Article Text

Abstract
Objective The Fragile X mental retardation (FMR) syndrome is a frequently inherited intellectual disability caused by decreased or absent expression of the FMR protein (FMRP). Lack of FMRP is associated with neuronal degradation and cognitive dysfunction but its role outside the central nervous system is insufficiently studied. Here, we identify a role of FMRP in liver disease.
Design Mice lacking Fmr1 gene expression were used to study the role of FMRP during tumour necrosis factor (TNF)-induced liver damage in disease model systems. Liver damage and mechanistic studies were performed using real-time PCR, Western Blot, staining of tissue sections and clinical chemistry.
Results Fmr1null mice exhibited increased liver damage during virus-mediated hepatitis following infection with the lymphocytic choriomeningitis virus. Exposure to TNF resulted in severe liver damage due to increased hepatocyte cell death. Consistently, we found increased caspase-8 and caspase-3 activation following TNF stimulation. Furthermore, we demonstrate FMRP to be critically important for regulating key molecules in TNF receptor 1 (TNFR1)-dependent apoptosis and necroptosis including CYLD, c-FLIPS and JNK, which contribute to prolonged RIPK1 expression. Accordingly, the RIPK1 inhibitor Necrostatin-1s could reduce liver cell death and alleviate liver damage in Fmr1null mice following TNF exposure. Consistently, FMRP-deficient mice developed increased pathology during acute cholestasis following bile duct ligation, which coincided with increased hepatic expression of RIPK1, RIPK3 and phosphorylation of MLKL.
Conclusions We show that FMRP plays a central role in the inhibition of TNF-mediated cell death during infection and liver disease.
- liver damage
- septic shock
- Fragile X syndrome
- TNF receptor signaling
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Viral infection, septic shock and cholestasis can trigger liver damage.
Tumour necrosis factor (TNF) pathway is well studied in promoting liver damage and fibrosis.
What are the new findings?
Fragile X mental retardation protein (FMRP) is critical for preventing TNF-induced cell death.
FMRP negatively regulates RIPK1 to prevent cell death.
FMRP-deficient mice developed severe pathology during acute cholestasis, septic shock and virally induced hepatitis.
The RIPK1 inhibitor Necrostatin-1s could alleviate disease in the absence of FMRP.
How might it impact on clinical practice in the foreseeable future?
Patients exhibiting sequence alterations in the Fmr1 gene may exhibit increased liver cell death in severe hepatotoxic situations.
Introduction
Liver damage can be triggered by hepatic infections, immune activation and intoxication.1–3 Prolonged tissue damage can result in fibrosis, cirrhosis and liver failure, which affects more than one million casualties worldwide.1 Tumour necrosis factor α (TNF) is a central cytokine during liver damage.1 3 Consistently, injection of TNF can cause severe liver damage and septic shock, when applied in combination with D-Gal, which is dependent on TNFR1.3 Notably, TNF has been shown to promote liver damage and fibrosis in a variety of animal models including acute cholestasis and toxic liver damage.4 5 Mechanistically, TNF binds to TNFR1, which can mediate cell activation via NF-κB, and apoptosis via caspase activation.6 On TNF binding, TNFR1 forms a complex with TRADD and RIPK1 with latter being the central molecular switch guided by its ubiquitination status.6 Non-ubiquitinated RIPK1 dissociates from the cell membrane to form cytosolic complexes termed IIa, IIb or IIc that promote cell death rather than NF-κB activation.6 Hence, either the lack of cIAPs that can ubiquitinate RIPK1 or the deubiquitination enzyme cylindromatosis (CYLD) results in reduced K63-linked polyubiquitin chains conserving non-ubiquitinated RIPK1 and promoting cell death.7–9 Non-ubiquitinated RIPK1 can recruit RIPK3, which is an elementary switch from apoptosis to necroptosis.10–12 Necroptosis depends on the kinase activity of RIPK3, which can be alleviated by caspase-mediated cleavage of the kinase domain.13 Accordingly, inhibition of caspase activity by c-FLIPS cannot inactivate RIPK3 and promotes necroptosis.14 15
During viral infections of the liver, viral-specific cytotoxic CD8+ T cells target infected hepatocytes to limit viral replication and induce tissue damage rather than direct cytolytic effects of the virus. Consistently, the murine, poorly cytolytic, lymphocytic choriomeningitis virus (LCMV) model system induces a CD8+ T-cell-mediated hepatitis, which is associated with increased liver cell enzymes in the blood stream.16 Hence, depletion of CD8+ T cells results in absent tissue damage during LCMV infection, although high viral load can be detected in the liver.16 CD8+ T-cell-mediated cytokine production such as secretion of TNF triggers liver damage following viral hepatitis.17 Consistently, viral-specific CD8+ T-cells induce liver cell death coinciding with activation of caspase-3 (Casp3).18
The leading cause for inherited intellectual disability is the Fragile X mental retardation (FMR) syndrome, caused by CGG repeats in the locus Xq27.3.19 20 Insertion at the folate-sensitive fragile site causes genetic instability of the Fmr1 gene, leading to reduced or absent expression of the FMR protein (FMRP).21 22 Fmr1 knockout (KO) mice exhibit learning deficits and deregulation of presynaptic and postsynaptic proteins.23 Additionally, Fmr1null mice exhibit macroorchidism and increased ovarian weight.23 24 FMRP is an RNA-binding protein known to regulate translation of over 40 proteins in the central nervous system (CNS). Additionally, FMRP was shown to bind over 6000 transcripts.24–26 Interestingly, increased expression of Fmr1 mRNA levels and FMRP correlates with prognostic indicators denoting aggressive breast and lung cancers.27 However, despite its ubiquitous expression, the physiological role of FMRP outside the CNS remains poorly understood.
