Article Text

Abstract
Objectives Bacteriome and virome alterations are associated with colorectal cancer (CRC). Nevertheless, the gut fungal microbiota in CRC remains largely unexplored. We aimed to characterise enteric mycobiome in CRC.
Design Faecal shotgun metagenomic sequences of 184 patients with CRC, 197 patients with adenoma and 204 control subjects from Hong Kong were analysed (discovery cohort: 73 patients with CRC and 92 control subjects; validation cohort: 111 patients with CRC, 197 patients with adenoma and 112 controls from Hong Kong). CRC-associated fungal markers and ecological changes were also validated in additional independent cohorts of 90 patients with CRC, 42 patients with adenoma and 66 control subjects of published repository sequences from Germany and France. Assignment of taxonomies was performed by exact k-mer alignment against an integrated microbial reference genome database.
Results Principal component analysis revealed separate clusters for CRC and control (p<0.0001), with distinct mycobiomes in early-stage and late-stage CRC (p=0.0048). Basidiomycota:Ascomycota ratio was higher in CRC (p=0.0042), with increase in Malasseziomycetes (p<0.0001) and decrease in Saccharomycetes (p<0.0001) and Pneumocystidomycetes (p=0.0017). Abundances of 14 fungal biomarkers distinguished CRC from controls with an area under the receiver-operating characteristic curve (AUC) of 0.93 and validated AUCs of 0.82 and 0.74 in independent Chinese cohort V1 and European cohort V2, respectively. Further ecological analysis revealed higher numbers of co-occurring fungal intrakingdom and co-exclusive bacterial–fungal correlations in CRC (p<0.0001). Moreover, co-occurrence interactions between fungi and bacteria, mostly contributed by fungal Ascomycota and bacterial Proteobacteria in control, were reverted to co-exclusive interplay in CRC (p=0.00045).
Conclusions This study revealed CRC-associated mycobiome dysbiosis characterised by altered fungal composition and ecology, signifying that the gut mycobiome might play a role in CRC.
- colorectal cancer
- fungi
- gut microbiota
- pathogen
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Donor gut microbiota from patients with colorectal cancer (CRC) has been shown to induce tumourigenesis in germ-deficient mice models.
Bacteriome and virome alterations are associated with CRC.
The involvement of gut fungal microbiota in CRC is largely unexplored.
What are the new findings?
We identified CRC-associated faecal fungal dysbiosis with increased Basidiomycota:Ascomycota ratio in patients with CRC compared with healthy subjects.
Fungal class Malasseziomycetes was enriched in CRC while classes Saccharomycetes and Pneumocystidomycetes were depleted.
Abundances of 14 fungal biomarkers distinguished CRC from controls with an area under the receiver-operating characteristic curve (AUC) of 0.93 and validated AUCs of 0.82 and 0.74 in two independent cohorts.
Ecological analysis revealed a higher number of co-occurring fungal intrakingdom correlations and more co-exclusive correlations between fungi and bacteria in CRC compared with control indicating that synergistic intrafungal and antagonistic bacterial–fungal ecological associations may play role in colorectal carcinogenesis.
How might it impact on clinical practice in the foreseeable future?
Our analysis highlights the role of fungi in colorectal tumourigenesis and the potential use of fungi faecal markers in diagnosing CRC.
Introduction
Gut microbiota is a complex ecosystem that plays an important role in the pathogenesis of colorectal cancer (CRC), a leading cause of cancer-related deaths worldwide.1 2 Nevertheless, the role of microbial components other than gut bacteria, such as fungi, is largely unexplored in CRC, partly due to their relatively lower abundance and lack of well-characterised reference genomes.3 4 As a result, the understanding of their contributions towards colorectal carcinogenesis is limited. Increased capacity of high-throughput sequencing methods has provided access to non-bacterial components of the gut microbiota. In this regard, we recently reported on virome signatures associated with CRC based on shotgun metagenomic analyses.5 Although the fungal composition of the mammalian microbiota was estimated to comprise less than 1% of all commensal microbial species by genomic equivalence,4 emerging studies have demonstrated its importance in the gut.6–11 It assumes a stable role in the development and maintenance of host immune system, but can be altered in both intestinal and extraintestinal diseases.6–11 Thus, investigation into enteric mycobiome in patients with CRC as compared with healthy subjects is worthwhile and would provide greater understanding of yet unknown mechanisms by which the gut microbiota contributes to colon tumourigenesis beyond the bacterial kingdom.
