Article Text

Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Neoadjuvant therapy followed by surgical resection cures less than half of patients with resectable oesophageal adenocarcinoma (OAC).
Response rates to neoadjuvant platinum-based chemotherapy are low at 15%.
Recent molecular landscape studies in OAC have indicated the presence of a DNA damage response impaired subgroup of tumours.
What are the new findings?
A 44-gene DNA damage immune response (DDIR) assay can successfully be applied to formalin-fixed paraffin-embedded pretreatment endoscopic biopsies with a success rate of >98%.
The DDIR assay is predictive of response and survival benefit following DNA-damaging neoadjuvant chemotherapy and surgery.
DDIR-positive patients have increased pathological response, lower nodal burden and reduced resection margin involvement.
DDIR positivity is associated with an inflammatory microenvironment characterised by the presence of CD8-positive tumour-infiltrating lymphocytes and high programmed death ligand 1 expression.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The ability to select the appropriate neoadjuvant therapy for individual patients with OAC could increase pathological response rates and survival.
Ineffective therapy could be avoided in patients with OAC unlikely to respond.
Insights into the molecular biology of the DDIR subgroup will allow novel combinations of conventional therapy with DNA repair inhibitors or immunotherapy to be explored.
Introduction
The incidence of oesophageal adenocarcinoma (OAC) in the Western world has risen sixfold in the last 40 years with the highest incidence occurring in the UK.1–3 In resectable cases, the addition of neoadjuvant or perioperative therapy provides a modest improvement in overall survival (OS), but only 15% of patients demonstrate a histopathological response to therapy in the resected tumour.4–7 Despite improvements in oncological and surgical management, the majority of patients relapse and die of their cancer.4–6 Therefore, there is a pressing need to identify biomarkers capable of predicting response in order to select the appropriate neoadjuvant therapy for individual patients.
Imaging and molecular features of OAC have been studied in an attempt to identify predictive biomarkers to neoadjuvant therapy. For example, serial [18F]−2-fluoro-2-deoxy-d-glucose (18FDG) positron emission tomography (FDG-PET) scans can detect changes in tumour metabolism with the aim of predicting pathological response.8–10 A 35% reduction in standard uptake value (SUV) 14 days after baseline has been correlated with a higher rate of tumour regression, R0 resection and improved survival in a prospective study of resectable OAC.9 However, 42% of FDG-PET responders identified by a reduction in SUV did not in fact achieve a pathological response, highlighting the pressing need to identify more accurate molecular predictive biomarkers. Various proposed single gene predictive biomarkers, such as nuclear-factor-κβ, epidermal growth factor receptor, TP53, ERCC1 and thymidylate synthase, have met with limited success as they fail to capture the complex biology of OAC.11–18 Recent advances in the molecular understanding of OAC have demonstrated that it is a disease characterised by a high level of mutations and copy number changes giving rise to prominent intratumoural heterogeneity.19–22 To encapsulate the biology underpinning response to chemotherapy in OAC, a number of studies have applied gene expression profiling to pretreatment endoscopic biopsies to identify a predictive gene signature.23–25 However, these signatures rely on fresh frozen tissue, which is not routinely available, and have been developed in small discovery cohorts without independent validation.
The DNA damage immune response (DDIR) assay, formerly known as the DNA Damage Response Deficiency assay, was previously developed in breast cancer using an unsupervised hierarchical clustering approach.26 When tested in an independent breast cancer dataset (n=203), DDIR positivity was associated with an OR for pathological response following neoadjuvant chemotherapy of 3.96 (95% CI 1.67 to 9.41; p=0.002), and in a cohort of 191 patients with node-negative breast cancer, the assay predicted 5-year disease-free survival following adjuvant chemotherapy with an HR of 0.37 (95% CI 0.15 to 0.88; p=0.025). Further validation in 664 chemo-naive patients indicated that the DDIR assay was not prognostic and only predicts outcome in the context of DNA-damaging chemotherapy. Biologically, the DDIR assay indicates constitutive activation of the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway in response to endogenous DNA damage.27 Deficiencies in DNA repair and the Fanconi anaemia/BRCA pathway in particular have been reported to activate this pathway. Importantly, the 44-gene DDIR assay includes well-known immune checkpoint targets, such as programmed death ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase 1, as well as several inflammatory cytokines. Immune activation via the STING pathway results in infiltration of the tumour by T lymphocytes and upregulation of immune checkpoints to create an inflammatory microenvironment associated with chemosensitivity. However, pathological response (tumour regression grade [TRG] 1/2) to DNA-damaging chemotherapy and chemoradiotherapy occurs in only 15% and 23% of OAC tumours, respectively.6 7 We hypothesised that pathological tumour response and improved survival may be due to pre-existing deficiencies in DNA repair pathways with associated activation of an innate immune response. An assay that could identify this subgroup of OAC tumours would predict benefit from neoadjuvant chemotherapy.
We, therefore, assessed the ability of the DDIR assay to predict pathological response and prognosis following DNA-damaging neoadjuvant chemotherapy in OAC. We demonstrate that the DDIR assay can be applied to routine diagnostic clinical specimens to allow the selection of patients for whom DNA-damaging chemotherapy would be beneficial. DDIR positivity is also strongly correlated with the presence of tumour-infiltrating lymphocytes (TILs) and PD-L1 expression indicating an association between deficiencies in DNA damage repair mechanisms and a proinflammatory microenvironment in OAC.
