Article Text

Abstract
Decades of intense research in molecular biology and biochemistry are fructifying in the emergence of therapeutic messenger RNAs (mRNA) as a new class of drugs. Synthetic mRNAs can be sequence optimised to improve translatability into proteins, as well as chemically modified to reduce immunogenicity and increase chemical stability using naturally occurring uridine modifications. These structural improvements, together with the development of safe and efficient vehicles that preserve mRNA integrity in circulation and allow targeted intracellular delivery, have paved the way for mRNA-based therapeutics. Indeed, mRNAs formulated into biodegradable lipid nanoparticles are currently being tested in preclinical and clinical studies for multiple diseases including cancer immunotherapy and vaccination for infectious diseases. An emerging application of mRNAs is the supplementation of proteins that are not expressed or are not functional in a regulated and tissue-specific manner. This so-called ‘protein replacement therapy’ could represent a solution for genetic metabolic diseases currently lacking effective treatments. Here we summarise this new class of drugs and discuss the preclinical evidence supporting the potential of liver-mediated mRNA therapy for three rare genetic conditions: methylmalonic acidaemia, acute intermittent porphyria and ornithine transcarbamylase deficiency.
- genetics
- liver metabolism
- molecular biology
Statistics from Altmetric.com
Introduction
The exponential growth in the knowledge of the molecular bases of diseases has paved the way to treatments based on the replacement of defective proteins. The use of recombinant proteins in human therapy widened the therapeutic arsenal that previously relied mostly on small molecules. However, recombinant proteins have some drawbacks for treatment of chronic diseases such as the high production cost, immunogenicity and the limited number of amenable targets. Protein/enzyme replacement therapy is mainly limited to secreted proteins as well as to a few proteins of bacterial origin or synthetic peptides where protein–protein interaction or receptor binding is needed to produce a response.
Alternative strategies for drug development came from the central dogma of molecular biology. Based on this dogma, DNA stores the genetic information that is transcribed into messenger RNA (mRNA). Then, mRNA is translated into the therapeutic protein. The first strategy that has led to the approval of advanced medicines was based on the use of genes. The introduction of a gene into eukaryotic cells has been possible by the use of engineered viruses. Viruses are specialised in inserting their genetic material into cells and hijacking the cell’s replication machinery to persist. The strategies developed by viruses have been harnessed for the design of non-viral vectors or used directly to introduce the gene of interest by modifying the viral genome. The use of viral vectors is leading to a slow but constant approval of advanced medicines by regulatory agencies. In 2016, the European Medicines Agency approved the use of ex vivo transduced haematopoietic stem cells for the treatment of adenosine deaminase-deficient severe combined immunodeficiency. The viral vector used in this case was a gamma-retroviral vector.1 A related type of vector based on lentiviruses is used for the transduction of chimeric antigen receptors into autologous T cells. Two products based on this strategy received Food and Drug Administration approval in 2018 for the treatment of haematological malignancies.2 Also in the oncology field, a herpes virus encoding granulocyte-macrophage colony-stimulating factor was approved for the treatment of advanced melanoma.3 Finally, adeno-associated viruses (AAV) have been first approved for the treatment of inherited retinal disease due to mutations in both copies of the RPE65 gene,4 although this vector could also be used for the treatment of genetic liver diseases. A number of clinical studies are currently ongoing, with haemophilia A and B spearheading these assays with four products being tested in phase III clinical trials.5 Despite these breakthroughs, certain hurdles of gene therapy are still difficult to overcome. Viral vectors induce a potent humoral immune response that prevents readministration. Interindividual variability of transduction efficiency leads to an inherent variability in the expression levels of the therapeutic protein that may range from an inactive low dose to high toxic doses. This toxicity may be especially relevant for those proteins with a long half-life that may accumulate in intracellular compartments. Finally, due to the complexity of the production process and regulatory approval, it is unlikely that combined treatments with different transgenes will be developed.
