmid (n=4 per condition) and 36 hours post-transfection stimulated with 100 ng/mL LPS for 3 hours and levels of TREM-2, MCP-1 and IL-6 determined by qRT-PCR. (F,G) WT and Trem-2-/- KCs were treated with 100 ng/mL LPS (F) or 2×107 CFU/mL heat-killed E. coli (G) for the indicated time points and phosphorylation of ERK1/2, p38, JNK, p65 and IκB-α degradation was determined by western blotting. Data represent mean±SEM and *, **, ***, **** denote a P value of ˂0.05, ˂0.01, ˂0.001 and <0.0001, respectively versus WT at the same time point (one-way analysis of variance, followed by Tukey’s posthoc test). Data in (C,D) are representative of two independent experiments. Cxcl1, C-X-C motif chemokine ligand 1; ERK, extracellular regulated kinase; HSCs, hepatic stellate cells; JNK, Jun N-terminal kinase; KCs, Kupffer cells; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein-1; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
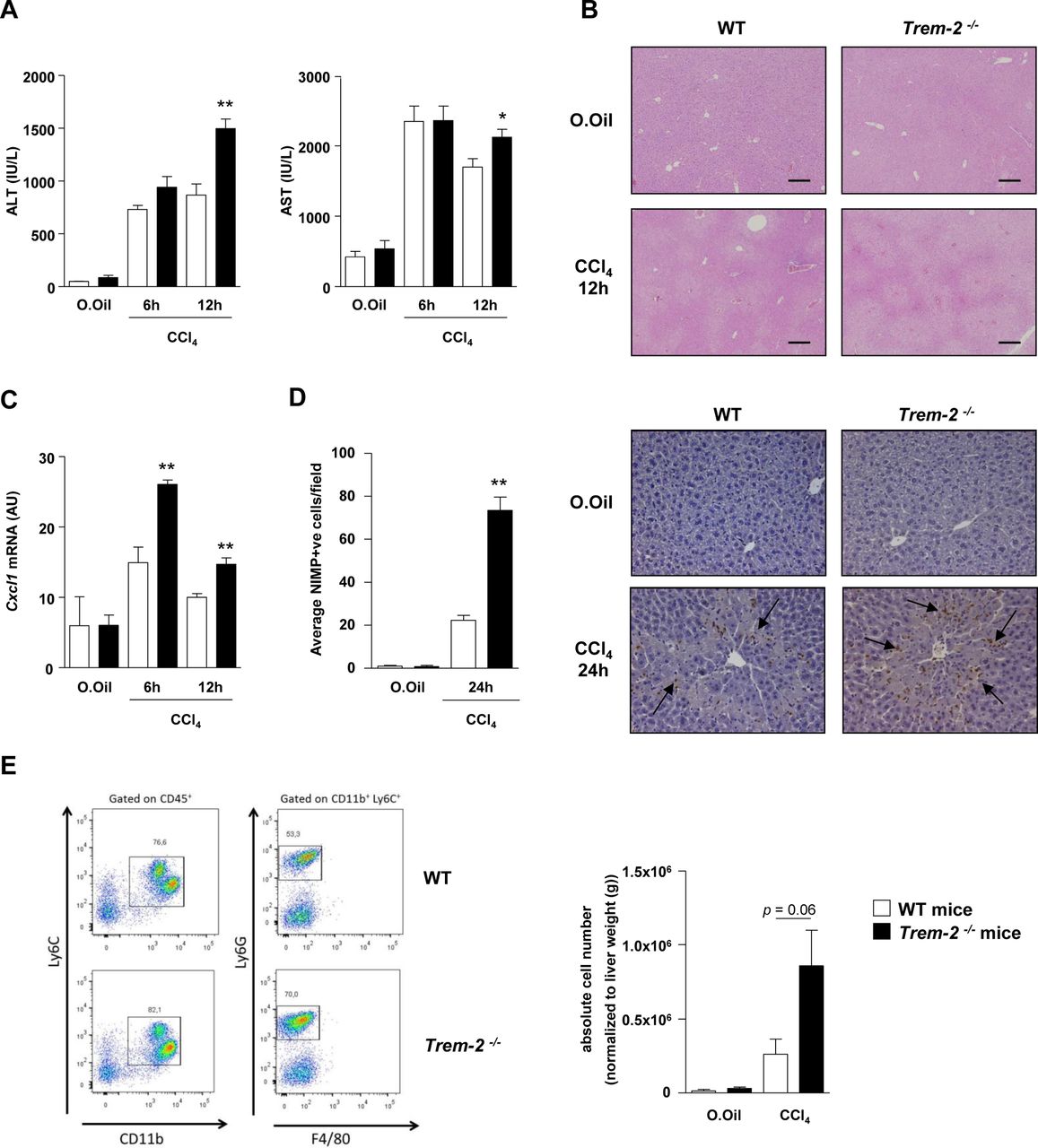
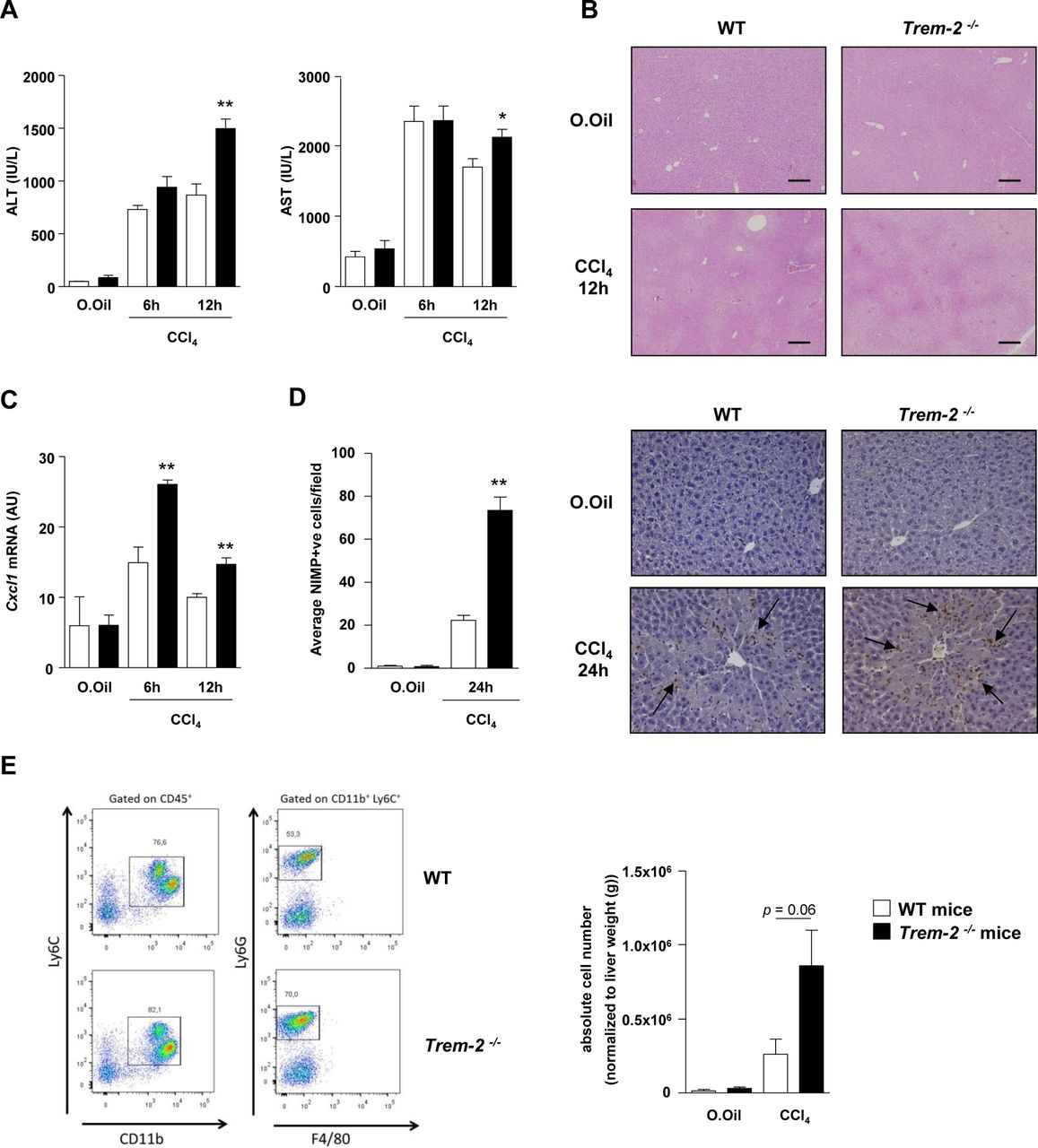
TREM-2 blunts acute CCl4 induced hepatic inflammation and injury. (A–C) Serum AST/ALT (A), representative H&E stains of liver (B) and qRT-PCR of liver Cxcl1 (C) postacute CCl4 treatment of WT and Trem-2-/- mice for the indicated times. (D) Representative images of liver sections immunostained for neutrophils (anti-neutrophil marker (NIMP)) of WT and Trem-2-/- mice 24 hours after acute CCl4 treatment. Arrows denote positively stained cells. Manual counts for NIMP-positive cells in livers post-CCl4 treatment are depicted. (E) Representative flow cytometry plot of hepatic neutrophils of WT and Trem-2-/- mice 24 hours postacute CCl4 treatment. Total number of neutrophils normalised to liver weight are indicated. All data represent mean±SEM and *, ** denote a P value of ˂0.05 and ˂0.01, respectively versus WT at the same time point. n=5 mice per condition (A–C), three mice per condition (D) and n=6–10 mice per condition and are pooled data from two independent experiments (E). Statistical analysis used was unpaired Student’s t-test. Scale bar in (B) indicates 200 µm. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CCl4, carbon tetrachloride; Cxcl1, C-X-C motif chemokine ligand 1; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
KC and HSC expressed TREM-2 dampens TLR4-mediated inflammation