Materials and methods
This study was performed according to the REporting recommendations for tumour MARKer prognostic studies (REMARK) as outlined in the criteria checklist (online supplementary table 1) and REMARK study design diagram (online supplementary figure 1 and online appendix A)
Supplemental material
Supplemental material
Supplemental material
Patient samples
FFPE prechemotherapy endoscopic biopsies from 273 patients with resectable OAC, treated with neoadjuvant chemotherapy followed by surgical resection, were collected at four UK centres in the Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) consortium between 2003 and 2014 (online supplementary table 2). Follow-up was performed according to local institutional guidelines. Patients with localised histologically confirmed adenocarcinoma of the oesophagus or gastro-oesophageal junction were included, and all patients were followed up for at least 2 years. Pathological response was assessed in the matched resection specimens according to the method described by Mandard et al with a responder defined as TRG ≤2.7 28 Assuming a marker positive rate of 21% (estimated from preliminary data), a sample set of 273 patients had an 80% power to detect a HR of 2.
For independent in silico validation, a publicly available dataset of 57 OAC resections that did not receive DNA-damaging chemotherapy (GSE19417) was assessed (online supplementary table 3). All tumour samples were collected and snap-frozen from patients undergoing potentially curative surgical resection at the Bristol Royal Infirmary between 1992 and 2000. Gene expression profiling was performed using a custom-made Agilent 44K 60-mer oligomicroarray as previously described.29
Gene expression profiling from FFPE tissue
Biopsies were reviewed for pathological subtype prior to marking for macrodissection and samples containing at least 50% adenocarcinoma tissue by area were taken forward. Where tumour material was limited endoscopic biopsy fragments from the same patient were pooled. Total RNA was extracted using the Recoverall Total Nucleic Acid Isolation Kit for FFPE (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and amplified using the NuGen Ovation FFPE Amplification System v3 (NuGen San Carlos, California, USA). The amplified product was hybridised to the Almac Diagnostics Xcel array (Almac, Craigavon, UK), a cDNA microarray-based technology optimised for archival FFPE tissue and analysed using the Affymetrix Genechip 7G scanner (Affymetrix, Santa Clara, California, USA) as previously described.26 30 Functional enrichment was performed using the Database for Annotation, Visualization and Integrated Discovery. Raw expression data are available at the Array Express repository (Accession Number E-MTAB-6969).
Immunohistochemistry (IHC)
Matched FFPE OAC resection specimens were available for 126 patients who received neoadjuvant chemotherapy prior to surgical resection at the Northern Ireland Cancer Centre. Pathological staging was defined according to International Union Against Cancer TNM Staging, 7th Edition, and the cases had a median follow-up time of 48.8 months (online supplementary table 4). All cases were represented in triplicate, and the Tissue Microarray (TMA) was constructed as previously described.31 32
Antibodies to CD8 (C8/144B, M7103, Dako) and PD-L1 (SP142, Roche) were used as previously described.27 TMA sections were scored by two independent observers (EEP and EM) who were blinded to the clinical data. A semiquantitative scoring system was used for CD8+ expression with a score of 3 indicating strong CD8+ expression, 2 moderate expression, 1 weak expression and 0 absence of expression. For PD-L1, tumour and stroma were scored for percentage of cells with positive expression and previously published cut-offs of 1% or greater and 5% or greater were used for analysis.33
Whole genome sequencing
Matched whole genome sequencing data were available for 44 patients who received neoadjuvant chemotherapy prior to surgical resection at three OCCAMS centres (Cambridge, Edinburgh and Southampton; online supplementary table 4).
Whole genome sequencing was performed, and mutational signatures were identified using the non-negative matrix factorisation (NMF) methodology as previously described.22 34
Statistical analysis
Microarray data were preprocessed using the Robust Multi-array Average model for the Almac Diagnostics Xcel array with DDIR signature scores calculated and predefined cut-points applied as previously described.26 A threshold of 0.3403 was optimised in an independent technical study of n=45 OAC samples and applied independently to the validation cohort dichotomising patients as DDIR positive (>0.3403) or DDIR negative (≤0.3403). Cox proportional hazards regression was used to investigate the prognostic effects of the DDIR signature on relapse-free survival (RFS) and OS defined as the time from surgical resection to relapse of disease or death from any cause, respectively. The estimated effect of the signature was adjusted for factors available at the time of diagnosis (clinical tumour status, clinical nodal status and tumour grade) by fitting a multivariate model.
Further details are available in the online supplementary methods.