Expression of FMRP in secondary lymphoid organs raised the question whether it affects anti-viral immunity. We therefore infected Fmr1null mice with LCMV and monitored increased CD8+ T-cell-mediated liver damage. No gross immune defects in the anti-LCMV defence pointed to an increased sensitivity of targeted liver cells. Consistently, we found increased liver damage following exposure of Fmr1null mice to D-galactosamine (D-Gal)/TNF along with increased presence of cell death markers. Mechanistically, increased expression of RIPK1 resulted in activation of apoptosis and necroptosis pathways. Consistently, treatment with the RIPK1 inhibitor Necrostatin-1s (Nec-1s) could reduce liver cell death and liver damage. During acute cholestasis following bile duct ligation (BDL) in mice, we observed increased presence of RIPK1, RIPK3 and phosphorylation of MLKL with severe RIPK1-dependent pathology.
Materials and methods
Animals
All experiments were performed under the authorisation of LANUV in accordance with German law for animal protection. Fmr1 KO (Fmr1null ) mice were purchased from Jackson Laboratories (USA) and maintained under specific pathogen-free conditions. Fmr1null mice were on a C57Bl/6 background (F8-F10) and compared with control C57Bl/6 mice. Key experiments were performed with littermate controls. For BDL, laparotomy was performed predominantly on male mice at 10–14 weeks of age, animals were anaesthetised by isoflurane and placed on a heating pad. Animals were shaved and the skin was disinfected with 70% ethanol and povidone-iodine. A midline incision in the upper abdomen was made and the common bile duct and the bile bladder were identified, isolated and ligated with three surgical knots using silk. Abdomen and peritoneum were closed with a running silk suture. Sham treatment was performed similarly but without ligation of the bile duct and bile bladder. Animals were monitored during recovery and treated with carprofen (0.05 mg/kg) after surgical intervention. Mice exhibiting severe disease symptoms where sacrificed and considered as dead. For blood and tissue collection, mice were anaesthetised, bled and serum was collected by retro orbital vein puncture. The organs were stored at −80°C for histology, RNA and protein extraction. For injections, age-matched and sex-matched control and Fmr1null mice were intraperitoneally injected with 10 mg D-Gal (Sigma, dissolved in phosphate-buffered saline (PBS) at a concentration of 100 mg/mL) and 15 min later intravenously injected with the indicated doses of rTNFα (R&D Systems, dissolved in PBS at a concentration of 100 µg/mL). For R-7-Cl-O-Nec-1 (Nec-1s) treatment experiments, mice were intraperitoneally injected with 10 mg D-Gal (Sigma) and 6 µg/g Nec-1s (Abcam, dissolved in dimethyl sulfoxide (DMSO) at a concentration of 20 mg/mL) or vehicle (DMSO, Sigma), then 15 min later intravenously injected with 100 ng/mouse rTNFα (R&D Systems). Mice were intraperitoneally injected with FS-7-associated surface antigen (Fas) (clone Jo2, 0.5 mg/mL, BD Pharmingen). Mice were intraperitoneally injected with 100 µg/mouse Etanercept (Pfizer, dissolved at a concentration of 10 mg/mL in PBS) 1 day before LCMV infection, then three times/week during LCMV infection.
Virus
LCMV strain W.E. (WE) was originally obtained from F. Lahmann-Grube (Heinrich Pette Institute). Virus was inoculated via intravenous tail vein injection. Virus titres were measured with a plaque-forming assay as previously described.16
Primary hepatocytes isolation
Primary hepatocytes were isolated from C57BL/6J control and Fmr1null mice using a collagenase perfusion technique. Briefly, livers were perfused with Hanks’ balanced salt solution (HBSS) buffer with 1% glucose and 2.5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) for 4 min. Then, liver tissue was perfused with 0.03 mg/mL collagenase for 6–8 min in HBSS buffer with 1% glucose and 5 mM Cacl2. The isolated primary hepatocytes were cultured in William medium.
Histology
Histological analysis of snap-frozen tissue was performed as previously described.16 Snap-frozen tissue sections were stained with anti-alpha smooth muscle actin (αSMA; abcam), anti-F4/80, Ly6G (eBioscience), anti-active Casp3 (BD Biosciences), anti-proliferating cell nuclear antigen (PCNA; Cell Signaling). Casp3 activity was performed with a fluorescence assay according to the manufacturer’s instructions (Cell Signaling). Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining was performed on liver sections according to the manufacturer’s instructions (Roche). Fluorescence images were taken with an Axio Observer fluorescence microscope (Zeiss). Quantification of fluorescence staining was analysed by ImageJ, with either measurement of the mean fluorescent intensity (MFI) or the numbers of positive signals.