In the meantime, a second strategy also based on the central dogma of molecular biology has entered into the clinical trial arena and is an emerging transformative therapy: the mRNA-based therapies. mRNA is demonstrating to be a viable option in several therapeutic areas including infectious and cardiovascular diseases as well as oncology.6 7 Moreover, recent studies are highlighting the potential of mRNA as a therapeutic tool for liver-targeted monogenic disorders, where protein replacement is needed to overcome the inability of mutated genes to produce a fully active protein.8 9 mRNAs offer the opportunity to use the human body to translate and produce any type of proteins including transmembrane, intramitochondrial, intracellular and secreted (figure 1). This therapeutic strategy can preserve the natural localisation of the therapeutic protein in different organelles driven by the protein localisation signals, which also favour natural post-translational modifications. mRNA-based therapy is founded on the introduction of chemically modified mRNA into specific cell population. The mRNA released into the cytoplasm is then translated into the therapeutic protein (figure 1). mRNA is therefore an intermediate information unit that connects the genome and the proteome, and its activity is transient by definition. This transient expression is the main difference to gene therapy, an approach that may introduce a stable modification into the host genome. In the case of mRNA-based therapies, an mRNA mode of action is to transfer information from the genome to the ribosome for translation and not to alter host genetic information. The transient nature of this technology is an advantage because it avoids the inherent genotoxicity of gene therapy, but also a potential disadvantage since repeated administration is required for the treatment of chronic diseases. The emergence of this technology required a careful optimisation of all the components of an mRNA vector.

LNPs deliver the therapeutic mRNA into hepatocytes, the target cells. mRNAs released from endosomes are translated and protein products eventually reach their intracellular or extracellular locations. LNP, lipid nanoparticles; PEG, polyethylene glycol.
mRNA-based therapy is composed of two main elements: the mRNA itself and the lipid nanoparticle (LNP) that can deliver the mRNA into the cytoplasm. From the 5′ to the 3′ end, the mRNA is composed of a 5′ cap structure, a 5′ untranslated region (UTR), the open reading frame, the 3′ UTR and, finally, the mRNA polymer is closed by a poly(A) tail (figure 2). The 5′ cap stabilises the polymer against exonuclease degradation and allows its engagement with the ribosomal machinery.10 The 5′ and 3′ UTRs are not translated into protein but are essential to stabilising the whole polymer. Standardised sequences are used in this region such as the α or β-globin 3′ UTRs.11 Tandem repeats of artificial microRNA target sites may also be incorporated into the 3′ UTR of the mRNA leading to its degradation in cells expressing the corresponding microRNA.12 This strategy would downregulate the levels of the mRNA in certain cell types, such as splenocytes, that could be involved in the initiation of an immune response against the protein of interest. Translation initiation can be enhanced using an optimum Kozak sequence and improved secondary structure, therefore translating more efficiently the designated protein.13 Another strategy implemented to increase the protein yield is the use of modified nucleotides such as N1-methylpseudouridine or 5-methoxyuridine. The incorporation of these modified nucleotides increases ribosome density on the mRNA. An additional advantage to using modified mRNAs is the reduced activation of immune RNA sensors such as Toll-like receptors (TLR3, TLR7, TRL8) and retinoic acid-inducible gene I receptors. The deimmunisation of the mRNA is crucial to allow the repeated administrations required for the treatment of chronic diseases.14

Therapeutic mRNAs: structural features and modifications aimed at optimising stability and translatability. ORF, open reading frame; UTR, untranslated region.
The second element that requires a finely tuned design is the LNP, which protects the mRNA from nuclease-mediated degradation and shields it from the immune system (figure 3). The most advanced LNPs in clinical trials are those taken up by the liver parenchyma. These LNPs are composed of phospholipids, sterols, ionisable lipids and polyethylene glycol (PEG)-conjugated lipids. PEG is essential to escape from early capture by phagocytic cells. LNPs are internalised into the hepatocytes through a process mediated by an interaction between the low-density lipoprotein (LDL) receptor and apolipoprotein E and other opsonins.15 Once in the endosome, the ionisable lipids induce the endosome escape of the LNP with the mRNA cargo, which will be finally released into the cytoplasm.16 17 Optimisation of all LNP components has led to the development of LNPs that mediate robust transduction of modified mRNA into non-human primate hepatocytes.16 These LNPs are characterised by high delivery rates, better endosomal escape once invaginated to the cell and high rate of biodegradability resulting in low accumulation of lipids in the targeted organs.16 Such features are important for rare genetic disorders where safety is paramount for young patients affected by these diseases. Avoiding lipid accumulation and improving the safety profile of the mRNA encapsulated in LNPs is an important aspect for a safe therapeutic intervention with the scope of replacing a fully functional protein.