We next focused on the effects of deletion of TREM-2 on the inflammatory properties of resident KCs and HSCs (figure 2) following TLR activation. While there was no effect of TREM-2 deficiency within KCs on LPS-driven Mcp1 transcription (online supplementary figure 7), LPS stimulated TREM-2 deficient KCs displayed increased induction of Il6, Tnf, Il1b and C-X-C motif chemokine ligand 1 (Cxcl1), as early as 1 hour post-treatment (figure 5A). As dysbiosis of gut bacteria is important in CCl4-induced liver injury and inflammation,22 23 we next set to more closely mimic this scenario by treating KCs with the intestinal bacterium Escherichia coli (E. coli). E. coli treatment also resulted in increased proinflammatory cytokine transcription in Trem-2 -/- KCs compared with WT (figure 5B). ELISA data verified that lack of TREM-2 enhanced the induction of inflammatory mediators following challenge with either LPS or E. coli (online supplementary figure 8). As we had observed increased Mcp1 production in vivo following chronic injury (figures 3 and 4), but no effect of Trem-2 expression within KCs on LPS-driven induced MCP-1 production (online supplementary figure 7), we speculated that HSCs might be a source for MCP-1 production in vivo. Indeed, examining TLR4-driven inflammation in both genotypes of activated HSCs revealed that LPS and E. coli treatment resulted in augmented Cxcl1 and MCP-1 levels in activated Trem-2-/- HSC versus WT (figure 5C,D). Augmented inflammation in Trem-2-/- opposed to WT HSC was specific for TLR4-dependent signalling as LPS but not interleukin (IL)-1β treatment resulted in enhanced inflammation (online supplementary figure 9A). While IL-1β and TNF-α treatment of KCs was in general less proinflammatory compared with LPS, TREM-2 deficiency only led to augmented inflammation in response to LPS, further suggesting that this receptor does not impinge directly on TNF-α and IL-1β signalling cascades in non-parenchymal cells and ratifying specificity for TLR4-mediated signalling (online supplementary figure 9B). To corroborate that TREM-2 could dampen TLR4 responses within HSCs, we next performed gain of function experiments. TREM-2 overexpression dampened LPS-induced MCP-1 and IL-6 levels within human LX2 HSCs (figure 5E). We conclude that TREM-2 attenuates TLR4-mediated inflammatory responses of both KC and HSC.