Supplemental material
Results
Assessment of the DDIR assay and survival following neoadjuvant chemotherapy and surgical resection in OAC
To assess the ability of the DDIR assay to predict survival following neoadjuvant DNA-damaging chemotherapy and resection in OAC, it was applied to a retrospective dataset of 273 FFPE biopsy samples. A total of 66 OAC samples (24%) were characterised as DDIR positive with the remaining 207 (76%) being DDIR negative. Reductions in lymph node and circumferential resection margin (CRM) involvement, a more proximal tumour location and older age were observed for DDIR positive tumours (table 1). DDIR assay positivity was associated with improved RFS (HR 0.58, 95% CI 0.38 to 0.90; p=0.015) and OS (HR 0.62, 95% CI 0.41 to 0.95; p=0.029) following surgical resection (figure 1). When evaluated as a continuous variable, higher DDIR scores were associated with both improved RFS (HR 0.34, 95% CI 0.13 to 0.93; p=0.036) and OS (HR 0.32, 95% CI 0.12 to 0.87; p=0.026). Univariate analysis confirmed associations between survival and presurgical clinical N stage as well as postsurgical factors such as pathological T and N stage, differentiation, lymphovascular invasion and CRM status (online supplementary table 5). Applying a published cut-off of ≥15 lymph nodes to indicate an adequate lymph node yield, we found that there was no association between the DDIR status and lymph node yield and neither was there an association between adequate lymph node yield and RFS (HR 0.94, 95% CI 0.66 to 1.39; P=0.847) or OS (HR 1.02, 95% CI 0.7 to 1.48; p=0.916). While the Lauren classification is known to be prognostic in OAC, it was not available for the whole cohort and so it is unclear how the DDIR assay relates to intestinal versus diffuse type adenocarcinomas.35 Also, there was no association between the administration of postoperative chemotherapy and DDIR status (HR 0.74, 95% CI 0.39 to 1.4; p=0.354), but there was a trend towards improved OS in DDIR-positive patients when no adjuvant chemotherapy was administered (HR 0.39, 95% CI 0.15 to 1.02; p=0.55) (online supplementary figure 2).
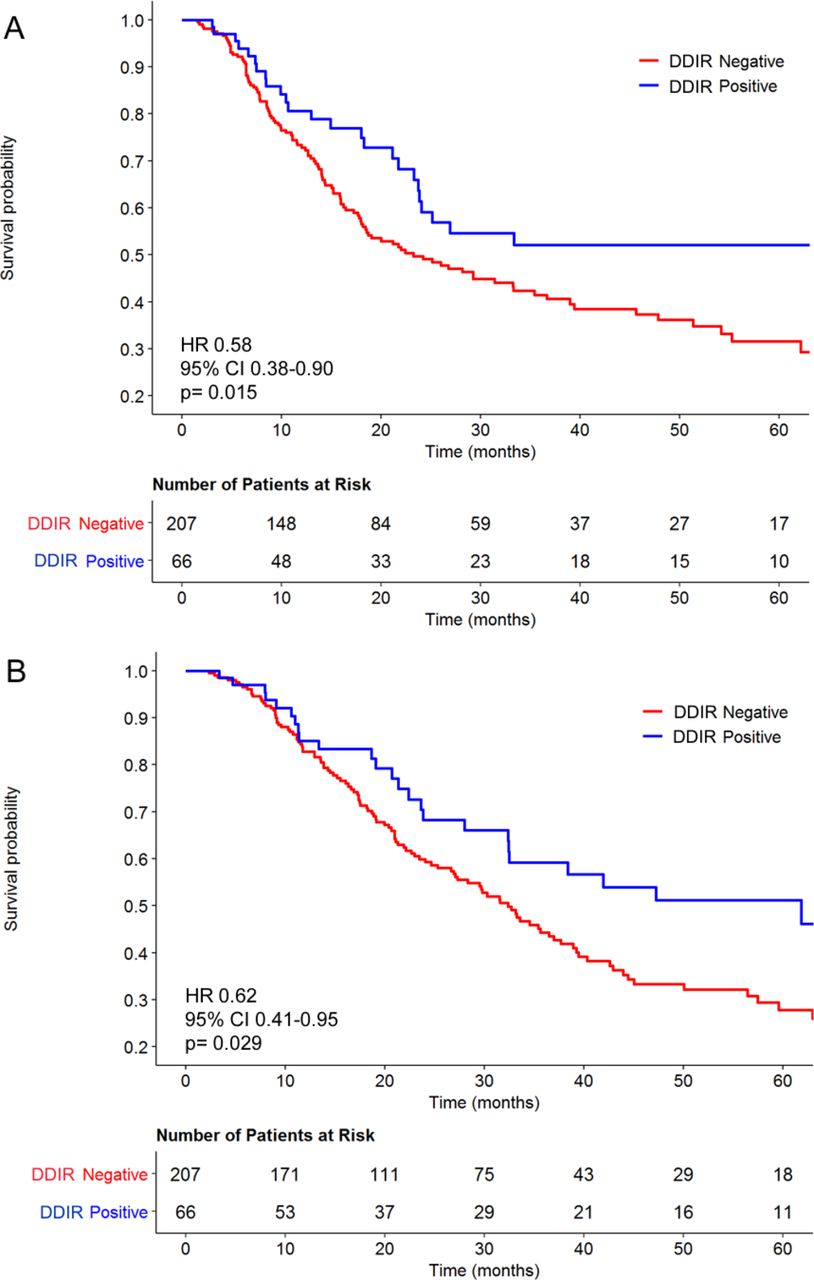
Kaplan-Meier curves stratified by the DDIR assay for (A) relapse-free and (B) overall survival for 273 patients with oesophageal adenocarcinoma treated with cisplatin-based neoadjuvant chemotherapy followed by surgical resection. DDIR, DNA damage immune response.
Association of clinicopathological characteristics with DDIR status in the OAC cohort
Multivariate analysis and combined model of clinicopathological factors, DDIR status, relapse-free and overall survival in OAC
Multivariable analysis was performed to test the association between DDIR status and each survival endpoint following adjustment for factors available at diagnosis (table 2). DDIR-positive patients had improved RFS relative to DDIR negative patients (HR 0.61, 95% CI 0.38 to 0.98; p=0.042), and assay positivity was also independently associated with improved OS (HR 0.52, 95% CI 0.31 to 0.88; p=0.015).