Flow cytometry
Different immune populations were identified from single cell solutions harvested from organ as indicated. Experiments were performed on BD LSRFortessa and analysed using FlowJo software. For tetramer staining, singly suspended cells were incubated with tetramer-glycoprotein (gp)33 and nucleoprotein (np)396 for 15 min, or incubated with tetramer-gp61 for 30 min at 37°C. After incubation, surface anti-CD8 (eBioscience) or anti-CD4 (eBioscience) antibodies were added for 30 min at 4°C. For intracellular cytokine staining, single suspended cells were stimulated with LCMV-specific peptide gp33, and gp61 for 1 hour after which Brefeldin A (eBiocience) was added for another 5 hours and incubated at 37°C, followed by permeabilisation and addition of anti-interferon gamma (IFNγ) or anti-TNFα antibodies (eBioscience).
Bile acids analysis
Bile acids and their glycine and taurine derivatives were analysed by ultra performance liquid chromatography - tandem mass spectrometer (UPLC-MS/MS). The system consists of an Acquity UPLC-I Class (Waters) coupled to a Waters Xevo-TQS tandem mass spectrometer equipped with an electrospray ionization source in the negative ion mode. Data were collected in the multiple reaction monitoring mode.
Casp3 activity assay
Casp3 activity assay kit (Cell Signaling, #5723) was used on liver tissue lysates by following the manufacturer’s instructions.
Quantitative real-time PCR
RNA was isolated using Trizol (Invitrogen; 15596018) and real-time PCR analyses were performed according to the manufacturer’s instructions (Applied Biosystems). For analysis, expression levels were normalised to GADPH (∆Ct). Gene expression values were then calculated based on the ∆∆Ct method, using the mean naive mice as a control to which all other samples were compared. Relative quantities (RQ) were determined using the equation: RQ=2∧-∆∆Ct.
Immunoblotting
Liver tissue was lysed in PBS containing 1% Triton X-100 lysis buffer, protease inhibitors cocktail and PhosSTOP (Roche). Equal amounts of the total protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane, after blocking probed with specific primary and secondary antibodies. The blots were developed using enhanced chemiluminescence (ECL) western blotting substrates (Thermo Fisher) or Li-COR. The following antibodies from Abcam were used: ASK1, FMRP, PARP, Phospho-MLKL. The following antibodies from Santa Cruz were used: FLIPS/L and cylindromatosis. The following antibodies from Cell Signaling were used: cleaved-caspase-8 (Casp8), Casp3, IκBα, c-IAP1, XIAP, SMAC, FMRP, Phospho-ASK1, MKK4, Phospho-MKK4, SAPK/JNK, Phospho-SAPK/JNK, Phospho-c-Jun, Phospho-Erk1/2, RIPK1, PCNA, RIPK3 and MLKL.
Immunoprecipitation
To monitor phosphorylation and ubiquitination of RIPK1, we used TNF-Flag treatment.28 Liver tissue was homogenised in buffer containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-Potassium hydroxide pH7.5, 0.2% NP-40, 120 mM NaCl, 0.27M sucrose, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 2 mM Na3VO4, cOmpleteTM Protease Inhibitor Cocktail, 2 mM phenylmethylsulfonyl fluoride and 10 mM N-Ethylmaleimide. Samples were either incubated with anti-FLAG M2 magnetics beads (Sigma) for 4 hours or RIPK1 antibody (Cell Signaling) overnight followed by 2 hours incubation with magnetics beads (Bio-rad) at 4°C. The beads were washed and proteins were eluted in 70°C with sample buffer and the eluted proteins were fractionated by SDS-PAGE gels. Proteins were detected by immunoblotting as described above.
Bioinformatic analyses
Target genes of FMRP were selected on the basis of enrichment in Rip-chip data published previously.24 The biological classification of associated genes in terms of their biological processes was obtained by Gene Ontology (GO) analysis using the Protein Analysis through Evolutionary Relationships (PANTHER) classification system (PANTHER V.9.0; http://www.pantherdb.org).29 30
Statistical analyses
Data are expressed as mean±SEM. Statistically significant differences were determined with a Student’s t-test, a one-way analysis of variance (ANOVA), two-way ANOVA and Log-rank (Mantel-Cox). All the quantifications were analysed by ImageJ.