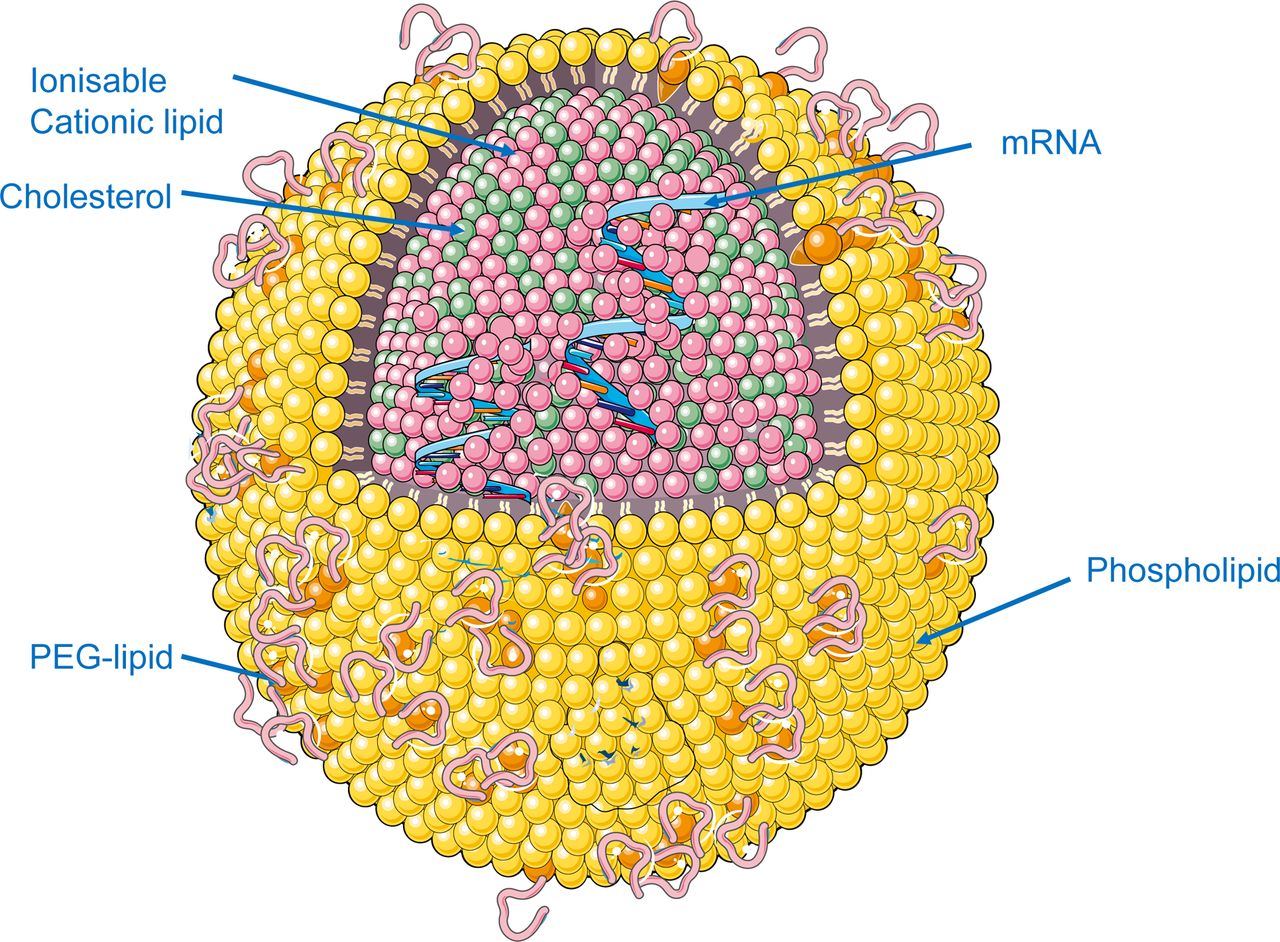
Encapsulation in lipid nanoparticles (LNP) allows mRNA stabilisation and hepatocyte delivery. PEG, polyethylene glycol.