To assess whether the DDIR assay was prognostic, independent of DNA-damaging chemotherapy treatment, it was applied to a publicly available dataset of 57 OAC resections, which did not receive neoadjuvant chemotherapy (online supplementary table 3). No significant difference in overall survival was noted between the DDIR-positive and DDIR-negative populations (HR 0.86, 95% CI 0.48 to 1.55; p=0.61) (online supplementary figure 3). However, further confirmatory results in a larger cohort are required. Taken together, these results indicate that the DDIR assay is a strong predictor of survival benefit following surgical resection in OAC, but only in the context of neoadjuvant DNA-damaging chemotherapy.
The DDIR assay is predictive of pathological response in OAC
The ability to predict pathological response to neoadjuvant chemotherapy would improve patient stratification and treatment selection in OAC. TRG was available for 228 patients in the OAC cohort with 24 (11%) of cases having a TRG ≤2, indicating a pathological response, and 203 (89%) TRG 3–5, in keeping with limited or no response to chemotherapy. Pathological response was observed in 16.7% and 6.8% of DDIR-positive and DDIR-negative cases, respectively (p=0.025) (table 1). DDIR scores were grouped by response status and one-way analysis of variance analysis demonstrated significantly higher DDIR scores in responders compared with non-responders (p=0.033). This indicates that the DDIR score was significantly enriched for tumours that respond to neoadjuvant chemotherapy in OAC (figure 2).

Boxplot of DDIR scores grouped by response status. DDIR, DNA damage immune response.
DDIR assay positivity and tumour mutational load
Recent sequencing studies have stratified OAC into subtypes defined by the pattern of somatic mutations. Secrier et al 22 identified three subgroups (C>A/T dominant, DNA damage response [DDR] impaired and mutagenic through the application of mutational signatures to WGS data from a cohort of 129 chemotherapy-naïve OAC samples. We sought to assess the overlap between cases defined as DDIR positive by our gene expression assay and DDR impaired by mutational signature analysis. A total of 44 cases had matched gene expression and WGS data available and demonstrated higher clinical nodal staging and different neoadjuvant chemotherapy regimens compared with the whole cohort and the tissue microarray (TMA) subset (online supplementary table 4). This may reflect differing staging methodologies used at the largest contributing centre to the WGS cohort (University of Cambridge; 29 [66%] of patients) and the increased use of cisplatin and oxaliplatin doublet neoadjuvant regimens due to clinical trials recruiting at that centre at the time of sample collection (OEO536 and LEO37).
NMF was applied to cluster the patients into the three subgroups (online supplementary figure 4). No association was observed between the DDIR status and the predominant mutational signature (online supplementary table 6; p=0.83). Although the size of the cohort limits the statistical power of the analysis, DDIR-positive patients did display a trend towards higher tumour mutational burden and a higher mutation rate (online supplementary figure 5). However, no significant differences were observed in the mean copy number or total number of deleterious somatic mutations or indels in multiple DDR pathways between the DDIR-positive and DDIR-negative samples. Neither were there any differences observed in the copy number of genes involved in the homologous recombination, double and single strand break repair pathways (data not shown). While both assays are related to loss of DNA repair, the assessment of differing biologies represented by immune activation in response to DNA damage measured by the DDIR assay, as opposed to the pattern of mutations caused by deficiencies in DNA repair mechanisms may lead to the lack of association.
DDIR assay positivity is associated with CD8+ T lymphocytes and expression of PD-L1
We hypothesised that increased DNA damage in DDIR-positive tumours may be associated with increased lymphocytic infiltration and upregulation of immune checkpoint genes. A list of 45 genes differentially expressed between DDIR-positive and DDIR-negative patients, with a fold change of >2, was generated (online supplementary table 7). As expected, this list included the genes from the DDIR signature, with 5 out of 44 genes represented, but it also included a number of genes encoding inflammatory cytokines and mediators of an immune response. Chemokines such as CXCL9 and CXCL13 showed 5.5-fold and 4.58-fold upregulation, respectively, and 29 of the 45 genes (64.4%) have a role in the immune response. Pathway analysis demonstrated enrichment of a wide range of biological processes related to immune activation and viral response (online supplementary table 8), further strengthening the association of DDIR-positive status with a proinflammatory microenvironment.