Distinct gut fungal communities and diversities were recently described as being associated with IBDs and liver cirrhosis in humans.8 9 12 Moreover, the potential of commensal fungi to functionally replace intestinal bacteria in maintaining host immune responses in the gut by reversing disease susceptibility induced by bacterial depletion has been demonstrated in a mouse model.13 Consistently, a recent study identified fungal compositional differences in tissue biopsies of 27 subjects with colorectal adenoma and adjacent tissues,6 implicating altered host-associated fungal populations in the development of CRC. Understanding mycobial diversity and abundance in CRC compared with healthy subjects is therefore important to elucidate the potential contribution of fungal species in colorectal tumourigenesis. Importantly, faecal fungal dysbiosis might open up new opportunities for using fungal species as non-invasive diagnostic biomarkers for CRC.
Here, we characterised CRC-associated mycobiome by analysing faecal metagenomic sequences of 73 patients with CRC and 92 healthy subjects with validation in two independent cohorts of a total of 201 patients with CRC, 239 patients with adenoma and 178 controls. For the first time, we show that faecal mycobiome profile is predictive of CRC, independent of bacterial and other clinical parameters. Additionally, ecological co-occurrence network analysis demonstrates CRC-specific fungal intrakingdom and fungi–bacteria interactions.
Materials and methods
Patient cohorts
We analysed faecal shotgun metagenomic sequences from 585 individuals comprising faecal shotgun metagenomic sequences of 184 patients with CRC, 197 patients with adenoma and 204 control subjects from Hong Kong (discovery cohort: 73 patients with CRC and 92 control subjects; independent Chinese validation cohort V1: 111 patients with CRC, 197 patients with adenoma and 112 controls from Hong Kong) who had undergone standard colonoscopy examinations at the Prince of Wales Hospital at the Chinese University of Hong Kong.14 Presentations of digestive symptoms to the outpatient gastroenterology clinics and asymptomatic individuals ages over 50 years were the recruitment criteria. All CRC subjects had intact colonic lesions as at the time of stool collection. Stool samples were collected and stored at −20°C within 4 hours and −80°C within 24 hours for long-term storage. Qiagen QIAmp DNA Stool Mini Kit (Qiagen) was used for DNA extraction according to the manufacturers’ instructions. The diagnostic potential of our fungal markers was further demonstrated in faecal metagenomics dataset comprising 90 CRC, 42 adenoma and 66 healthy subjects from Germany and France designated as European cohort V2.15 The demographic and clinical details of all samples are shown in table 1.
Demographic and clinical details of samples
Metagenomics sequencing
Whole-genome shotgun sequencing of Chinese discovery and validation samples were carried out on Illumina HiSeq 2000 (Illumina, San Diego, California, USA), and sequences from Germany and France were sequenced on HiSeq 2000/2500 platform (Illumina, San Diego, California, USA),15 which are available on public repository. Briefly, bases with Phred quality of less than 3, reads with average of quality less than 15 for every 4 bases and reads with lengths less than 36 bases were discarded using Trimmomatic software (V.0.36).16 Any adapter sequences in paired-end reads were clipped by checking for simple and palindromic matches of 10 and 30 bases with a library of universal Illumina Nextera and Truseq adapter sequences if an adapter-read pair had a mismatch count of 2 or less. Mammalian genomes (hg38, felCat8, canFam3, mm10, rn6, susScr3, galGal4 and bosTau8; UCSC Genome Browser)17 and 6877 bacterial plasmids (NCBI RefSeq database accessed on 12 May 2016), 2116 complete plastomes (NCBI RefSeq database accessed on 12 May 2016) and 6093 UNiVec sequences (NCBI RefSeq database accessed on 12 May 2016), which are potential habitat-associated/laboratory-associated or extrachromosomal sequence contaminants, were removed after alignment using Bowtie2 V.2.2.9 with ‘very-sensitive’ default settings.