Combining all these advances, mRNA therapy allows for reproducible dose-responsive protein production now shown in multiple animal models of disease. Importantly, and as will be discussed later, chronic treatment with mRNA shows the ability to maintain therapeutic protein expression levels in time. Lack of immediate immune response can also favour readministration of the mRNA. Moreover, the ability to combine several mRNAs simultaneously in a single treatment as implemented in vaccination strategies can increase the likelihood to treat different diseases including multigenic disorders that would not be addressable with the current technologies.18 19
The natural tropism of these LNPs for the LDL receptor has facilitated applications focused on the liver. Immunohistochemical analysis of the hepatocyte distribution on intravenous administration of mRNA encapsulated in LNPs in mouse models has demonstrated that virtually all hepatocytes can be reached.9 An alternative liver-targeting technology dubbed the hybrid mRNA technology delivery system (HMT) has been recently proposed. In this case, the injected product consists of a mixture of a non-targeted LNP containing the mRNA and a polymer micelle with N-acetylgalactosamine and a chemical structure that allows for mRNA release only in the acidic environment of the endosome. Through this(,) moiety micelles target the asialoglycoprotein receptor and attain specific cytoplasmic delivery in hepatocytes, while the LNPs protect mRNA from degradation by nucleases.20
The biosynthesis of a variety of macromolecules is one of the functions of the liver, and this capacity can be used to overexpress heterologous proteins. In some cases, the protein must remain in the hepatocytes for the therapeutic activity in different subcellular localisations, while in other cases the main target is in distant organs and the protein must be secreted (figure 1). This can be achieved by incorporation of a signal peptide in the N terminus of the recombinant protein. This signal peptide will incorporate the nascent polypeptide into the secretory pathways being cleaved before secretion. Therefore, the liver can be envisioned as a factory for systemic therapies using mRNAs and as a target for mRNA therapies designed for the treatment of liver monogenic diseases. In this review we discuss recent experimental studies of mRNA-based therapies that are paving the way for the treatment of rare genetic metabolic diseases of high morbidity and limited therapeutic options.
Rare genetic metabolic diseases: difficult to treat conditions and potential for mRNA therapy
Case 1: mRNA therapy for methylmalonic acidaemia
Methylmalonic acidaemia (MMA, MIM 251000) is a rare metabolic disease affecting 1 in 50 000–100 000 individuals.21 This is a severe condition primarily caused by mutations in the gene coding for the vitamin B12-dependent enzyme methylmalonyl-CoA mutase (MUT, EC 5.4.99.2) causing a partial or complete loss of enzymatic activity. In the mitochondrial matrix, MUT catalyses an essential reaction that channels the entry of carbon skeletons from branched amino acids, odd-chain fatty acids and cholesterol into the Krebs cycle via the formation of succinyl-CoA (reviewed in ref 22). This central role in intermediary metabolism explains why impaired MUT activity is associated with profound biochemical derangements, including the accumulation of toxic metabolites and the alteration of mitochondrial oxidative phosphorylation.22 The MUT gene is ubiquitously expressed, and its deficiency results in multisystemic complications which are more evident in tissues and organs with high metabolic demand such as the brain, eye, kidney and heart.22 Acute metabolic crises may occur in patients with MMA already in the neonatal period or at any age in adulthood.23 These acute crises may be started by a range of events that trigger catabolism, and typically present with nausea, vomiting and encephalopathy.23 24 Currently there are no therapies available for MMA, and patients are managed through careful dietary interventions with protein-limited diets and by carnitine supplementation.22 These strategies have significantly reduced mortality in patients with MMA, however mortality still remains at around 20%, and surviving individuals develop numerous systemic complications.24 25 The clinical phenotype of these patients can be severe, and neurological symptoms with motor disorders, optic nerve and retinal degeneration, and renal deterioration and failure are commonly observed.22 24 Liver transplant and combined liver-kidney transplant have been performed in patients with MMA with severe disease, and organ replacement is particularly favoured for young patients.22 26–28 Solid organ transplantation certainly improves the metabolic perturbations of patients with MMA and diminishes the risk of mortality. However, these procedures are not exempt from risks, and patients have to face lifelong immune suppression. These factors, together with the shortage of liver grafts, limit their widespread application in patients with MMA.