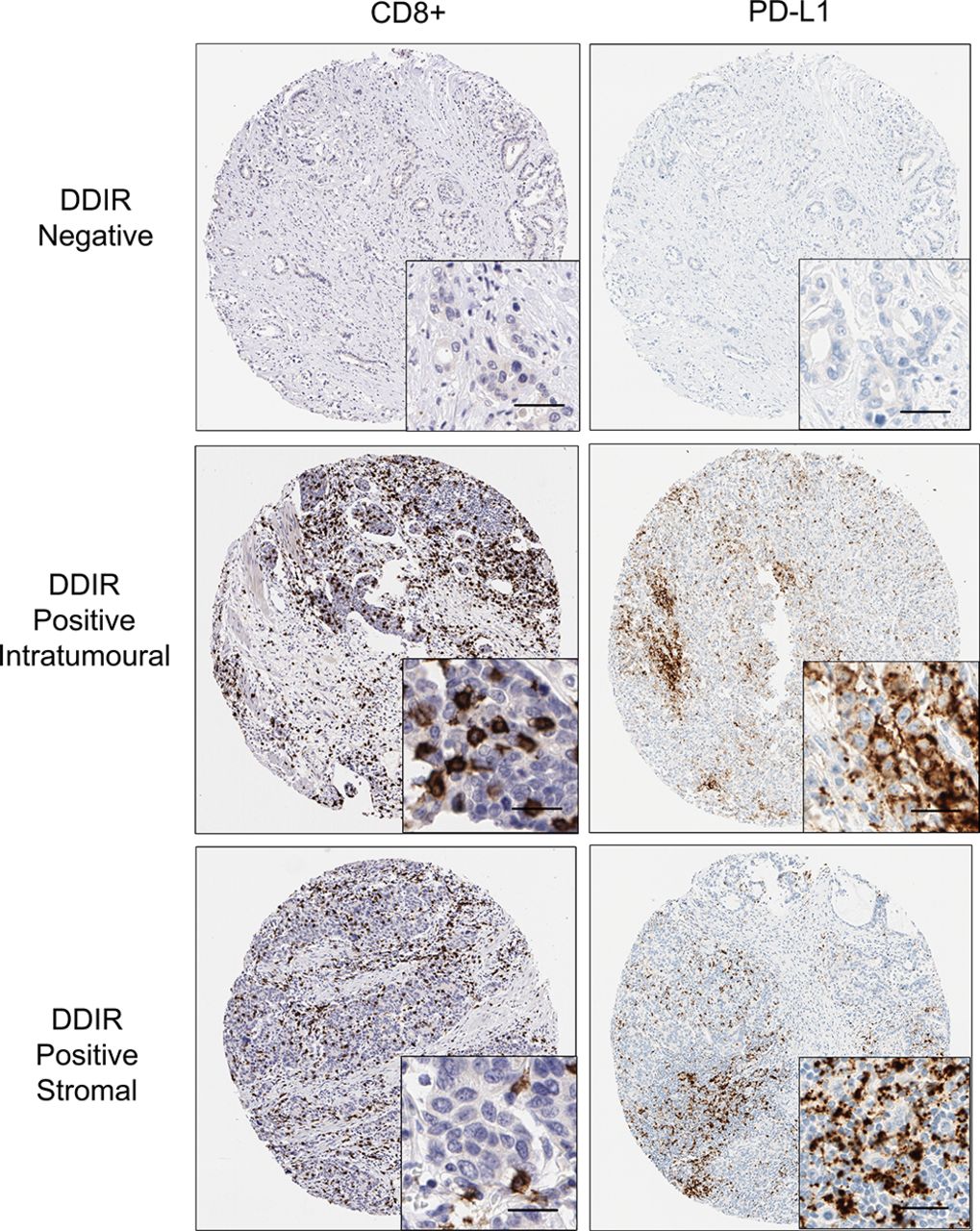
To assess the relationship between DDIR status, PD-L1 expression and the presence of TILs, we performed IHC analysis on 126 resection specimens matched to patients in the gene expression cohort (figure 3, table 3, online supplementary tables 9 and 10). Previously, published cut-offs of 1% or greater and 5% or greater were used to define PD-L1 positivity. A statistically significant association was observed between DDIR assay positivity and intratumoural and stromal PD-L1 expression at the 5% cut-off (p=0.047; p=0.25, respectively). The presence of both intratumoural and stromal CD8+ TILs was also associated with DDIR assay positivity (p<0.001; p=0.026, respectively).
{kind=link}
{kind=link}
{kind=link}
Immunohistochemistry images (×10; inset ×40) showing absence of CD8+ lymphocytes and PD-L1 staining in DDIR assay-negative tumours. Both intratumoural and stromal CD8+ lymphocytes were observed in DDIR assay-positive tumours along with PD-L1 tumours. Scale bar represents 50 µM. DDIR, DNA damage immune response; PD-L1, programmed death ligand 1.
CD8+ intratumoural and stromal lymphocytic infiltrate and PD-L1 staining assessed by IHC in DDIR-positive and DDIR-negative tumours
Discussion
We have demonstrated that the DDIR assay is predictive of response and independently prognostic following DNA-damaging neoadjuvant chemotherapy and surgical resection in OAC. DDIR assay positivity was associated with improved survival following chemotherapy and surgery and identified those patients with a higher probability of obtaining a pathological response, reduced nodal burden and clear resection margins. When assessed alongside clinical factors available at the time of diagnosis, DDIR status demonstrated superior prognostic ability compared with standard clinicopathological factors. Application of the DDIR assay to a cohort of patients who did not receive neoadjuvant therapy demonstrated no difference in survival according to DDIR status indicating that the DDIR assay may not be prognostic in its own right but only in the context of DNA-damaging therapy.
Our study has a number of advantages compared with prior attempts to identify a predictive biomarker to neoadjuvant therapy in OAC. Previous biomarker studies have relied on fresh frozen tissue, which is not routinely collected, and suffered from high attrition rates for samples analysis. However, our study used FFPE diagnostic tissue with a success rate of 95.8% in samples submitted for analysis, allowing the assay to be readily applied to clinical practice. Other attempts to develop a predictive classifier have also been limited by small sample size and lack of suitable validation sets.23–25 38 We were able to validate the DDIR assay in a sufficiently powered real-world cohort of patients to assess its predictive ability, and the assay has also undergone extensive analytical validation enabling it to be reproducibly applied to clinical samples.