Sequence taxonomic annotation
Taxonomy was assigned to metagenomic reads using k-mer-based algorithms implemented in Kraken taxonomic annotation pipeline.18 A custom database comprising 7505 bacterial and 1083 fungal reference genomes from NCBI (http://www.ncbi.nlm.nih.gov), FungiDB (http://fungidb.org), Ensemble (http://fungi.ensembl.org) and Broad Institute (http://broadinstitute.org) (accessed on 22 October 2017) was built using Jellyfish program by counting distinct 31-mer in the reference libraries, with each k-mer in a read mapped to the lowest common ancestor of all reference genomes with exact k-mer matches. Each query was thereafter classified to a taxon with the highest total hits of k-mer matched by pruning the general taxonomic trees affiliated with mapped genomes. The final metagenomic read counts were normalised by cumulative sum scaling method using metagenomeSeq R/Bioconductor package.19
Statistical analysis
MaAsLin (multivariate association with linear models), a multivariate statistical framework,20 was used to identify associations between clinical metadata and microbial abundance while controlling for confounders, namely age, gender and Body Mass Index (BMI). Co-occurrence and co-exclusion relationships within fungal and between fungal and bacterial kingdoms were estimated using SparCC algorithm,21 known for its robustness to gut microbiome data compositionality. P values less than 0.05 after multiple comparisons correction using false discovery rate (FDR) method were considered significant. The inclusion criterion for plotting features was having a minimum absolute magnitude of interkingdom or intrakingdom correlation of 0.6 and FDR of less than 0.05 in either one or both of control and CRC. For visualisation, correlations with absolute coefficients below 0.3 were masked to show all signals that are exclusively significant. Taxonomies were sorted by the levels of phylum and each phylum was clustered separately by classes. Areas under receiver-operating characteristic curves (AUCs) were determined with standard linear regression using one-standard error rule method, and adjusted for age, gender and BMI. Two-tailed Mann-Whitney U test was used to determine statistically significant differences between case and control.
Results
Enteric fungal dysbiosis is associated with CRC
The discovery cohort comprised 165 subjects, namely 73 patients with CRC and 92 healthy control subjects (table 1). About 10.34% of reads were filtered out by Trimmomatic trimming and contaminant removal steps leaving an average of 27 640 420 high-quality paired reads per sample for microbial taxonomic classification (online supplementary table 1). To validate the quality of our reference database, we analysed alpha and beta diversities of bacterial fraction of the microbiota. In agreement with our previous study,14 alpha diversity indices were significantly reduced in CRC compared with control while beta diversity analysis showed separate clusters for CRC and control (p<0.005, online supplementary figure 1). Rarefaction plots reached plateau for fungal species in both CRC and control samples (online supplementary figure 2). We then assessed and compared the enteric fungal composition in our cohort. Both unsupervised principal component analysis and supervised redundancy analysis (RDA) revealed that the gut mycobiome discriminated CRC and control into two significantly distinct groups (p=0.0006 and p=0.001, respectively; figure 1A,B), indicating that CRC exhibits unique fungal profiles from healthy subjects. Interestingly, RDA also revealed that the fungal components formed different clusters for control, early-stage (TNM stage I and II) CRC and late-stage (TNM stage III and IV) CRC, depicting a CRC-stage-specific disruption in mycobiome homeostasis (p=0.001; figure 1C). The fungal alpha diversities as measured by species richness and Shannon diversity, however, were not significantly different between CRC and control (figure 1D), unlike the observation with bacteria. Taken together, these results indicate alterations in gut fungal composition in association with colorectal carcinogenesis.
Supplemental material

Alteration of enteric fungi in colorectal cancer (CRC). (A) Unsupervised principal component analysis of Bray-Curtis distance showing the stratification of CRC from control samples by fungi compositional profile. Control, n=92; CRC, n=73. PC1 and PC2 represent the top two principal coordinates that reflect most of the diversity, given as a percentage. Groups were compared using Mann-Whitney U test. (B) Supervised multivariate redundancy analysis of fungi compositional profile stratifying patients with CRC (n=73) from control (n=92). RDA1 and RDA2 represent the top two coordinates that reflect most of the diversity, given as a percentage. Groups were compared using Mann-Whitney U test. (C) Stage-specific enteric fungal compositional multivariate redundancy analysis in control, n=92; Early_CRC, n=20; Late_CRC, n=54. RDA1 and RDA2 represent the top two coordinates that reflect most of the diversity, given as a percentage. Groups were compared using Kruskal-Wallis test. (D) Richness and Shannon Index diversity indices between CRC (n=73) and control (n=92).