The clinical benefits of liver transplantation, such as improved growth and protection from metabolic crises, suggested that restoration of MUT activity in the liver could be a therapeutic strategy for the treatment of MMA. This notion elicited a number of preclinical gene therapy studies in Mut-/- mice using different vectors including AAVs harbouring the MUT cDNA.29–31 These works provided proof-of-concept evidence for the therapeutic value of hepatic MUT activity restoration, as Mut-/- mice were rescued from neonatal mortality and impaired growth in spite of a partial correction of plasma methylmalonic acid levels. With these precedents, the potential of mRNA treatment for MMA has also been recently explored.8 Systemic administration of human MUT (hMUT) mRNA formulated in LNPs as described above demonstrated a rapid (as early as 2 hours after intravenous injection) and sustained expression of hMUT protein in mouse livers (figure 4). The therapeutic capacity of this hMUT mRNA was tested in two clinically relevant mouse models: Mut-/- mice rescued from perinatal lethality by transgenic expression of MUT in skeletal muscle, and transgenic Mut-/- mice expressing the p.G715V MUT mutant variant found in patients with MMA.8 In the first model, hMUT mRNA administration resulted in a rapid reduction of methylmalonic acid levels in plasma and tissues, including liver, kidney, brain, heart and skeletal muscle. Importantly, repeat dosing of hMUT mRNA markedly improved mice growth and survival. In the hypomorphic p.G715V MUT-expressing mice, a fast reduction in plasma methylmalonic acid concentrations was also noticed after a single hMUT mRNA injection and this response lasted for at least 2 weeks. The pharmacokinetic properties of hMUT mRNA suggest that this therapy could be of high value for patients with MMA undergoing potentially lethal acute metabolic decompensations. Importantly, these effects were observed in the absence of signs of hepatic toxicity or inflammation, even on repeat mRNA dosing, and treated mice did not develop anti-hMUT antibodies, which could hamper hMUT mRNA efficacy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Potential treatment of rare genetic metabolic diseases with mRNAs. Lipid nanoparticles (LNP) deliver into hepatocytes the therapeutic mRNAs coding for the target enzymes. The segments indicate restoration of cytosolic porphobilinogen deaminase in acute intermittent porphyria (AIP), mitochondrial methylmalonyl-CoA mutase in methylmalonic acidaemia (MMA) or mitochondrial ornithine transcarbamylase (OTC) in OTC deficiency.
Case 2: mRNA therapy for acute intermittent porphyria
Acute intermittent porphyria (AIP, MIM 176000) is a rare metabolic disorder of haem metabolism affecting 5.9 per million people.32 The main symptoms are acute neurovisceral attacks caused by a dysregulation of hepatic haem homeostasis due to a deficiency of porphobilinogen deaminase (PBGD, EC 2.5.1.61), the third enzyme of its synthesis pathway. Although all the nucleated cells produce haem, this pathway is highly active in erythroblasts for haemoglobin synthesis and in the liver. As a cofactor of numerous proteins, haem is involved in diverse hepatic functions including energy metabolism, synthesis of steroid hormones and bile acids and oxidative metabolism of lipophilic compounds. However, despite its important metabolic role, haem and its biosynthetic intermediaries may have toxic effects. Thus, their concentrations are tightly regulated by pathway compartmentalisation (the first and last three steps occur in the mitochondrion), and by finely tuned, haem-dependent regulation at the level of its first step of synthesis (δ-aminolevulinate synthase 1, ALAS1) and its degradation (haem oxygenase, HO).
Strong upregulation of hepatic ALAS1 in PBGD-deficient individuals leads to a marked overproduction and accumulation of δ-aminolevulinic acid (ALA) and porphobilinogen (PBG), which are the haem intermediates between both enzymes. AIP symptoms are invariably accompanied by excess accumulation and renal excretion of these two haem precursors. Current treatment is based on infusions of haem33 and opioid analgesics to relieve pain.34 However, reduction of ALA and PBG levels occurs 3 days after intravenous haem infusion (3–4 mg/kg/day), with pain and nausea typically resolving on day 4.35
Approximately 5%–10% of patients suffer from recurrent attacks which persist for many years with a dramatic impact on their quality of life, predisposing them to chronic hypertension,36 renal damage37 and increased incidence of hepatocellular carcinoma.38 Prophylactic haem arginate administration appears to be beneficial to these patients, however progressive tolerance by the activation of HO, thromboembolic disease and iron overload can occur.33 Ultimately, degradation of the patient’s condition and advanced stages of neuropathy may leave liver transplantation,39 or combined kidney/liver transplantation,40 as the only therapeutic option. Thus, more efficacious and safer therapies need to be implemented before porphyria-related injuries become irreversible.