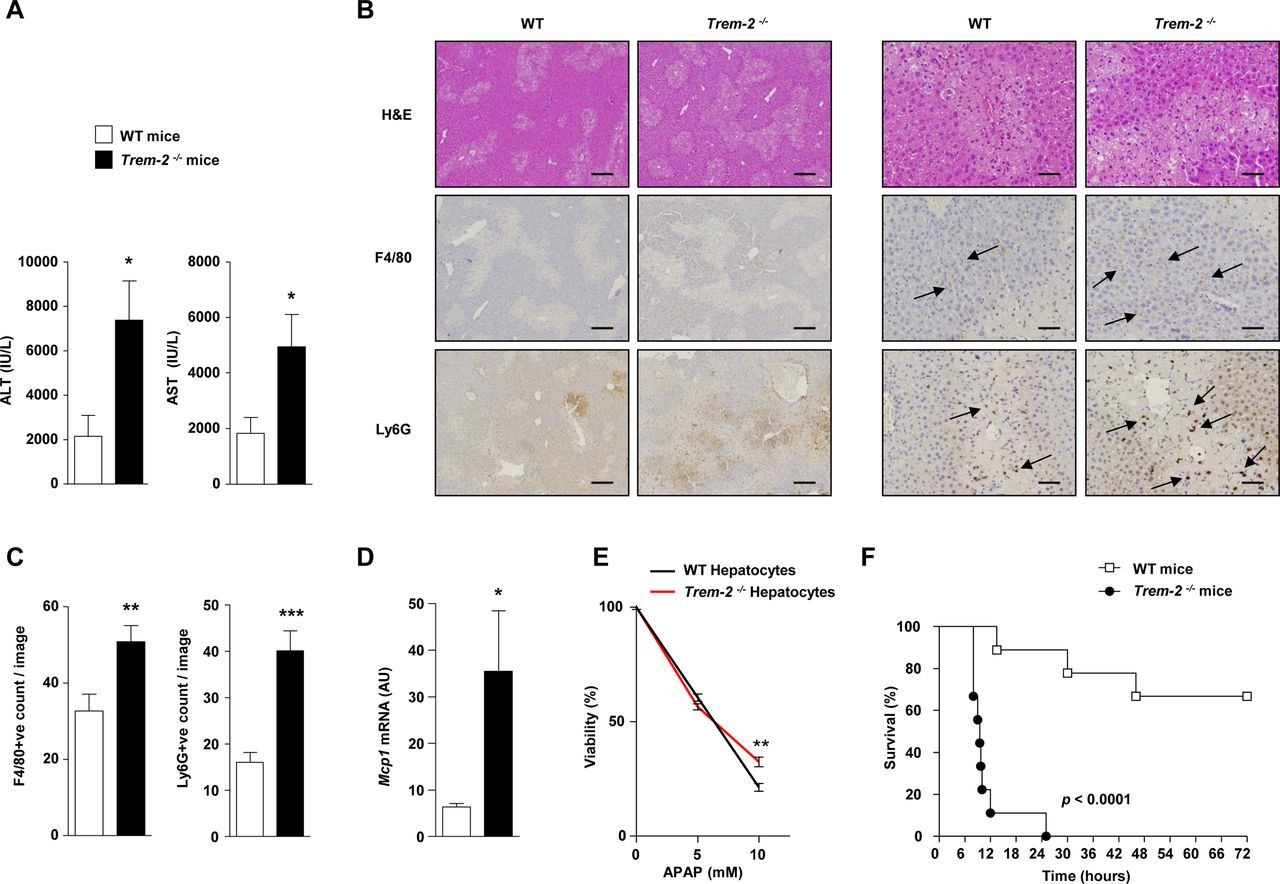
TREM-2 dampens acetaminophen-induced liver injury. (A) WT and Trem-2-/- mice were injected with 300 mg/kg APAP and 24 hours postinjury ALT and AST levels were determined. (B) Representative H&E, F4/80 and Ly-6G stains from the groups of mice are depicted. Trem-2-/- mouse liver exhibit more extensive parenchymal necrosis compared with WT liver. Arrows denote positively stained cells. (C) Manual counts for F4/80 and Ly6G positive cells in livers post-APAP treatment. (D) WT and Trem-2-/- mice were injected with 500 mg/kg APAP, and liver Mcp1 levels were determined. (E) WT and Trem-2-/- hepatocytes were treated with indicated doses of APAP for 24 hours and cellular viability evaluated. (F) WT and Trem-2-/- mice were injected with 750 mg/kg APAP and survival was monitored. Data represent mean±SEM and *, **, ***, **** denote a P value of ˂0.05, ˂0.01, ˂0.001 and <0.0001, respectively versus WT. n=5 mice per genotype (A–D), 6 hepatocytes per genotype (E) or 9 mice per genotype (F). Statistical analysis used was unpaired Student’s t-test (A–E) and Log-rank (Mantel-Cox) test (F). Scale bar in (B) indicates 50 µm (left panel) or 200 µm (right panel). Data in (E) are representative of two independent experiments. ALT, alanine aminotransferase; AST, aspartate aminotransferase; APAP, acetaminophen; TREM, triggering receptor expressed on myeloid cells; WT, wild type.