Taxonomic alterations of enteric fungi in CRC
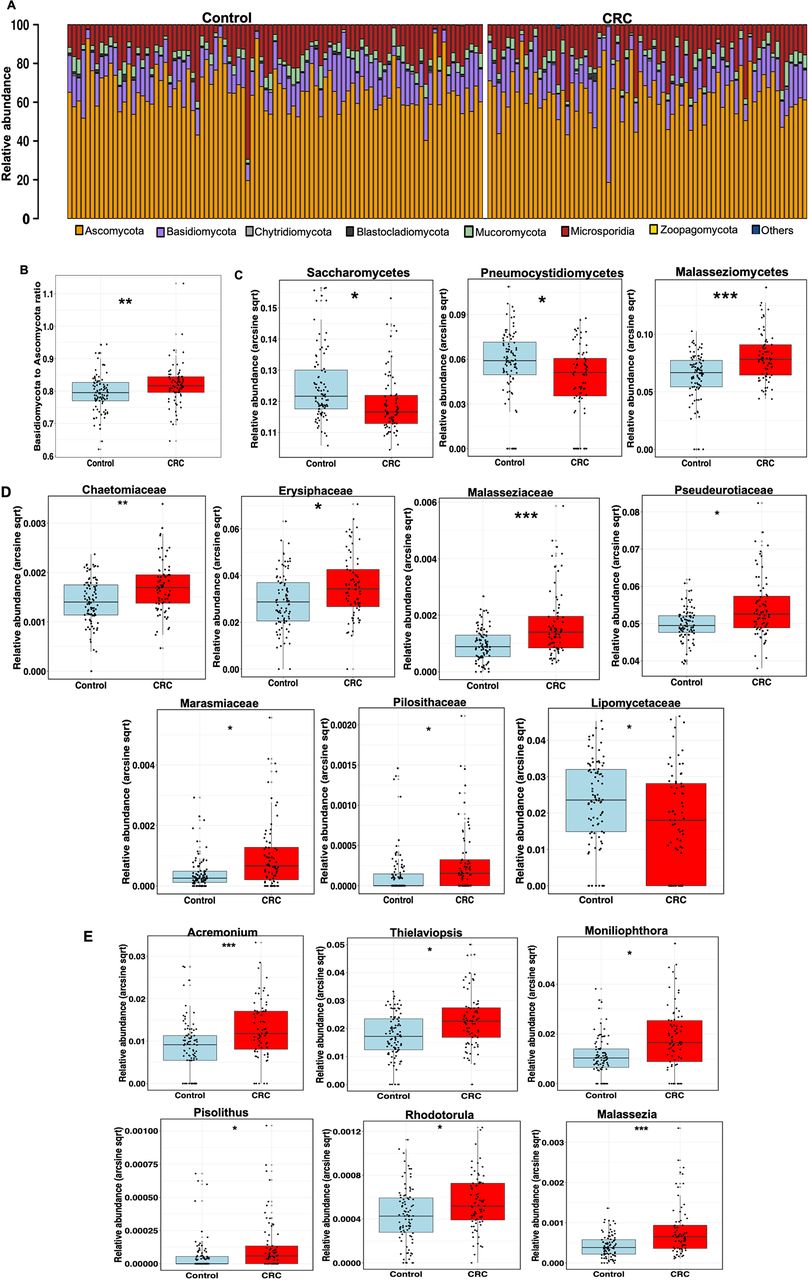
Consistent with previous studies and as a validation for our analysis, we observed enrichment and depletion of bacteria phyla Fusobacteria and Firmicutes, respectively, in CRC compared with control (online supplementary figure 1). Among the fungal taxa, the phylum Ascomycota dominated the mycobiota, while Basidiomycota was observed as the second most abundant phylum (figure 2A) in both case and control groups. Moreover, the Basidiomycota:Ascomycota ratio, which reflects fungal dysbiosis,12 was skewed with higher values in CRC than control (figure 2B, p=0.0042). Additional differences were observed at lower taxonomic levels, including significant depletion of Saccharomycetes and Pneumocystidomycetes classes and enrichment of Malasseziomycetes class in CRC compared with control (figure 2C, online supplementary table 2, supplementary data). At the family level, Lipomycetaceae was significantly reduced in CRC while Pisolithaceae, Marasmiaceae, Malasseziaceae, Erysiphaceae, Pseudorotiaceae and Chaetomicaeae were increased (figure 2D, online supplementary table 3). Furthermore, at the genus level, six fungal features were enriched in CRC, including Malassezia, Moniliophthtora, Rhodotorula, Acremonium, Thielaviopsis and Pisolithus (figure 2E, online supplementary table 4).