Results
Absence of FMRP results in increased CD8+ T-cell-mediated liver damage during viral hepatitis
FMRP expression in spleen, liver and lymph node tissue prompted us to investigate whether Fmr1 also plays a critical role during viral infection (online supplementary figure 1). On infection of control and Fmr1null mice with LCMV-WE, we observed higher activity of the enzyme alanine aminotransferase (ALT) along with increased bilirubin concentrations in the serum of Fmr1null mice (figure 1A, B). Liver tissue harvested from Fmr1null animals exhibited increased presence of highly nucleated cells when compared with liver tissue from control animals (figure 1C). Furthermore, we observed increased expression of αSMA, an indicator of fibrosis, in the liver tissue of Fmr1null mice compared with control animals (figure 1D). During infection with LCMV, infected hepatocytes are targeted by viral-specific CD8+ T cells, and this is followed by cell death of infected cells.16 18 Liver sections of Fmr1null mice exhibited increased DNA fragmentation indicating cell death, following infection (figure 1E). Notably, the infiltration of macrophages or granulocytes into liver tissue following infection appeared similar between Fmr1null and control mice (online supplementary figure 2A). Due to the increased liver damage in Fmr1null mice, we wondered whether anti-viral CD8+ T-cell immunity was increased in the absence of FMRP. As expected, increased liver damage in Fmr1null mice was dependent on cytotoxic T-cell immunity as depletion of CD8+ T cells blunted liver damage in both control and Fmr1null mice (figure 2A). However, we did not observe a difference in the numbers of circulating viral-specific CD8+ or CD4+ T cells (figure 2B). Furthermore, the surface expression of interleukin 7 receptor which is associated with T-cell survival and programmed cell death-1 along with other surface molecules associated with T-cell exhaustion,31 was similar on anti-viral T cells, suggesting that anti-viral T-cell immunity was unaffected by FMRP (online supplementary figure 2B). Consistently, the proportion of effector, effector memory and central memory T cells was similar between control and Fmr1null mice (online supplementary figure 2C). IFNγ and TNF production in CD8+ T cells following re-stimulation with LCMV-specific epitopes was not dependent on the presence of FMRP in spleen or liver tissue, which was consistent with similar frequencies of LCMV-specific CD8+ T cells in both groups (figure 2C,D, online supplementary figure 2D, E). These data indicate that cytotoxic T-cell immunity was not affected by lack of FMRP. In further support of this, CD4+ T-cell frequencies were similar in spleen and liver tissue between control and Fmr1null animals (online supplementary figure 2F). Hence, we concluded that FMRP is dispensable for induction of viral-specific immunity against LCMV and wondered whether FMRP might influence LCMV replication. However, we observed no significant difference in viral titres in organs harvested from control and Fmr1null mice following infection (figure 2E). The presence of LCMV nucleoprotein (NP) was similar between liver tissue of control and Fmr1null mice (figure 2F). Since T cells secrete TNF during LCMV infection, and TNF can contribute to aggravated LCMV-induced liver damage,17 we speculated that blockage of TNF signalling might reduce severe liver damage in absence of FMRP. Consistently, when we applied TNFR2-Fc fusion protein (Etanercept) as TNF inhibitor, we found reduced presence of liver enzymes including ALT, aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) in the sera of infected Fmr1null mice when compared with untreated Fmr1null animals (figure 2G). Taken together, these data indicate that FMRP is important for reducing liver damage during viral infection but does not affect the induction of anti-viral immunity in this setting.
Supplemental material

FMRP alleviates virally induced hepatitis LCMV infection. (A–E) Mice were infected with 2×106 pfu of LCMV-WE. (A) ALT activity was measured in serum of control and Fmr1null (Fmr1 KO) mice at the indicated time points (n=5–7). (B) Bilirubin was measured in the serum of control and Fmr1null mice infected with LCMV-WE at the indicated time points (n=4). (C) Haematoxylin and eosin staining was performed on liver sections of control and Fmr1null mice at day 12 post-infection (n=4, scale bar=50 µm). Representative sections are shown. Right panel indicates quantification. (D) Liver tissue sections harvested from control and Fmr1null mice were stained for αSMA at day 12 post-infection (n=4, scale bar=100 µm). Representative sections are shown. Right panel indicates quantification. (E) Sections from liver tissue harvested from control and Fmr1null mice at day 12 post-infection were stained for TUNEL (n=4, scale bar=100 µm). Representative sections are shown. Right panel indicates quantification. *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; αSMA, alpha smooth muscle actin; FMRP, Fragile X mental retardation protein; KO, knockout; LCMV, lymphocytic choriomeningitis virus; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.

Increased liver damage in absence of FMRP is triggered by CD8+ T cells and TNF. (A) CD8+ T cells were depleted in control and Fmr1null (Fmr1 KO) mice. ALT activity and total bilirubin concentration were measured after infection at the indicated time points (n=4). (B) Anti-viral T-cell response was measured in blood samples using T-cell-specific tetramers of gp33, np396 and gp61 in control and Fmr1null mice at indicated time points after infection (n=5-10). (C) Spleen and (D) liver tissue were harvested from control and Fmr1null mice at day 12 post-infection. Single cell suspensions were stimulated with gp33 and np396 peptides followed by staining with anti-IFNγ and anti-TNFα antibodies in CD8+ T cells (n=12). (E) LCMV titres were measured in different organs from control and Fmr1null mice at day 12 post-infection (n=12). (F) Liver tissue sections harvested from control and Fmr1null mice were stained for LCMV-NP (clone VL4) (n=3–6, scale bar=50 µm). Representative sections are shown. (G) Control and Fmr1null mice were treated with Etanercept. ALT, AST and LDH activities were measured after infection as indicated (n=3–4). *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; FMRP, Fragile X mental retardation protein; gp33, glycoprotein 33; IFN, interferon; KO, knockout; LCMV, lymphocytic choriomeningitis virus; LDH, lactate dehydrogenase; NC, negtive control; np396,nucleoprotein 396; TNF, tumour necrosis factor.