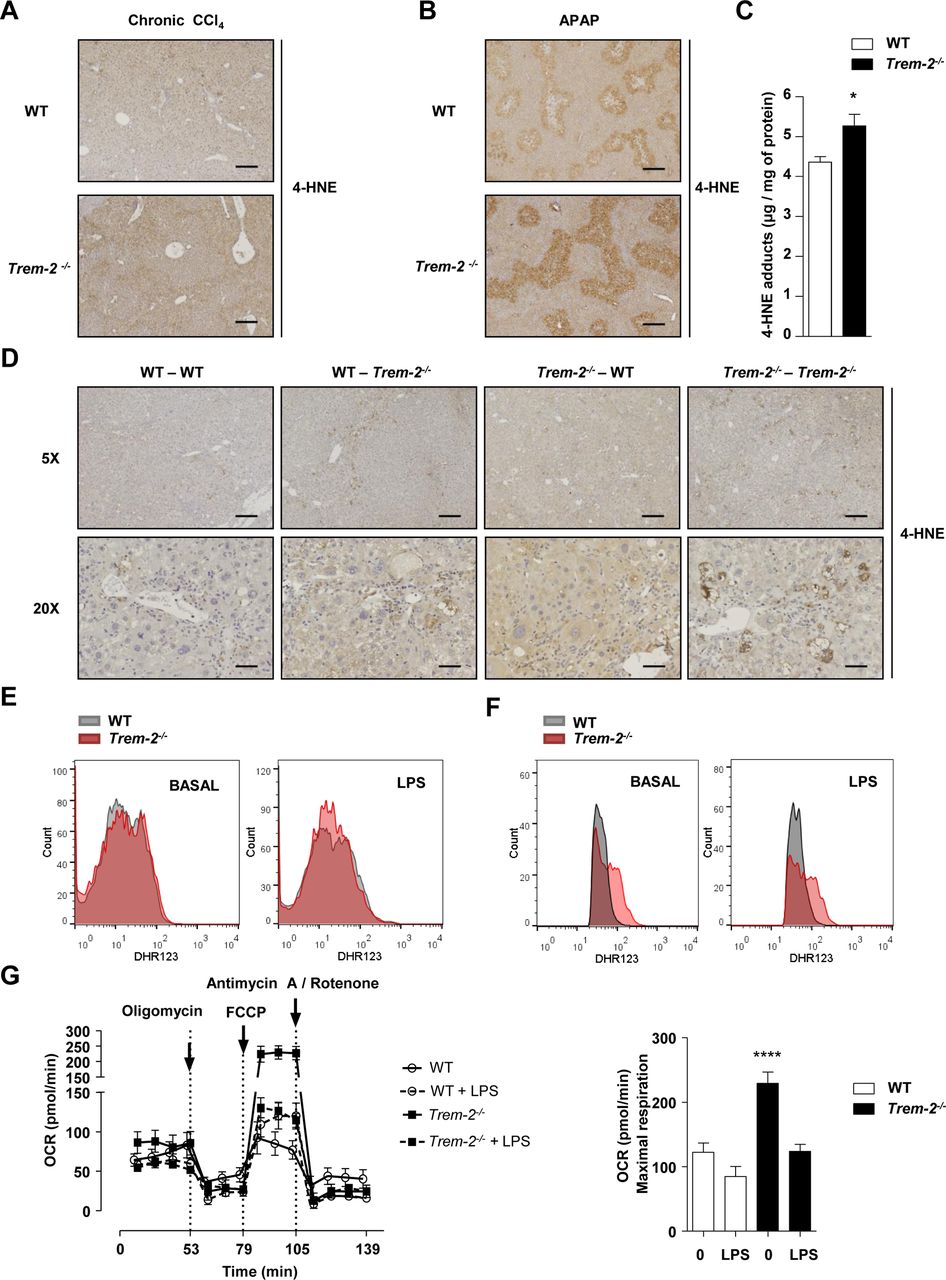
TREM-2 impacts hepatic lipid peroxidation and macrophage ROS levels. (A,B) Representative hepatic 4-HNE stain from WT and Trem-2-/- mice that were either treated with CCl4 for 8 weeks and sacrificed 1 day after the last CCl4 injection (A) or injected with 300 mg/kg APAP for 24 hours (B). n=3 mice per genotype (olive oil) or 4–8 mice per genotype (CCl4) or 5 mice per genotype (APAP). (C) 4-HNE content was determined by ELISA in livers of mice injected with 300 mg/kg APAP for 24 hours. n=5–7 per genotype. (D) Representative hepatic 4-HNE stain from WT mice reconstituted with WT-GFP+BM (WT-WT), Trem-2-/- mice reconstituted with Trem-2-/- -GFP+BM (Trem-2-/--Trem-2-/- ) or chimeric mice (WT-Trem-2-/- ) and (Trem-2-/- -WT) that were treated with CCl4 for 8 weeks and sacrificed 1 day after the last CCl4 injection. n=3 per genotype (olive oil) and 3–5 per genotype (CCl4). Scale bar 5× indicates 50 µm and 20× indicates 200 µm. (E,F) WT and Trem-2-/- KCs (E) or BMDM (F) were treated with 100 ng/mL LPS for 3 hour and total cellular ROS levels were determined using flow cytometry for dihydrorhodamine 123. n=3–4 per genotype and condition and a representative histogram is shown. (G) Oxygen consumption rate of naive and LPS-treated BMDM was evaluated. n=4–5 per condition. Data in (C,G) represent mean ±SEM and * and **** denote a P value of< 0.05 and <0.0001 versus WT. Data in (E) are representative of 3 and (F,G) of two independent experiments. Scale bar in (A,B) indicates 50 µm. APAP, acetaminophen; BMDM, bone marrow derived macrophages; CCl4, carbon tetrachloride; GFP, green fluorescent protein; 4-HNE, 4-hydroxynonenal; KCs, Kupffer cells; LPS, lipopolysaccharide; ROS, reactive oxygen species; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
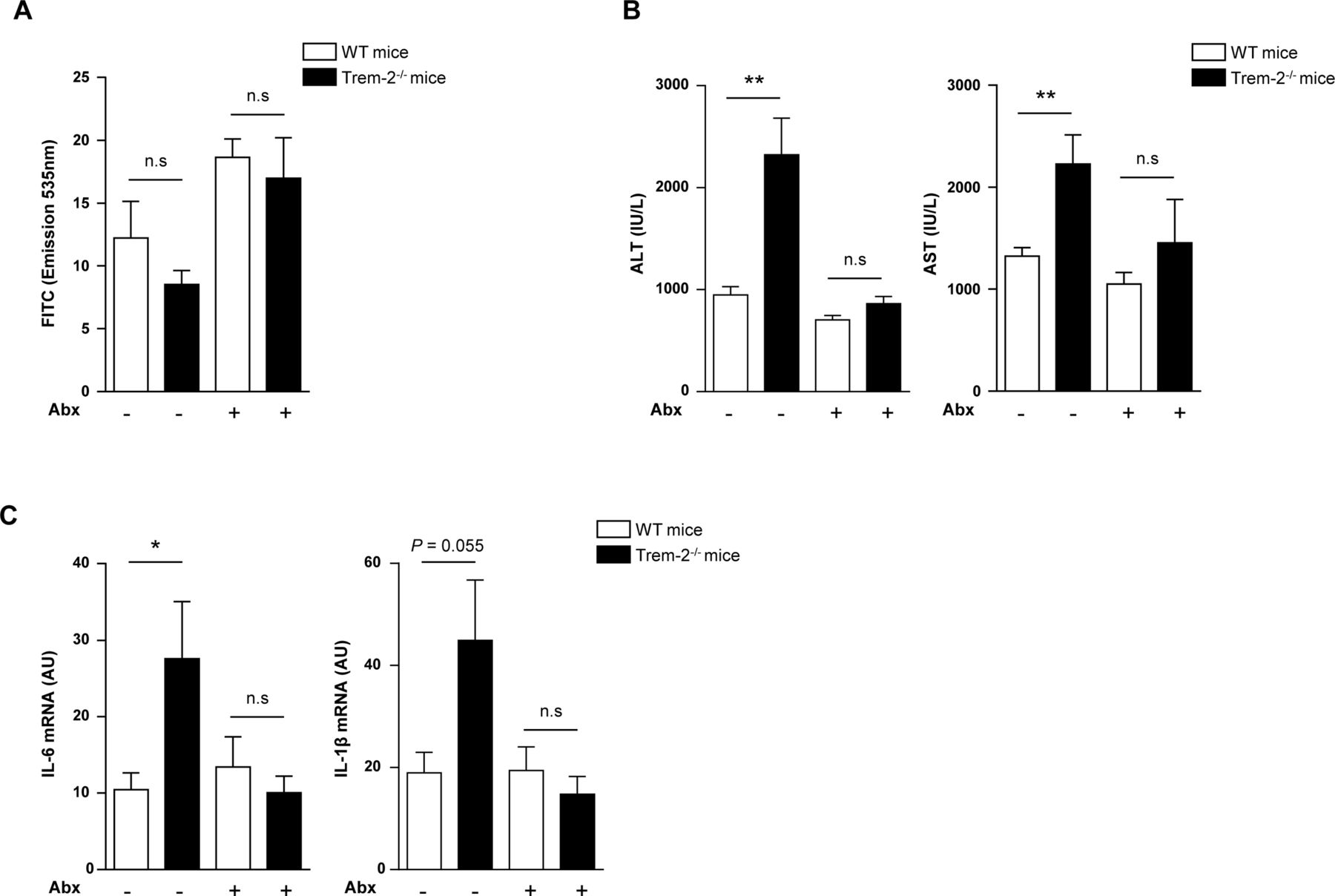