Supplemental material
Supplemental material
Supplemental material
Supplemental material

Taxonomic distribution of enteric fungi in colorectal cancer (CRC). (A) Relative abundance of dominant enteric fungi phyla in control, n=92 and CRC, n=73. The dominant phyla are Ascomycota, Basidiomycota and Mucoromycota in both groups. (B) Basidiomycota:Ascomycota relative abundance ratio, an index of fungal dysbiosis, is higher in CRC, n=73 than control, n=92 (**p<0.001, Mann-Whitney U test). (C) Differentially abundant fungal classes between CRC (n=73) and control (n=92). Differences in abundance were detected using multivariate statistical model and corrected for confounders including age, Body Mass Index (BMI) and gender (*Q<0.20, **Q<0.10, ***Q<0.05, arcsin-sqrt transformation of relative abundance are derivatives of MaAsLin). (D) Differentially abundant fungal families between CRC, n=73 and control, n=92. Differences in abundance were detected using multivariate statistical model and corrected for confounders including age, BMI and gender (*Q<0.20, **Q<0.10, ***Q<0.05, arcsin-sqrt transformation of relative abundance are derivatives of MaAsLin). (E) Differentially abundant fungal genera between CRC, n=73 and control, n=92. Differences in abundance were detected using multivariate statistical model and corrected for confounders including age, BMI and gender (*Q<0.20, **Q<0.10, ***Q<0.05, arcsin-sqrt transformation of relative abundance are derivatives of MaAsLin).
Fungal species as CRC diagnostic markers
To identify gut fungal taxa with potential value for CRC diagnosis, we investigated fungal alterations at the species level. After correction for confounders, namely age, gender and BMI, the abundances of 38 fungal species were observed to be significantly different in CRC when compared with control. Twenty-nine of these were CRC-enriched while nine others were CRC-depleted (figure 3A, online supplementary table 5). The diagnostic potential of species with greater than twofold abundance changes, including Aspergillus flavus, Kwoniella mangrovensis, Pseudogymnoascus sp. VKM F-4518, Debaryomyces fabryi, A. sydowii, Moniliophthora perniciosa, K. heavenensis, A. ochraceoroseus, Talaromyces islandicus, Malassezia globosa, Pseudogymnoascus sp. VKM F-4520, A. rambellii, Pneumocystis murina and Nosemia apis were investigated. We observed that these markers classified CRC and controls with an AUC of 0.93. They also distinguished control from early-stage CRC with an AUC of 0.91 (figure 3B).
Supplemental material

Fungal species as CRC diagnostic markers. (A) Heatmap showing the relative fold change of fungal species in colorectal cancer (CRC), n=73 against mean normalised relative abundance of control group, n=92 in discovery cohort. Species with absolute log2 fold change greater than 1 were selected as CRC fungal metagenomics markers. (B) Diagnostic performance of fungal markers for classification of CRC from control by logistic regression, corrected for confounders including age, Body Mass Index (BMI) and gender in discovery cohort. The markers achieved an area under receiver-operating characteristic curve (AUC) of 0.93 for the classification of control, n=92 from CRC, n=73, and AUC of 0.91 for the classification of control, n=92 from early CRC, n=20. .
Validation of faecal fungal diagnostic markers of CRC in independent cohorts
The performance of the fungal classifiers identified in the discovery cohort was investigated in independent Chinese cohort V1, comprising 112 control, 197 adenoma and 111 CRC subjects. Interestingly, our fungal markers distinctly stratified CRC subjects from the control with an AUC of 0.82 and control from early-stage CRC, with an AUC of 0.81 (figure 4A). Furthermore, our fungal markers were validated in an ethnically different European cohort V2 (German and French; control, n=66; CRC, n=90; early-stage CRC, n=32), from which AUCs of 0.74 and 0.72 were obtained for CRC versus control and early-stage CRC versus control, respectively (figure 4B). However, the fungal markers could only classify adenoma subjects from control with AUCs of 0.63 and 0.6, respectively, in cohorts V1 and V2 (figure 4A,B).

Validation of faecal fungal diagnostic markers of colorectal cancer (CRC) in independent cohorts. (A) The markers achieved an area under receiver-operating characteristic curve (AUC) of 0.82 for the classification of control, n=112 from CRC, n=111; AUC of 0.81 for the classification of control, n=111 from early_CRC, n=61; and AUC of 0.63 for the classification of control, n=111 from adenoma, n=197 in Chinese cohort V1. (B) The markers achieved an AUC of 0.74 for the classification of control, n=66 from CRC, n=90; AUC of 0.71 for the classification of control, n=66 from early_CRC, n=32; and AUC of 0.6 for the classification of control, n=66 from adenoma, n=42 in German & France cohort V2.