The administration of the small interfering RNA givosiran (Alnylam Pharmaceuticals) directed against hepatic ALAS generates a reduction of ALA and PBG accumulation 2–3 days post-treatment, and the effect is maintained over weeks.41 Another emerging approach involves the use of gene therapy in which the wild-type PBGD gene is delivered to hepatocytes using viral vectors such as AAVs (rAAV2/5-PBGD).42 Several studies demonstrated the efficacy of gene therapy in a clinically relevant model of AIP, the AIP mouse, an engineered strain that retains about 30% of normal liver PBGD activity in which acute porphyria attacks can be induced.43 Metabolic and symptomatic correction of the disease was observed in AIP mice on AAV-mediated delivery of PBGD, findings that suggested potential clinical efficacy.44 45 However, porphyrin precursor overexcretion was not modified in eight patients enrolled in a phase I open-label, multicentre clinical trial,46 indicating that sufficient delivery of the transgene was not achieved in humans and that more efficient vectors are needed. A recent preclinical study demonstrated that the intravenous administration of a human PBGD mRNA encapsulated in biodegradable LNPs (figure 4) induced high protein expression and enzyme activity in mouse livers in less than 2 hours.9 The therapeutic efficacy of this strategy was put forward in AIP mice and a rabbit model of the disease where the rapid expression of PBGD protein in the liver induced normalisation of both ALA/PBG excretion in hours and a full protection against pain and motor neuropathy. A homogenous distribution of the human protein over the liver parenchyma in both small and large animal models was observed. Lack of hepatic ALAS1 induction in AIP mice treated with hPBGD mRNA during the acute attack indicated normal regulatory haem levels and the restoration of its pathway feedback regulation throughout the liver. Prompt normalisation of various liver functions was supported in AIP mice by measurements of serum pharmacokinetic profile of phenobarbital, respiratory chain function and protection against phenobarbital-induced hypertension.
Human PBGD showed therapeutic levels in the liver of AIP mice for 7–10 days at the doses tested.9 From a translational perspective, since the mean duration of an acute attack was reported between 5 and 7 days in patients with AIP,47 hPBGD mRNA offers a promising alternative to treat acute attacks. Moreover, administration of equivalent doses of the mRNA was well tolerated and reached the same efficacy in rat, rabbit and monkey (75% increase over endogenous activity).9 In the case of maintaining the same gain in humans (patient’s liver shows 50% of healthy livers’ endogenous activity), normal PBGD activity would be achieved and the accumulation of neurotoxic precursors could be avoided.
Case 3: mRNA therapy for ornithine transcarbamylase deficiency
Urea cycle disorders result in the accumulation of ammonia causing devastating neurological sequelae in long-term survivors.48 The liver is primarily responsible for ammonia detoxification and mitochondrial ornithine transcarbamylase (OTC, EC 2.1.3.3) catalyses the incorporation of carbamoyl phosphate, involved in ridding the body of excess nitrogen, into the urea cycle. OTC deficiency (MIM 311250) is the most frequent inborn error of the urea cycle affecting 6 in 100 000 individuals.49 This deficiency results in increased ammonia levels in the bloodstream (hyperammonaemia) which may lead to neurological damage, coma and potential lethality.50 A protein-restricted diet for life, arginine and citrulline supplementation and currently available ammonia scavengers are effective interventions. However, liver transplantation is the only therapeutic option in cases of severe OTC deficiency in the neonatal form or on recurrent hyperammonaemia episodes.51 The central metabolic role of the OTC enzyme, the neonatal onset of the disease and its X linked inheritance pattern explain why it has concentrated numerous efforts in developing new therapies. OTC deficiency has been modelled in Otcspf-ash mice, which harbour a mutation in the Otc gene resulting in impaired splicing and 5%–10% residual OTC activity.52 This mutation results in a mild phenotype, with elevated orotic acid levels in urine but no clinically meaningful hyperammonaemia. Elimination of residual OTC activity by knocking down the remaining OTC mRNA levels with AAV-mediated delivery of short hairpin RNA (shRNA) targeting OTC mRNA results in severe hyperammonaemia, neurological impairment and lethality.53 Experimental gene therapy approaches with AAV vectors using these Otcspf-ash mice showed promising results that prompted clinical studies.53 54 Among the 16 clinical trials registered for OTC deficiency two assayed hepatocyte transplantation and five aimed to use recombinant AAV vectors to transfer a copy of OTC cDNA to liver cells (ClinicalTrials.gov/EudraCT database). Low doses of recombinant AAVs showed no correction of the OTC deficiency and high dose caused severe hepatitis and lethal multiorgan failure in a patient.55 Three clinical trials for OTC using different self-complementary AAV8 vector are ongoing in adults with late-onset OTC deficiency (NCT02991144, EudraCT2018-000156-18 and EudraCT2016-001057-40). Given that OTC deficiency is a chronic disorder requiring sustained transgene expression throughout the life of the patient, repetitive administration of the same therapeutic product is required. In this way, gene therapy for OTC deficiency faces the same general limitations of this technology previously mentioned, plus the fact that enzyme replacement therapy should be started early in neonates to avoid lethality in severe cases.