Limitations of the study include the use of a retrospective clinical cohort that may influence survival outcomes due to the absence of standardised follow-up procedures and so the DDIR assay will require further validation in a randomised controlled trial dataset and by a prospective study. Also, all patients were treated with neoadjuvant platinum-based chemotherapy prior to surgical resection. Considering neoadjuvant chemoradiotherapy is standard practice in the USA and many parts of Europe; further validation is required in a sample set treated with this modality. An additional challenge for many biomarker studies is the heterogeneity demonstrated by OAC. A high level of intratumoural heterogeneity has been correlated with response to neoadjuvant chemotherapy in OAC and has indicated the limitations of a single biopsy to develop a predictive biomarker.39 This may be partially mitigated in our study by the pooling of endoscopic biopsy fragments with sufficient tumour material, but only samples from multiple sites within the tumour could encompass the underlying clonality of OAC tumours. The limited amount of tumour tissue available in the biopsy samples also precluded their use in the analysis of TILs and PD-L1 expression and so matched resection specimens were used. However, the prior administration of neoadjuvant chemotherapy may have influenced the amount of TILs present and the expression levels of PD-L1 in these specimens.
With regard to the clinical applicability of the assay, a number of factors should be taken into consideration. The response rate of 16.7% observed in DDIR-positive patients was significantly higher than that observed in DDIR-negative patients (6.8%), but is comparable with unselected published retrospective and clinical trial cohorts.4 7 This may limit the utility of the assay as a tool to enhance pathological response following neoadjuvant chemotherapy. Conversely, a response rate of 6.8% in DDIR-negative patients may not be low enough to dissuade clinicians from using neoadjuvant cisplatin-based chemotherapy in this patient population. Data from other cancer types indicating an increase in response following taxane treatment in tumours with intact DNA repair mechanisms may provide a rationale for the use of the docetaxel, oxaliplatin, fluorouracil/leucovorin chemotherapy (FLOT) regimen in DDIR-negative patients. For example, ovarian cancer patients with low/intermediate levels of BRCA1 have improved survival following treatment with platinum-based chemotherapy, whereas high levels of BRCA1 expression correlate with improved OS following the use of taxane-contatining chemotherapy.40 Similarly, in breast cancer cell lines, exogenous expression of BRCA1 increased sensitivity to spindle poisons, such as paclitaxel and vinorelbine.41 We would hypothesise that DDIR-positive patients may benefit from the direct damage to DNA induced by cisplatin or radiotherapy, whereas the DDIR-negative cases may also require the addition of inhibitors of microtubule formation, such as docetaxel. Testing of the assay in sufficiently powered randomised trial cohorts containing suitable treatment regimens could answer such a question. Further considerations regarding the utility of the assay include the association of DDIR positivity with older patients that could indicate an increased prevalence of this proinflammatory subgroup with increasing age. Also, the trend towards increased survival for DDIR-positive patients who do not receive adjuvant chemotherapy should be interpreted with caution as it is likely to be confounded by patients who had an excellent pathological response not going on to receive further chemotherapy.
The biology of a DNA repair-deficient subgroup should be examined in the context of recent publications from collaborative sequencing efforts that have characterised the molecular landscape of oesophagogastric adenocarcinoma.19 20 42 43 Multiple platform analysis by The Cancer Genome Atlas has identified four subgroups within oesophagogastric adenocarcinoma with tumours of the distal oesophagus and gastro-oesophageal junction characterised by chromosomal instability, a paucity of oncogenic driver mutations and frequent amplifications of upstream activators of signalling pathways.19 42 43 Within the stomach, tumours may also be of the genomically stable or mismatch repair subtype with the final subgroup of Epstein-Barr virus (EBV)-positive tumours occurring in the distal stomach.42 43 Mutational signature analysis of whole genome sequencing data from 129 chemotherapy-naïve OAC samples has revealed three subgroups demonstrating either deficiencies in DNA damage repair, high mutational burden or a C>A/T mutational pattern.22 The DDR-impaired subgroup constitutes 20% of patients with OAC and, while this is in keeping with a DDIR positive rate of 24%, our analysis has shown no significant overlap between the two subgroups. Reasons for this discrepancy could include the differing methodologies used to define DNA repair defects between the sequencing and gene expression dataset, the lack of a defined cut-point to call DDR impaired status in the WGS data and the limited sample size. Also, the DDIR assay takes a functional approach, capturing the inflammatory response activated by DNA damage, whereas analysis of the sequencing data assesses the pattern of mutations that occur as a result of loss of DNA repair. Furthermore, Janjigian et al performed prospective sequencing of 295 patients with metastatic oesophagogastric cancer using a capture-based next-generation sequencing (NGS) platform capable of detecting mutations, copy number alterations and selected rearrangements in up to 468 cancer genes. No single mutant allele or gene with a role in DNA repair was associated with immune response, and a surrogate marker of homologous recombination deficiency, termed the large scale transition (LST) score, was not associated with improved progression-free survival (HR 0.99, p=0.947) following first-line platinum-based chemotherapy. Higher LST scores were not observed in patients with response to first-line therapy lasting over 24 months (p=0.6) and neither did the majority of patients with prolonged responses harbour somatic alterations in know HR genes. Conversely, Smyth et al showed that assessment of homologous recombination deficiency using a genomic signature for loss of heterozygosity (LOH) derived from an NGS panel could identify a high LOH group of patients with prolonged survival following platinum-based chemotherapy.44 However, this study was limited by a high attrition rate for LOH inference (47% of samples successfully scored) and small sample numbers. A possible explanation for these results is the limitations imposed by targeted platforms that are unable to detect alterations in genes absent from the panel as well as epigenetic and transcriptional consequences of somatic mutations. Previous studies in breast cancer have shown that while BRCA1/2 mutations may confer sensitivity to DNA-damaging chemotherapy, this is not true for all cases as not all mutations may affect DNA repair or may be compensated for by alternate mechanisms.45 Conversely, BRCA1/2 wild-type tumours can possess an abnormal DNA damage response due to epigenetic silencing of BRCA1/2.46 47 Therefore, it is likely that the transcriptome-based DDIR assay is capable of capturing the downstream effects of genomic and epigenetic changes and so detect a broader range of mechanisms of DDR impairment. It is clear that a subgroup of patients with DNA repair deficiencies exists within OAC, and further work is needed to accurately characterise this patient group.