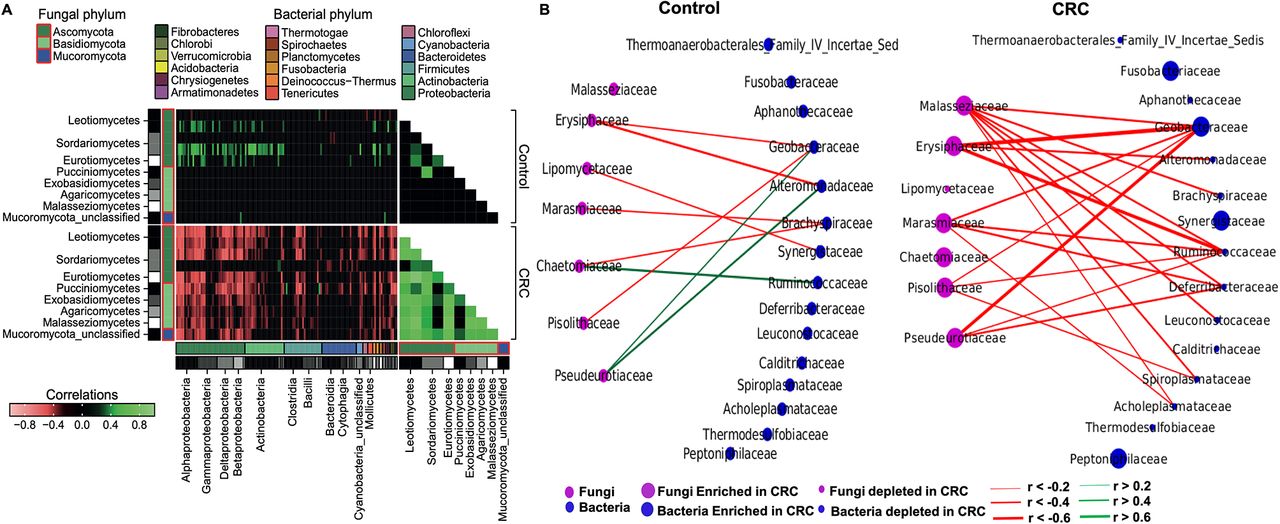
Perturbed intrakingdom and interkingdom ecological networks in CRC
Due to the complex and multifactorial nature of CRC, we investigated the potential alterations of polymicrobial ecological interactions in colorectal tumourigenesis by estimating fungal intrakingdom and bacterial–fungal interkingdom correlations. We observed a considerable difference within the fungal kingdom network, with significantly more positive and stronger interactions in CRC (p<0.0001, figure 5A, online supplementary tables 6–8). At the phylum level, only Ascomycota and Mucoromycota were significantly correlated in control. New correlations among the Ascomycota, Basidiomycota and Mucoromycota phyla of the fungal kingdom were observed in CRC. Furthermore, markedly different interkingdom ecological networks were observed between CRC and control. Correlations between fungi and bacteria were both fewer and mostly positive in control than in CRC, in which more and stronger co-exclusive relationships were observed (p<0.00001, figure 5A, online supplementary tables 6–10). In control, classes of Ascomycota phylum, including Leotiomycetes, Sordariomycetes, Eurotiomycetes and an unclassified class of Mucoromycota, were found to correlate synergistically with Alphaproteobacteria, Gammaproteobacteria, Deltaproteobacteria and Betaproteobacteria of the bacterial Proteobateria phylum. Conversely, additional fungal classes of Basidiomycota and Mucoromycota phyla were observed to participate in new interkingdom interactions in CRC, such as the Pucciniomycetes, Exobasidiomycetes and Malasseziomycetes, which formed significant co-exclusive network with major bacterial classes including Alphaproteobacteria, Gammaproteobacteria, Deltaproteobacteria and Cytophagia (figure 5A). Moreover, interkingdom co-occurrence interactions involving fungal classes Leotimycetes, Sodariomycetes and Eurotiomycetes in control were reversed to become co-exclusive interactions in CRC (figure 5A), indicating that gut microbiota homeostasis may involve a balanced and synergistic interkingdom relationship in healthy subjects which becomes disrupted in CRC. The enhanced intrafungal co-occurrence and bacteria–fungi co-exclusive correlation networks in CRC compared with control were further validated in cohort V1 (p=0.004414 and p<0.00001, respectively; online supplementary tables 11–15) and V2 (p=0.00006 and p<0.00001, respectively; online supplementary tables 16–20). Taken together, these results suggest a complex relationship between fungi and bacteria in the gut, in which their antagonistic associations may contribute to colorectal tumourigenesis.