PAMPs are upstream of TREM-2 during liver injury (A–C) WT and Trem-2-/- mice that either received antibiotics or not in their drinking water for 4 weeks were injured acutely with CCl4 and 8 hours postinjection were orally gavaged with 4KDa-fluorescein isothiocyanate (FITC) Dextran and sacrificed 4 hours later. Serum levels of FITC Dextran (A), AST/ALT (B) and qRT-PCR of liver Il6 and Il1b (C) were determined. Data represent mean±SEM and *, ** denote a P value of ˂0.05 and ˂0.01, respectively versus the indicated genotype and condition (Student’s t-test). n=8–9 mice per genotype and condition. Abx, antibiotics; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CCl4, carbon tetrachloride; IL, interleukin; PAMPs, pathogen-associated molecular patterns; TREM, triggering receptor expressed on myeloid cells; WT, wild type.
TREM-2 has been reported to modulate MAPK signalling and phosphorylation including ERK, p38 and Jun N-terminal kinase (JNK) phosphorylation, which are early and critical signal transduction events in TLR4-induced cytokine production.11 24 25 We next examined effects of TREM-2 on TLR4-mediated MAPK activation. E. coli-induced phosphorylation of ERK and p38 was potentiated in Trem-2-/- KCs, while there was a minor decrease in JNK activation, compared with WT KCs (figure 5G). Whereas decreases in JNK activation were more pronounced in response to LPS in Trem-2-/- compared with WT KCs, LPS-induced ERK phosphorylation was similarly augmented in Trem2-/- KC, suggesting TREM-2 functions to dampen the activation of ERK downstream of TLR4 (figure 5F,G). However, we observed no effect of TREM-2 deletion on TLR4-mediated nuclear factor kappa B (NF-κB) activation (figure 5F,G). We conclude that within resident liver macrophages, TREM-2 dampening of TLR4-driven inflammation is associated with an early attenuation of MAPK phosphorylation, particularly ERK activation.