TNF-mediated liver damage can be alleviated by FMRP
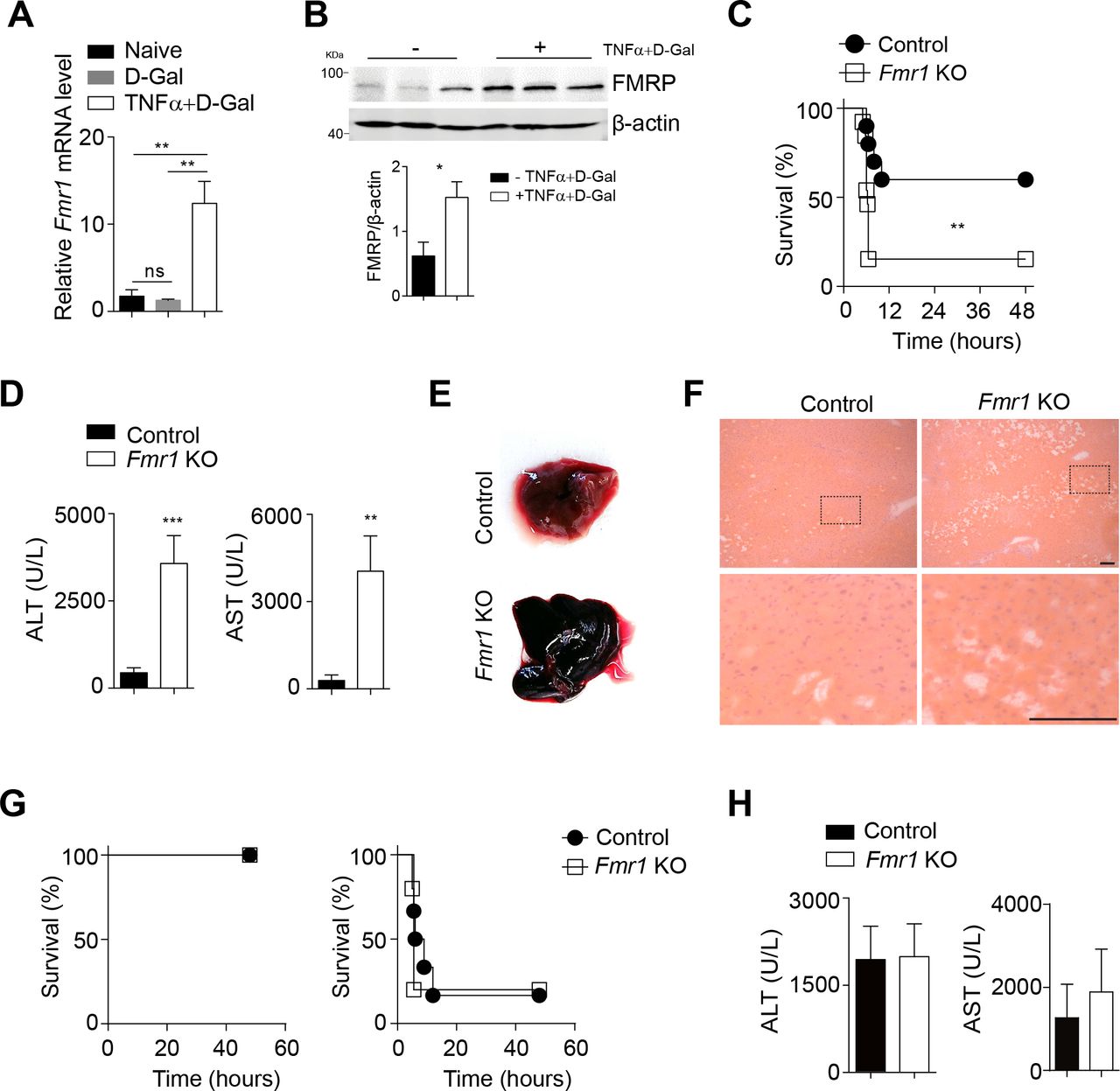
TNF can induce liver damage via TNFR1 signalling when injected in combination with D-Gal.3 Following injection of TNF and D-Gal into C57Bl/6 mice, we observed increased expression levels of Fmr1 in liver tissue (figure 3A). Consistently, protein expression of FMRP was increased in liver tissue following TNF/D-Gal treatment (figure 3B). We therefore hypothesised that FMRP might be upregulated as a protection mechanism against TNF-mediated damage. When challenged with D-Gal/TNF, the survival of Fmr1null mice was reduced compared with control mice (figure 3C), while exposure to D-Gal alone did not cause severe pathology (online supplementary figure 3A). ALT and AST activities in the serum of Fmr1null mice were increased when compared with control mice (figure 3D). The livers of Fmr1null mice appeared macroscopically enlarged and dark red when compared with control liver tissue (figure 3E). Moreover, histological analysis showed the disruption of hepatic organisation and reduced presence of nuclei in liver tissue from Fmr1null mice when compared with control mice (figure 3F).32 We did not observe a difference in the infiltration of macrophages or granulocytes (online supplementary figure 3B). Next, we wondered whether FMRP inhibits liver damage induced by other receptors than that for TNF such as the well-studied death receptor CD95.33 When we exposed control and Fmr1null mice to an anti-CD95 activating antibody, there was no difference between survival or ALT/AST activity in the sera between the two groups (figure 3G, H). Taken together, while we determined that FMRP is important in reducing TNF-mediated liver damage and incurring protection from lethal septic shock, it remained unclear whether these effects could affect cell death.