{kind=link}
![[gutjnl-2018-317178-SP2.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/11/23/gutjnl-2018-317178/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Perturbed intrakingdom and interkingdom ecological networks in colorectal cancer (CRC). Correlations were estimated by SParCC algorithm. Absolute correlations below 0.3 were masked to show all signals that are exclusively significant. (A) Stronger co-occurrence ecological network in CRC than control (p<0.0001, Mann-Whitney U test). New correlations among Ascomycota, Basidiomycota and Mucoromycota phyla of the fungal kingdom emerged in CRC. (B) Ecological network among differentially abundant fungi and bacterial families in CRC compared with control.
Ecological interactions among differentially abundant bacterial and fungal families
To understand the potential interplay among differentially abundant bacteria and fungi in CRC and control, we performed additional bacterial differential abundance analysis (online supplementary table 21) and estimated the ecological network at the family taxonomy level. In control, co-occurrence interactions were observed between fungal Chaetomiaceae and bacterial Ruminicoccaceae, and between fungal Pseudeurotiaceae and bacterial Alteromonadaceae and Geobacteraceae (figure 5B), which reflect the contribution of synergistic interkingdom interactions to colon microbiota homeostasis. However, in CRC, with the enrichment of bacterial Geobacteraceae, Synergistaceae, Peptoniphilacea and Fusobacteriaceae, the positive correlations between CRC-enriched fungal Chaetomiaceae and CRC-depleted bacterial Ruminicoccaceae, and between CRC-enriched fungal Pseudeurotiaceae and CRC-depleted bacterial Alteromonadaceae no longer existed while that between fungal Pseudeurotiaceae and bacterial Geobacteriaceae was reversed to negative, despite both families being CRC-enriched. Moreover, only Geobacteraceae was involved in significant co-exclusive relationship with fungi in CRC among the CRC-enriched bacterial families, while other observed associations involved CRC-depleted bacterial families (figure 5B). Notably, our network analysis revealed that, in CRC, depleted bacterial families formed significant co-exclusive interactions with enriched fungal families, while enriched bacterial families such as Fusobacteriaceae, Synergistaceae and Peptoniphilaceae did not correlate significantly with enriched or depleted fungal families. This suggests that dysbiosis in bacterial kingdom may enhance colonisation by certain fungi families in CRC as previously described.22 On the other hand, within the fungal kingdom, extensive synergistic network formed among the CRC-enriched families, including Erysiphaceae, Malasseziaceae, Marasmiaceae, Pseudeurotiaceae, Chaetomiaceae and Pisolithaceae and several other undifferentiated fungal families in CRC compared with control (p<0.0001). In particular, Aspergillaceae was observed to have relatively strong positive ecological interactions with all CRC-enriched fungal families, a phenomenon that was non-existent in control (online supplementary figure 3). Thus, our analysis shows that co-occurrence interkingdom ecological relationships may be important for gut homeostasis in a healthy gut, while dysbiosis might provide a suitable environment for enhanced positive ecological interactions within the fungal kingdom, which, in turn, can play a role in colorectal carcinogenesis.
Supplemental material
Discussion
In this study, faecal shotgun metagenomic sequences were analysed to investigate gut fungal compositional and ecological alterations in association with CRC. To avoid overestimation of fungal role in CRC, which is possible with targeted marker gene sequencing,23 we assigned taxonomies simultaneously to bacterial and fungal DNA reads of metagenomic data24 using exact k-mer alignment metagenomic sequence classifier. This enabled the elucidation of differences in the interkingdom interactions between patients with CRC and control subjects. For the first time, we show that the enteric mycobial profile in CRC cases is distinct from healthy controls. In accordance with reported human gut mycobiome profiles, both CRC and control subjects were dominated by Ascomycota and Basidiomycota phyla25–27 with CRC being associated with a higher Basidiomycota:Ascomycota ratio, an index that defines fungal dysbiosis in an ecosystem.28 29 Our analysis clearly underscores the disruption of enteric fungal community in CRC.