TREM-2 blunts acute CCl4 and APAP-induced liver inflammation and injury
Given the influence of TREM-2 on acute inflammatory responses of hepatic non-parenchymal cells, we determined if the receptor is functionally involved in controlling the response to acute toxic liver injury. We therefore performed two distinct acute liver injury models, administering mice with either a single dose of CCl4 or APAP overdose, the latter being a common cause of acute human liver failure associated with mitochondrial dysfunction, stress, lipid peroxidation and liver necrosis.26
Twelve hours postacute CCl4 injury, Trem-2-/- mice displayed elevated liver transaminases compared with WT, this effect was associated with elevated levels of the neutrophil chemoattractant Cxcl1 and increased hepatic neutrophil recruitment 24 hours postinjury, as determined using immunohistochemistry (figure 6A–D). By contrast, there were unremarkable effects of TREM-2 deletion on macrophage recruitment (data not shown). To confirm these data, we next isolated inflammatory cells from the livers of both genotypes of mice 24 hours post CCl4 injury and using flow cytometry again observed a strong tendency (P=0.06) for elevated hepatic neutrophil numbers in Trem2-/- mice compared with WT controls (figure 6E).
Similar to acute CCl4 injury, following sublethal APAP intoxication, Trem-2-/- mice displayed augmented transaminase levels, more extensive parenchymal necrosis and increased neutrophil content compared with WT (figure 7A–C). However, in this model, Trem-2-/- mice also displayed elevated macrophage content, which was associated with elevated levels of Mcp1 (figure 7B–D). These data suggested that enhanced pathogen-associated molecular pattern (PAMP)-driven inflammation from non-parenchymal liver cells in Trem-2-/- mice might be a driver for the elevated liver damage. To exclude a toxic effect of APAP on parenchymal cells, we isolated both genotypes of hepatocytes, treated them with APAP and evaluated cell viability. While APAP dose dependently killed hepatocytes, cell death was not elevated in TREM-2 deficient hepatocytes, in fact, surprisingly there was a small although statistically significant tendency that viability was increased (figure 7E). To further examine the consequences of the enhanced liver damage in Trem-2-/- mice, we administered both genotypes of mice with a lethal dose of APAP and monitored survival. Strikingly, while 70% of WT mice survived up until 72 hours, all Trem-2-/- mice died within 25 hours (figure 7F). We conclude that TREM-2 is critical for appropriate dampening of the acute liver damage response and its associated inflammatory reaction, with the immune receptor therefore being essential for preventing lethal APAP intoxication.
TREM-2 suppresses injury-induced hepatic ROS and lipid peroxidation
Previous observations from our laboratory indicate that TREM-2 can modulate peroxisome proliferator-activated receptor (PPAR) and nuclear receptor activities that regulate lipid-associated pathways.27 Inflammation, particularly that mediated by CCl4 and APAP intoxication is associated with enhanced lipid peroxidation, which can result in hepatocyte death.28–30 In search for a unified mechanism behind the elevated liver injury of Trem-2-/- mice following chronic CCl4 and acute APAP intoxication, we next monitored levels of 4-hydroxynonenal (4-HNE), a surrogate for lipid peroxidation.30 Both, at peak injury following chronic CCl4 treatment and APAP intoxication, elevated 4-HNE levels were observed in the livers of Trem-2-/- mice (figure 8A,B). To confirm these data, in an independent experiment, we next isolated livers from both genotypes of mice 24 hours post APAP treatment and could indeed observe significantly elevated 4-HNE levels as determined using ELISA (figure 8C). Examining 4-HNE staining revealed that lipid peroxidation appeared to be most intense around hepatic venules, where inflammatory cells reside, suggesting that elevated lipid peroxidation might occur through enhanced reactive oxygen species (ROS) levels within resident or infiltrating immune cells. Supporting this idea, elevated lipid peroxidation following chronic CCl4 treatment was only observed when TREM-2 was deficient within resident and infiltrating immune cells (figure 8D). As the ability of TREM-2 to dampen liver damage following chronic CCl4 treatment depended on TREM-2 expression in both resident and infiltrating cells (figure 4), these data further linked elevated lipid peroxidation to injury and additionally suggested that this effect could be associated with enhanced ROS levels within Trem-2-/- resident and liver infiltrating immune cells. However, examining ROS levels within both genotypes of KCs revealed similar levels in both basal and LPS-driven ROS (figure 8E). Considering increased lipid peroxidation in Trem-2-/- mice was associated with elevated MCP-1 and newly recruited monocyte derived hepatic macrophage levels, we next asked if elevated liver damage in Trem-2-/- mice could be associated with differential ROS levels within injury-associated inflammatory monocytes that repopulate the liver. We used BMDM to model this. Strikingly, Trem-2-/- BMDM exhibited higher baseline and LPS-induced ROS levels (figure 8F). Mitochondria via the process of oxidative phosphorylation and the NADPH oxidase complex produce ROS, so we next set out to validate these findings as well as determine the cellular source for the elevated ROS in Trem-2-/- macrophages. Intriguingly, when assessing mitochondrial activity, we observed that Trem-2-/- BMDM exhibited higher baseline oxidative phosphorylation and maximal respiration, suggesting that this metabolic signature might be responsible for the enhanced ROS (figure 8G). These data suggest that TREM-2 affects lipid peroxidation and subsequent damage of hepatocytes by skewing metabolic pathways and ROS generation within newly recruited liver macrophages following liver injury.