Lack of FMRP triggers increased susceptibility towards TNF-induced septic shock. (A) Fmr1 gene expression was quantified in liver tissue of C57Bl/6 mice 3 hours post D-Gal (10 mg/mouse) and rTNF (100 ng/mouse) injection (n=4–5). (B) Liver lysates were probed for FMRP at 3 hours post D-Gal/rTNF injection. Bottom panels indicate quantification. (C–F) Control and Fmr1null (Fmr1 KO) mice were treated with D-Gal (10 mg/mouse) and rTNF (100 ng/mouse). (C) Survival of control and Fmr1null mice was monitored (n=14). (D) ALT (n=15) and AST (n=6) activities were measured in serum samples of control and Fmr1null mice 5 hours post-injection. (E) Morphology of whole liver tissue from control and Fmr1null mice was assessed 5 hours after treatment. (F) Haematoxylin and eosin staining of liver tissue sections from control and Fmr1null mice 5 hours after treatment (n=3, scale bar=50 µm). Representative sections are shown. (G) Control and Fmr1null mice were treated with 0.28 µg/g (left panel, n=3) and 0.56 µg/g (right panel, n=6-7) Fas antibody and survival was monitored. (H) ALT (n=9) and AST(n=3) activities were measured in serum samples of control and Fmr1null mice 3 hours post-Fas antibody treatment (0.56 µg/g). *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; FMRP, Fragile X mental retardation protein; KO, knockout; TNF, tumour necrosis factor.
Absence of FMRP results in increased TNF-mediated cell death
A higher degree of DNA fragmentation was observed in liver tissue harvested from Fmr1null mice when compared with control animals after D-Gal/TNF stimulation (figure 4A). DNA damage can be repaired by the enzyme Poly(ADP—ribose)-Polymerase 1 (PARP).34 PARP is cleaved during cell death to prevent DNA repair.35 We observed increased PARP cleavage in protein lysates harvested from liver tissue of Fmr1null animals when compared with controls (figure 4B, online supplementary figure 4A). PARP cleavage is mediated by Casp3, a common effector caspase shared by the intrinsically and extrinsically induced cell death pathways.6 As expected, we observed increased cleavage and activity of Casp3 in liver tissue of Fmr1null mice when compared with control animals (figure 4B–D). Effector caspases are activated by initiator caspases such as Casp8 during TNFR1-mediated cell death.6 We observed increased Casp8 cleavage in D-Gal/TNF stimulated Fmr1null mice in comparison to control animals (figure 4E, F). These in vivo effects were corroborated in vitro as we also found more cleaved PARP and Casp8 in primary hepatocytes harvested from Fmr1null mice than in hepatocytes from control mice stimulated with cyclohexamide and TNF (figure 4G).

Fmr1null mice exhibit increased TNF-mediated cell death. (A) TUNEL staining of liver tissue sections from control and Fmr1null (Fmr1 KO) mice 5 hours post D-Gal (10 mg/mouse) and rTNF (100 ng/mouse) treatment. Right panel indicates quantification (n=5, one representative picture is shown, scale bar=100 µm). (B) Liver tissue homogenates from control and Fmr1null mice were assessed for expression of cleaved-PARP and Casp3 (one representative immunoblot of n=3 is shown). Bottom panel indicates quantification for cleaved-Casp3. (C) Casp3 activity in liver tissue homogenates was performed in control and Fmr1null mice 5 hours post D-Gal/rTNF treatment (n=5). (D–E) Sections of liver tissue from control and Fmr1null mice were stained for active Casp3 (D) and cleaved-Casp8 (E) 5 hours post D-Gal/rTNF treatment (n=5, scale bar=100 µm). Representative sections are shown. (F) Liver tissue homogenates harvested from Control and Fmr1null mice were analysed for expression of cleaved-Casp8 at the indicated time points (one representative set of n=3 immunoblots is shown). Bottom panel indicates quantification. (G) Primary hepatocytes were isolated from naive control and Fmr1null mice and treated with cyclohexamide (10 µg/mL) and rTNF (40 ng/mL) for 8 hours. Cell lysates were analysed for the presence of cleaved-Casp8 and PARP (one representative immunoblot of n=4 is shown). Right panels indicate quantification. (H) Liver tissue homogenates from control and Fmr1null mice were assessed for expression of IκBα (one representative immunoblot of n=3 is shown). (I) Expression levels of genes as indicated were quantified in control and Fmr1null mice at indicated time points after D-Gal/rTNF treatment (n=3–4). *p<0.05, **p<0.01, ***p<0.001. Casp3, caspase-3; Casp8, caspase-8; IκBα; inhibitor of κB α; KO, knockout; PARP, poly(ADP—ribose)-polymerase 1; TNF; tumour necrosis factor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.