A recent study addressed the efficacy and tolerability of mRNA-based therapy for OTC deficiency using the HMT two-nanoparticle mRNA delivery technology described above20 (figure 4). Administration of a single dose of human HTM/OTC mRNA (hOTC) to Otcspf-ash mice resulted in widespread mitochondrial hOTC protein expression across the liver parenchyma and in increased OTC enzymatic activity up to 10 days after mRNA injection. Interestingly, biweekly injection of HTM/hOTC mRNA to Otcspf-ash mice that had been infected with AAVs harbouring Otc shRNA completely normalised plasma ammonia and urinary orotic acid levels. Most relevant, while all control animals died within 3 weeks of Otc shRNA AAV infection, mice receiving the HTM/hOTC mRNA survived the 35-day dosing period, and for at least 3 additional weeks after cessation of treatment. Importantly, no signs of inflammation or liver toxicity were appreciated in HTM/hOTC mRNA-treated mice, attesting to the safety of the procedure. Together, these findings provide proof of concept and lend further support to mRNA therapy as an option for the treatment of a severe metabolic condition by delaying or averting liver transplantation. Interestingly, a phase I/II clinical trial has recently been initiated and is recruiting OTC-deficient patients to test the safety and efficacy of single and multiple ascending doses of human OTC mRNA formulated in LNPs (NCT03767270). To our knowledge, this trial represents the first study of an mRNA-based therapy for the treatment of a genetic metabolic disease.
Conclusions and future perspectives
Protein replacement therapies represent a major milestone in medicine. Monogenic rare diseases are in some cases devastating life-threating conditions in which large investments by for-profit organisations are not possible due to the reduced numbers of patients. The exogenous administration of the deficient protein is a straightforward therapeutic strategy that improved the quality of life in several rare diseases.56 Unfortunately, the suboptimal pharmacokinetic and biodistribution properties of many recombinant proteins limit the extension of these strategies. Fusion proteins may in some instances partially solve these limitations. Still, the complexity of fusion proteins increases the risk of immunogenicity and production of recombinant proteins usually has high manufacturing costs.57
The promise of a single shot treatment spurred the interest in gene therapy with long-lasting viral vectors. This possibility became true with the recent approval of an AAV vector for the treatment of RPE65 mutation-associated retinal dystrophy.4 However, the interpatient variability of the transduction process may limit the applicability of this technology. Diseases that could benefit from gene therapy would be those responsive to a wide range of therapeutic protein expression levels and with no toxicity due to long-term accumulation of the therapeutic protein could be envisioned, haemophilia being a paradigmatic example. New engineered AAV vectors are being developed to reduce cross-reactivity with wild-type AAV serotypes, or to be administered in combination with immune modulators like rapamycin which would allow repeated doses of the same vector.58 59 This issue is especially relevant in the case of AAV therapy in paediatric patients, such as OTC-deficient neonates, in which proliferation of the developing hepatocytes may lead to loss of non-integrating viruses such as AAVs.60 61 Altogether, there are still important issues to be addressed in this field, as learnt from the previously mentioned gene therapy clinical trials for AIP and OTC deficiency.46 55 Thus, an urgent medical need exists for many devastating rare diseases that may not be manageable with protein replacement therapies and gene therapy. mRNA-based therapy may be an intermediate approach that can reach these neglected diseases, leading to better life quality and increased survival of the patients. However, although being a very promising strategy, mRNA-based therapy is still in its infancy and many uncertainties must be solved in clinical trials. A critical factor that will determine the success of the mRNA-based therapy will be the lack of toxicity due to accumulation of the LNPs, as slight differences in the tolerability of LNPs may dramatically influence the long-term safety.62 Active research in this area is leading to the development of innovative LNPs with very good pharmacokinetics and importantly with a favourable toxicity profile tested in non-human primates after repeated administrations.16 A comparative analysis of protein replacement therapy, gene therapy and mRNA-based therapy is presented in table 1.