PAMPs are upstream of TREM-2 during liver injury
Thus far our observations indicated an importance for TREM-2 in dampening hepatic injury and inflammation. In an effort to demonstrate that the upstream driver for acute liver injury in TREM-2 deficient mice was PAMPs and associated TLR4 signalling downstream of gut bacteria, we administered both genotypes of mice antibiotics and evaluated liver injury and inflammation post-CCl4 treatment versus animals that did not receive antibiotics. Eight hours postacute CCl4 injection, we orally gavaged all groups of mice with 4 kDa FITC-Dextran, to evaluate intestinal permeability as previously described22 and sacrificed all animals 4 hours later, that is, we administered CCl4 for a total of 12 hours. Although there was a trend that antibiotic treatment increased intestinal permeability, serum FITC levels were unaltered between genotypes in either the presence or absence of antibiotics post-CCl4 treatment (figure 9A). Trem-2 -/- mice exhibited exaggerated transaminase release compared with WT following acute CCl4 treatment, as previously observed (figure 7A) and notably antibiotics abolished this (figure 9B). Evaluating transcript levels of Il1b and Il6, revealed that antibiotic treatment also reverted the augmented hepatic inflammation of Trem-2 -/- mice to WT levels (figure 9C). Together, these important data show that following acute liver injury, differences in altered intestinal permeability of TREM-2 deficient mice do not contribute to their exaggerated liver damage. They further support the concept that TREM-2 dampens PAMP-derived signals that emanate from gut bacteria which perpetuate hepatic injury and inflammation.
Discussion
Unresolved inflammation is a pathogenic component of liver disease and contributes to progressive liver damage and fibrosis. Altered intestinal permeability during liver injury results in leakage of PAMPs that are recognised by non-parenchymal liver cells and injury recruited cells. These cells release a variety of proinflammatory mediators including cytokines, chemokines, lipids and ROS that contribute to the demise of hepatocytes. Illustrating the importance of innate immunity, mice deficient in key mediators of LPS signalling such as TLR4, CD14 and MyD88 display better outcomes during liver disease.4 31 32 Here, we tested a hypothesised anti-inflammatory role for TREM-2 in liver disease.
We show that TREM-2 expression dramatically rises during various forms of liver injury in both mice and humans and that hepatic mouse TREM-2 is expressed on non-parenchymal KCs and HSCs. Nonetheless, we cannot be certain that in other species, including humans, TREM-2 is not expressed in hepatocytes during liver injury. Notably, although TREM-2 receptor expression has previously been detected on a variety of resident macrophages, including KCs,8 10 13 27 to our knowledge, this is the first study demonstrating TREM-2 expression on quiescent and activated HSCs, expanding on studies showing TREM-1 expression on this important immune and fibrogenic cell type.12 Further, we disclose that during HSC activation, TREM-2 is dramatically increased in a conserved manner and is upregulated within HSCs during chronic liver injury. However, TREM-2 exerted unremarkable effects on HSC activation in vitro and in the context of chronic liver injury, effects consistent with minor effects of TREM-2 on fibrosis and the critical fibrogenic mediator, TGFβ−1 during chronic injury.20 21 Further, as the net amount of collagen deposition is controlled by matrix metalloproteinases and their inhibitors, augmented Mmp13 levels observed in Trem-2- /- mice during repetitive injury could play a role in fibrosis resolution.33 Interestingly, although HSCs are not professional phagocytes, phagocytosis of apoptotic bodies by HSCs has been documented and postulated to promote HSC activation, survival, TGFβ−1 and collagen production.34–36 In some cell types, TREM-2 has been described to be important for the clearance of bacteria and apoptotic cells.11 16 27 37 Thus, defects in the uptake of apoptotic bodies by Trem-2- /- HSCs could additionally contribute to the minor effects of TREM-2 on fibrosis observed during chronic CCl4 injury. To summarise, our data indicate that TREM-2 primarily limits hepatocellular injury, a process that appears to be linked to its negative regulatory role during inflammation.
Similar to BM macrophages,8 TREM-2 dampened TLR4-driven inflammation in KCs and HSCs. Higher inflammation in Trem-2- /- KCs correlated with effects of TREM-2 on TLR4-induced ERK activation, consistent with studies demonstrating effects of TREM-2 on the ERK but not the NF-κB pathway.11 24 During acute CCl4-mediated injury, Trem-2-/- mice exhibited enhanced numbers of liver neutrophils compared with WT, which correlated with elevated hepatic levels of the neutrophil chemoattractant CXCL1 and liver damage.38 The ability of TREM-2 to dampen CXCL1 and neutrophil recruitment following acute liver injury is consistent with how pulmonary expressed TREM-2 reacts during early pneumococcal infection.27 Similarly, following acute APAP intoxication, augmented neutrophil influx was observed in Trem-2-/- livers. However, unlike acute CCl4 treatment hepatic macrophage content was also elevated in Trem-2-/- livers. Although the relative contribution of macrophages versus neutrophils to the enhanced liver injury of Trem-2-/- mice in the various models of liver injury remains to be determined, we importantly show that (1) TREM-2 dampens liver damage in all models tested; (2) lack of TREM-2 is associated with worsened survival outcome following APAP challenge; (3) gut-derived PAMPs are a driver for acute liver injury of TREM-2 deficient mice.