TNFR1 activation can result in both Casp8-mediated apoptosis and NF-κB activation. NF-κB is retained in the cytosol by binding to an inhibitor of κB α (IκBα). Accordingly, proteasomal degradation of IκBα results into release of NF-κB, which can translocate to the nucleus to induce transcription of pro-inflammatory cytokines.6 Degradation of IκBα was observed in both FMRP-deficient and control mice following stimulation with TNF (figure 4H). Consistently, we monitored similar translocation of the NF-κB subunit p65 into the nucleus in liver tissue from Fmr1null and control mice over time (online supplementary figure 4B). Moreover, expression levels of genes typically induced by NF-κB were similar in liver tissue from control or Fmr1null mice (figure 4I). Notably, at later time points, we observed a slightly but significantly increased expression of Il2, Ccl2, Cxcl10 and Cxcl12 in Fmr1null liver tissue when compared with controls (online supplementary figure 4C). These data indicate that in the absence of FMRP while TNF-mediated cell death is highly increased, TNF-mediated NF-κB activation is only marginally affected.
Inhibition of RIPK1 reduces TNF induced cell death in absence of FMRP
Next, we investigated the molecular mechanism by which FMRP inhibits cell death. A previous study identified over 6000 transcripts that can potentially bind to FMRP through the binding consensus sequences ACU (G or U) or (A or U) GGA.24 By GO analysis using the PANTHER classification system, about 6% of these genes clustered in the ‘response to stimulus’ category (online supplementary figure 5A). Functional categories identified targets encoding for genes involved in cell death and the TNF signalling pathway (online supplementary figure 5B), with multiple binding consensus sites in genes involved in cell death (figure 5A). We found increased RNA expression levels of Casp8 in Fmr1null mice compared with control animals following stimulation with TNF, but not in other genes encoding for proteins critical for cell death, which were measured (figure 5B). At the protein level, we found protein expression of cIAP1, cIAP2, XIAP, SMAC and A20 to be comparable between both groups after TNF/D-Gal challenge (online supplementary figure 6A-E). As FMRP-binding sites were observed in genes involved in the MAPK signalling pathway, we next evaluated the effects of FMRP deficiency on this pathway following TNF stimulation (figure 5A). We observed increased expression and phosphorylation of ASK1, along with increased phosphorylation of MKK4 following stimulation with TNF (online supplementary figure 6F, G). Moreover, sustained JNK activation has been shown to be associated with increased TNF-dependent cell death and hepatitis.36–38 Consistently, we observed increased phosphorylation of JNK, and the transcription factor Jun after TNF exposure in absence of FMRP when compared with controls (figure 5C, D, online supplementary figure 6H), while activation of ERK was unaffected (figure 5D, online supplementary figure 6I). Activated JNK was previously shown to induce turnover of c-FLIPLONG, which inhibits TNF-mediated apoptosis.39 However, we did not see a significant difference in c-FLIPLONG (figure 5E, online supplementary figure 6J).
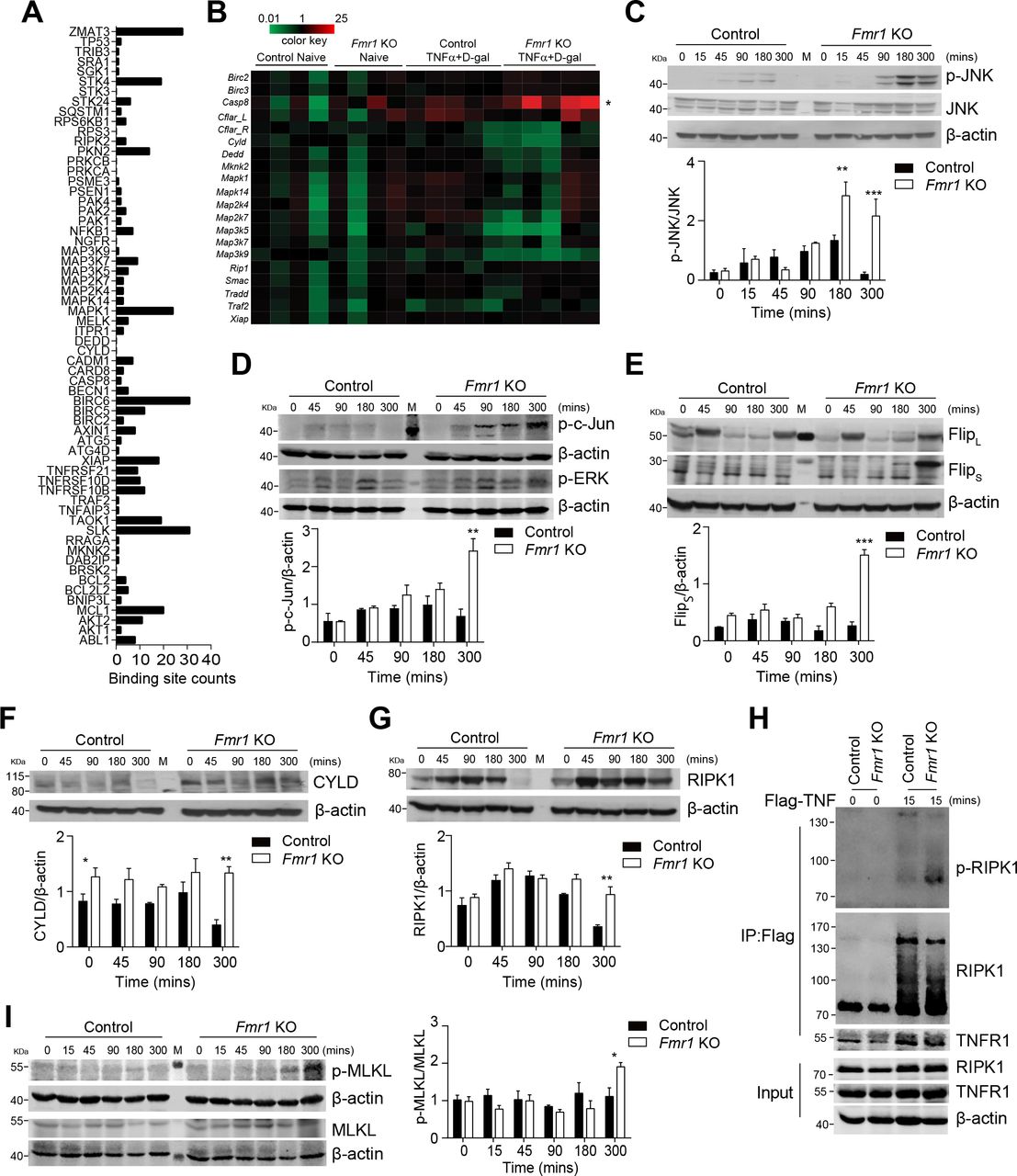