Recently, the field of DNA repair biology has enjoyed renewed interest due to its involvement in the immune response to cancer. Increased DNA damage within cancer cells has been shown to generate a highly immunogenic state within the tumours leading to the presence of TILs and the upregulation of suppressors of the immune response, such as PD-L1.48 Our data indicate a strong association between DDIR positivity and an immunogenic microenvironment. Indeed, our group has demonstrated the role of the cGAS-STING pathway in the response to DNA damage with the resultant upregulation of inflammatory cytokines such as CXCL10 and CCL5 as well as PD-L1.27 The STING pathway is activated by cytosolic DNA released from the nucleus in response to DNA damage, driving an innate immune type 1 interferon response and a subsequent upregulation of immune checkpoints including PD-L1, a key component of the DDIR signature. Furthermore, the cGAS-STING pathway has been shown to be a key player in response to immune checkpoint blockade.49 50 In keeping with this, we demonstrated increased CD8+ T cell infiltration and PD-L1 expression in DDIR-positive oesophageal tumours, both of which have been proposed as predictive biomarkers for immunotherapy agents.51 52 The presence of a DNA damage-deficient subgroup in oesophagogastric cancer may indicate sensitivity to conventional chemotherapy and response to immune checkpoint targeted agents.
In summary we have developed an array-based classifier using pretreatment FFPE biopsies to predict benefit from, and response to, neoadjuvant therapy in resectable OAC. The assay is readily applicable to routine pathological samples with potential for rapid translation into clinical use. The identification of a subgroup of tumours with deficiencies in their DNA repair mechanisms will enable these patients to be selected for more effective therapy and improve survival outcomes. Also, knowing the underlying biology of these tumours allows the possibility of further enhancing response to therapy through combinations with novel inhibitors of DNA repair and immunotherapy. Overall, the DDIR assay enables treatment selection and patient stratification in oesophagogastric adenocarcinoma and may improve response to therapy, resection rates and survival in this poor prognostic disease.
Acknowledgments
This work was supported by the Gastrointestinal Cancer Research Charitable Fund administered by the Belfast Health and Social Care Trust, the Cancer Research UK Experimental Cancer Medicine Centre Initiative, Invest Northern Ireland and Almac Diagnostics. Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) was funded by a programme grant from Cancer Research UK (RG66287). We would like to thank the Human Research Tissue Bank, which is supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre from Addenbrooke’s Hospital. Additional infrastructure support was provided from the CRUK funded Experimental Cancer Medicine Centre. RF has programmatic funding from the Medical Research Council and infrastructure support from the NIHR Biomedical Research Centre and the Cambridge Experimental Medicine Centre. Tissue samples used in this research were received from the Northern Ireland Biobank, which is funded by HSC Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast Cancer Research UK Centre and the Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre. The Northern Ireland Molecular Pathology Laboratory has received funding from Cancer Research UK, the Friends of the Cancer Centre and the Sean Crummey Foundation. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no 721906. The OCCAMS Study Group is a multicentre UK collaboration.
References
Footnotes
Contributors RCT, RK and RF contributed to the study concept, design and supervision. RCT had access to all of the data and takes responsibility for data integrity and data analysis. RCT and RK obtained funding for the study. LAK, JKB, MS, AMM, CJS and JP conducted the bioinformatic analysis. All other authors contributed to interpretation of data and critical revision of the manuscript for intellectual content.
Funding This work is funded in part by Belfast Health and Social Care Trust Gastrointestinal Cancer Research Fund, Almac Diagnostics, Medical Research Council, Cancer Research UK, HSC Research and Development Division of the Public Health Agency in Northern Ireland, National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre and Invest Northern Ireland.
Competing interests LAK, AMM, SMW, DPH and RK are employees of Almac Diagnostics and have patent declarations. GEL and CJS are employees of Almac Diagnostics.