In terms of mechanistic parallels between APAP intoxication and chronic CCl4 injury, we observed significantly enhanced Mcp1 levels in Trem-2- /- versus WT livers in both models, which was associated with more damage, macrophage levels and lipid peroxidation. Although resident KCs have a prenatal origin and are generated from the yolk sac and/or fetal liver, during liver injury, circulating Ly6C+ monocytes are recruited to the injured liver in a MCP-1/CCR-2 dependent manner, where they can contribute to injury.39–41 Indeed, it has recently been shown that hepatic infiltrating monocyte derived macrophages exert proinflammatory functions early after APAP and thereby promote liver injury. Consequently, reducing monocyte infiltration via MCP-1/CCR2 inhibition attenuates APAP-induced liver injury.42 Together, these data indicate that during acute toxic hepatic injury, the recruitment of monocytes to the liver perpetuates parenchymal damage, a hypothesis that is supported by studies indicating that KCs play a protective role in APAP-induced injury.43 We now add to this knowledge by highlighting the importance of TREM-2 signalling for suppressing recruitment of extrahepatic macrophages in toxic liver injury.
Our data suggest that TREM-2 dampens TLR4-dependent MCP-1 production within HSCs, impacting hepatic macrophage recruitment. Furthermore, TREM-2 expressed on these infiltrating macrophages blunts ROS production, thus limiting ROS-mediated hepatocellular lipid peroxidation and cell death. Effects of TREM-2 on ROS within infiltrating macrophages alone appears insufficient to dampen liver damage, as WT mice reconstituted with Trem-2-/- BM do not display more liver damage than WT mice with WT BM. Rather, our data suggest that TREM-2 effects on TLR4-dependent inflammatory events in HSCs is critical for setting the stage for hepatic macrophage recruitment, a process influenced by HSC-derived MCP-1 and by ROS and cytokine production from infiltrating immune cells. These observations are concordant with data demonstrating that liver steatosis sensitises to CCl4 hepatotoxicity, in part through oxidative stress.44 Supporting a cross-talk between infiltrating monocytes and HSCs in liver injury, purified Cd11b+F4/80+Ly6C+ cells derived from CCl4 treated mice can directly activate HSCs, promoting hepatocellular damage.45 In this regard, it is interesting that infiltrating inflammatory Ly6C+ macrophages downregulate Ly6C on uptake of apoptotic hepatocytes and this is associated with a tissue protective phenotype following alcoholic-induced liver injury.46 Although the exact nature of infiltrating macrophages during injury in Trem-2- /- mice in the models of liver injury requires further examination, our data, along with published observations, suggest they could be important for TREM-2 to limit hepatocellular damage following APAP intoxication and chronic CCl4 injury. Hepatocyte death would presumably release a variety of danger-associated molecular patterns, including heat-shock proteins, purine metabolites and mitochondrial DNA that can further provoke a vicious cycle of inflammation, macrophage recruitment and cell death.28 Such a hepatotoxic feed forward loop could be present and elevated in Trem-2- /- livers during chronic injury and APAP intoxication. Further, effects on TREM-2 on liver lipid metabolism are reminiscent with very recent studies indicating TREM-2 alters brain lipid metabolism and purogenic signalling in the context of cuprizone-induced oligodendrocyte injury and that TREM-2 can sense lipids.17 47 Highlighting the importance of TREM-2 in lipid metabolism, recent data indicate that TREM-2 promotes adipogenesis and in vivo high fat diet fed mice overexpressing TREM-2 were more obese and developed elevated hepatic steatosis versus controls.48 Although the authors of the aforementioned study did not perform an extensive analysis of TREM-2’s function in hepatic injury in this context, these data suggest TREM-2’s role in hepatocellular injury during non-alcoholic fatty liver disease may be distinct and complex, involving not just its immune but also adipogenic and lipid regulatory functions.47 48
This is the first study demonstrating a role for TREM-2 in toxic liver injury and is reminiscent of published effects of TREM-2 in injury responses in other organ and tissue systems. For example, in the colon, TREM-2 has been reported to be important in wound-healing.15 In the brain, TREM-2 functions to clear apoptotic neurons and limit microglial proinflammatory responses.11 16 Our work suggests that TREM-2 could be an attractive target for promoting the resolution of inflammation during liver injury and for the prevention of parenchymal cell death, this akin to events described for TREM-2 in neuroinflammatory diseases and sepsis.16 49
Acknowledgments
The authors thank the staff of the animal facilities at the Medical University of Vienna and the Biodonostia Research Institute for technical assistance. We also thank Anastasiya Hladik for help with bone marrow transplantation.
References
Footnotes
OS and DAM contributed equally.
Contributors MJP, FO, AE-B, IL, AK, AJ, JM, DT, AS-L, AA, RG, AS, AF, HE, RJ-A, HE, DS, LB, JMB and OS performed experiments. MJP, SK, OS and DAM discussed and interpreted the data. MJP, OS and DAM wrote the manuscript. OS and DAM share senior authorship.
Funding The present study was supported by grants from the Wellcome Trust (grant WT086755MA), European Commission FP7 program grant ’INFLA-CARE' (EC Contract No. 223151;), the Medical Research Council, UK (grant MR/K001949/1) and Cancer Research UK Programme Grant C18342/A2390. to DAM. The study was also funded by the Basque Government’s Department of Industry, Innovation, Commerce and Tourism, SAIOTEK Programme grant (SAIO13-PE13BN010) and the Spanish Ministry of Economy and Competitiveness, Instituto de Salud Carlos III grant (PI14/00399), both to MJP. MJP was funded by IKERBASQUE, Basque foundation for Science and the Ministry of Economy and Competitiveness, Ramón y Cajal Programme (RYC-2015-17755). AE-B was funded by the University of the Basque Country (UPV/EHU; PIF2014/11). IL was funded by the Department of Education, Language Policy and Culture of the Basque Government (PRE_2016_1_0152). RG was funded by the Austrian Science Fund (FWF; APW01205FW, within the Doctoral Program Cell Communication in Health and Disease, CCHD; to SK). OS and SK were funded by the Austrian Science Fund (OS, FWF-25801; SK, Grant I 289-B09).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Clinical Research Ethics Committees of the Basque Country and Donostia Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.