We identified CRC-specific shifts in fungal composition reflected by the enrichment of six genera, including Rhodotorula and Malassezia of the Basidiomycota phylum and Acremonium of the Ascomycota phylum. The pathogenic potentials of these genera in diseases have been reported. For instance, isolates of the Acremonium genus were recovered from human fluids, such as blood, sputum and tracheal aspirates, where they were recognised as opportunistic pathogens.30 Also, Rhodotorula is an emerging pathogen capable of colonising, infecting and disrupting the essential microflora of human digestive system.31 Moreover, Malassezia, a fungal genus commonly found on the human skin with the capability of gut colonisation,12 27 32 encodes some secreted enzymes similar to the human pathogen Candida albicans.33 The contribution of Malassezia-synthesised aryl hydrocarbon receptor ligands to basal cell carcinoma through UV radiation–induced carcinogenesis has also been suggested.34 Notably, four species of Aspergillus, namely A. flavus, A. rambellii, A. sydowii and A. ochraceoroseus, were found to be enriched in CRC with the highest fold change of about four for A. flavus, a major producer of a highly toxic carcinogen aflatoxin.35 While Aspergillus has been detected in the human gut mycobiome in different studies,7 36 37 we particularly observed that the species whose abundance correlates with CRC in this study are known producers of carcinogens.38 39 The higher abundance ratios of these genera and species in CRC therefore suggest their possible contribution towards CRC pathogenesis and warrants further investigations.
In this study, the fungal class Saccharomycetes and, at the species level, Lipomyces starkeyi and S. cerevisiae, were found to be depleted in CRC. S. cerevisiae is a major component of the human gut microbiota,32 40 41 which had been shown to reduce adherent-invasive E. coli–induced ileal colitis in a mouse model.42 S. cerevisiae was also found to exhibit regulatory and anti-inflammatory effects on the host by inducing interleukin-10 production.8 43 The depletion of S. cerevisiae in CRC as identified in this study highlights the potential beneficial role of S. cerevisiae in the gut. This phenomenon can be adapted as a therapeutic approach for CRC prevention or treatment. Interestingly, we demonstrated that the faecal fungi species could discriminate healthy subjects from patients with CRC with validation in independent and ethnically different cohorts, suggesting that fungal markers might help to improve CRC detection.
Commensal fungi and bacteria coexist in the gut.44 As such, their mode of interactions in the colon may be altered in disease state and could reflect their potential roles in CRC. Our analysis shows that there exists complex ecological network between fungi and bacteria in healthy gut, which is substantially disrupted in CRC. We show that both intrafungal and interkingdom bacteria–fungi correlation networks were altered in CRC. Intriguingly, while correlation networks within fungi became more positive in CRC, those between fungi and bacteria were largely negative. We also observed the disappearance of some co-occurring bacteria–fungi correlations in CRC. The CRC-associated co-occurring intrafungal network suggests that the roles of fungi in CRC may be interdependent, while the observed co-exclusive bacteria–fungi interkingdom ecological network suggests that antagonistic interkingdom interactions may contribute to colorectal tumourigenesis and that altered bacterial composition in CRC may provide fungi with a favourable condition for intrafungi interaction, which may mediate their effect on CRC development.
The gut mycobiota has been shown to be significantly influenced by food colonisation, with same fungal species detected in consumed food and faecal samples of subjects.26 45 As such, diet is an important factor to be considered in associating specific fungi with disease and may affect their universal application as diagnostic markers. Although we did not take diet into account in this study, the demonstrated diagnostic potential of fungal markers in two ethnicities with different dietary habits indicate the potential importance of our identified markers in CRC.
In conclusion, our study elucidates the disruption of gut mycobiome homeostasis in colorectal carcinogenesis and demonstrates the potential usefulness of faecal fungal markers in detection of CRC. Moreover, we unravel CRC-specific intrakingdom and interkingdom ecological network alterations, indicating that synergistic intrafungal and antagonistic bacteria–fungi interactions might contribute to CRC. Further investigations to identify the functional consequences of the disrupted fungal composition and ecology are warranted for deeper understanding of the roles of fungi in colon tumourigenesis.
Supplemental material
![[gutjnl-2018-317178-SP3.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/11/23/gutjnl-2018-317178/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
![[gutjnl-2018-317178-SP25.jpg]](https://gut.bmj.com/content/gutjnl/early/2018/11/23/gutjnl-2018-317178/DC25/embed/inline-supplementary-material-25.jpg?download=true){kind=link}
Supplemental material
References
Footnotes
Contributors OOC analysed data and drafted the manuscript. GN and RZD organised and analysed data. WKKW, SHW, SCN, FKLC and JJYS commented on the study and revised the manuscript. JY designed, supervised the study and wrote the paper.
Funding This project was supported by Science and Technology Program Grant Shenzhen (JCYJ20170413161534162), HMRF Hong Kong (17160862), grant from Faculty of Medicine CUHK on Microbiota Research, RGC-GRF Hong Kong (14106415, 14111216, 14163817), Vice-Chancellor’s Discretionary Fund CUHK and CUHK direct grant, Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.