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators On behalf of the OCCAMS Study Group: Ayesha Noorani (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Paul A W Edwards (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK, Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Nicola Grehan (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Barbara Nutzinger (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Caitriona Hughes (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Elwira Fidziukiewicz (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Jason Crawte (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Alex Northrop (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Gianmarco Contino (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Xiaodun Li (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Rachel de la Rue (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK); Maria O’Donovan (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK, Department of Histopathology, Addenbrooke’s Hospital, Cambridge, UK) Shalini Malhotra (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK, Department of Histopathology, Addenbrooke’s Hospital, Cambridge, UK); Monika Tripathi (Medical Research Council Cancer Unit, Hutchison/Medical Research Council Research Centre, University of Cambridge, Cambridge, UK, Department of Histopathology, Addenbrooke’s Hospital, Cambridge, UK); Simon Tavaré (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Andy G Lynch (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Matthew Eldridge (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Lawrence Bower (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Ginny Devonshire (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Sriganesh Jammula (Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge, UK); Jim Davies (Department of Computer Science, University of Oxford, Oxford, UK); Charles Crichton (Department of Computer Science, University of Oxford, Oxford, UK); Nick Carroll (Cambridge University Hospitals NHS Foundation Trust, Cambridge, Cambridge, UK); Peter Safranek (Cambridge University Hospitals NHS Foundation Trust, Cambridge, Cambridge, UK); Andrew Hindmarsh (Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK); Vijayendran Sujendran (Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK); Stephen J Hayes (Salford Royal NHS Foundation Trust, Salford, UK; Faculty of Medicaland Human Sciences, University of Manchester, Manchester, UK); Yeng Ang (Salford Royal NHS Foundation Trust, Salford, UK; Wigan and Leigh NHS Foundation Trust, Wigan, Manchester, UK; GI Science Centre, University of Manchester, Manchester, UK); Shaun R Preston (Royal Surrey County Hospital NHS Foundation Trust, Guildford, UK); Sarah Oakes (Royal Surrey County Hospital NHS Foundation Trust, Guildford, UK); Izhar Bagwan (Royal Surrey County Hospital NHS Foundation Trust, Guildford, UK); Vicki Save (Edinburgh Royal Infirmary, Edinburgh, UK); Richard J E Skipworth (Edinburgh Royal Infirmary, Edinburgh, UK); Ted R Hupp (Edinburgh Royal Infirmary, Edinburgh, UK); Olga Tucker (University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK; Heart of England NHS Foundation Trust, Birmingham, UK); Andrew Beggs (University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK; Institute of Cancer and Genomic Sciences, University of Birmingham, Birmingham, UK); Philippe Taniere (University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK); Sonia Puig (University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK); Jack Owsley (University Hospital Southampton NHS Foundation Trust, Southampton, UK); Hugh Barr (Gloucester Royal Hospital, Gloucester, UK); Neil Shepherd (Gloucester Royal Hospital, Gloucester, UK); Oliver Old (Gloucester Royal Hospital, Gloucester, UK); Jesper Lagergren (Guy’s and St Thomas’s NHS Foundation Trust, London, UK; Karolinska Institutet, Stockholm, Sweden) James Gossage (Guy’s and St Thomas’s NHS Foundation Trust, London, UK; King’s College London, London, UK); Andrew Davies (Guy’s and St Thomas’s NHS Foundation Trust, London, UK; King’s College London, London, UK); Fuju Chang (Guy’s and St Thomas’s NHS Foundation Trust, London, UK; King’s College London, London, UK); Janine Zylstra (Guy’s and St Thomas’s NHS Foundation Trust, London, UK; King’s College London, London, UK); Ula Mahadeva (Guy’s and St Thomas’s NHS Foundation Trust, London, UK); Vicky Goh (King’s College London, London, UK); Francesca D Ciccarelli (King’s College London, London, UK); Grant Sanders (Plymouth Hospitals NHS Trust, Plymouth, UK); Richard Berrisford (Plymouth Hospitals NHS Trust, Plymouth, UK); Catherine Harden (Plymouth Hospitals NHS Trust, Plymouth, UK); Mike Lewis (Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, UK); Ed Cheong (Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, UK); Bhaskar Kumar (Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, UK); Simon L Parsons (Nottingham University Hospitals NHS Trust, Nottingham, UK); Irshad Soomro (Nottingham University Hospitals NHS Trust, Nottingham, UK); Philip Kaye (Nottingham University Hospitals NHS Trust, Nottingham, UK); John Saunders (Nottingham University Hospitals NHS Trust, Nottingham, UK); Laurence Lovat (University College London, London, UK); Rehan Haidry (University College London, London, UK); Laszlo Igali (Norfolk and Waveney Cellular Pathology Network, Norwich, UK); Michael Scott (Wythenshawe Hospital, Manchester, UK); Sharmila Sothi (University Hospitals Coventry and Warwickshire NHS, Trust, Coventry, UK); Sari Suortamo (University Hospitals Coventry and Warwickshire NHS, Trust, Coventry, UK); Suzy Lishman (Peterborough Hospitals NHS Trust, Peterborough City Hospital, Peterborough, UK); George B Hanna (Department of Surgery and Cancer, Imperial College London, UK); Krishna Moorthy (Department of Surgery and Cancer, Imperial College London, UK); Christopher J Peters (Department of Surgery and Cancer, Imperial College London, UK); Anna Grabowska (Queen’s Medical Centre, University of Nottingham, Nottingham, UK); and Helen Coleman (Centre for Cancer Research and Cell Biology, Queen’s University Belfast, Northern Ireland, UK).
Correction notice This article has been corrected since it published Online First. The author correspondence details have been corrected.
Patient consent for publication Not required.