Comparative analysis of protein replacement therapy, gene therapy and mRNA therapy for the treatment of genetic metabolic diseases
Advances in mRNA and LNP technology and expansion of the indications will reduce the cost of production and development, making this technology affordable for a wide variety of rare and ultrarare diseases. Due to the liver-targeting properties of the LNPs developed so far, mRNA technology will be of great interest for those monogenic diseases for which liver transplantation is indicated,63 and even for other diseases with an established therapeutic alternative. Of special interest will be the case of haemophilia A and B, rare diseases with high prevalence. The deficiency of a coagulation factor can be tackled by administration of the recombinant protein. However, the hepatotropism of LNPs would allow the production of coagulation factors in the liver, where they are naturally produced.64 65 Table 2 provides a list of biopharmaceutical companies developing mRNA-based therapies targeting the liver for genetic metabolic diseases and haemophilia.
Biopharmaceutical companies developing mRNA therapeutics for metabolic diseases and coagulation disorders targeting the liver
Among the different conditions to be treated with mRNA therapy, AIP is an ideal candidate, as it presents with acute episodes. Other diseases in which the continued and unregulated expression of the transgene might generate metabolic imbalances could be also considered. One additional factor to take into account is that dosage of mRNA therapy will be marked by the half-life of the therapeutic protein. In this sense, the flexibility of the mRNA vector platform allows for the design of more stable engineered protein variants that could increase the efficacy of therapeutic mRNA. With all these premises the results of ongoing and upcoming clinical trials with this new class of drugs are eagerly awaited.
Key messages
Messenger RNA-based therapy is founded on the transient and controlled expression of a therapeutic protein in a target tissue.
Therapeutic proteins encoded by messenger RNA-based therapies can be retained in intracellular compartments, targeted to the cell membrane or secreted to the extracellular space or circulation.
Messenger RNA-based therapy relies on carefully designed lipid nanoparticles that prevent RNA degradation, mediate its precise intracellular delivery and allow safe repetitive administrations.
Preclinical data support the usefulness and safety of messenger RNA-based therapy for the treatment of acute intermittent porphyria, methylmalonic acidaemia and ornithine transcarbamylase deficiency.
Messenger RNA-based therapy holds promise as an alternative strategy to protein replacement therapy and gene therapy for the treatment of rare diseases.
References
Footnotes
Contributors All authors made an equal contribution.
Funding This study was supported in part by grants from the Fundación Mutua Madrileña (Madrid, Spain), Fundación Eugenio Rodríguez Pascual (Madrid, Spain), Hepacare Project Fundación Bancaria La Caixa (Barcelona, Spain) and Spanish Institute of Health Carlos III (FIS) cofinanced by European FEDER funds (grant numbers PI09/02639, PI12/02785, PI15/01951, PI16/00668 and PI18/00860). PB is supported by a Miguel Servet II (CPII15/00004) contract from Instituto de Salud Carlos III. Figures contain elements of the Servier Medical Art. The generous support of Mr Eduardo Avila and Mr Sergio Durá to the Hepatology Program of CIMA, University of Navarra, is acknowledged.
Competing interests PGVM is an employee of Moderna, focusing on the development of therapeutic approaches for rare diseases. PGVM receives a salary and stock options from Moderna, as compensation for his employment by the company.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.