Expression of RIPK1 is increased during TNF-induced liver damage in absence of FMRP. (A) FMR1-binding sites on apoptosis-related genes derived from the publically available FMR1 PAR-CLIP data6 were analysed. (B) Control and Fmr1null (Fmr1 KO) mice were treated with D-Gal (10 mg/mouse) and rTNF (100 ng/mouse). Gene expression was quantified as indicated in liver tissue 3 hours post-treatment (n=4–5). (C–G) Control and Fmr1null mice were treated with TNF and D-Gal and liver tissue was harvested at the indicated time points. Liver lysates were assessed for expression of JNK and phosphorylated JNK (C), phosphorylated c-Jun and p-ERK (D), FLIPL and FLIPS (E), CYLD (F), RIPK1 (G). One representative set of immunoblots of n=3 is shown. Bottom panels show quantification as indicated. (H) Control and Fmr1null mice were treated with Flag-mTNFα (200 ng/mouse). TNF-complex was immunoprecipitated using anti-Flag beads and RIPK1, p-RIPK1 and TNFR1 were analysed by immunoblotting. One representative of n=4 is shown. (I) Control and Fmr1null mice were treated with TNF and D-Gal and liver tissue was harvested at the indicated time points. Liver lysates were assessed for expression of p-MLKL and MLKL. One representative set of immunoblots of n=3 is shown. Right panel shows quantification as indicated. *p<0.05, **p<0.01, ***p<0.001. CYLD, cylindromatosis; FMRP, Fragile X mental retardation protein; TNF, tumour necrosis factor.
In turn, the deubiquitinase CYLD promotes TNF-induced JNK activation and might contribute to increased phosphorylation of JNK and Jun.40 CYLD also inhibits tumour cell proliferation and Cyld-/- mice suffer from increased tumour susceptibility.41 42 Consistent with enhanced JNK phosphorylation, increased protein expression of CYLD was observed in untreated and TNF stimulated liver tissue of Fmr1null mice when compared with controls (figure 5F). Furthermore, CYLD catalyses deubiquitination of RIPK1, which consequently dissociates from the TNFR1 complex and is released into the cytosol in order to form a complex with fas-associated protein with death domain (FADD) and Casp8 to promote cell death.6 7 RIPK1 protein expression was increased in the absence of FMRP following exposure to TNF when compared with controls (figure 5G). Increased RIPK1 expression correlates with a higher susceptibility to cell death.6 In addition, FLIPS is upregulated in FRMP-deficient cells (figure 5E). FLIPS is also known to promote formation of the riptoptosome and accordingly necroptotic cell death.14 15 In contrast to FLIPL, which forms a catalytic active heterodimer with Casp8 that cleaves and inactivates RIPK1, the heterodimer between Casp8 and FLIPS does not cleave RIPK1. This leads to FLIPS-dependent stabilisation of RIPK1 and RIPK1-mediated necroptosis induction.14 15 Hence, we speculated that increased presence of RIP1K resulted in enhanced susceptibility towards TNF-mediated apoptosis and necroptosis in the absence of FMRP. Consistently, we observed increased phosphorylation and reduced ubiquitination of RIPK1 following immunoprecipitation of TNF-Flag from liver tissue of Fmr1null mice compared with control mice after treatment (figure 5H). During necroptosis, the pseudokinase MLKL is phosphorylated.43 Accordingly, p-MLKL levels were increased in liver tissue harvested from Fmr1null mice compared with control mice (figure 5I). Taken together, these data indicate that absence of FMRP results in increased apoptosis and necroptosis pathways following TNF/D-Gal treatment.
RIPK1 can be inhibited by Necrostatin-1, and, with higher affinity by 7-Cl-O-Nec-1 (Nec-1s), which is also suitable for animal models.44 Following treatment of Fmr1null mice with Nec-1s, we observed reduced phosphorylation of RIPK1 (figure 6A). Accordingly, application of Nec-1s resulted in reduction of p-MLKL in liver tissue of Fmr1null mice (figure 6B). Moreover, reduced presence of active Casp3 was detected in liver tissue of Nec-1s treated Fmr1null mice compared with untreated Fmr1null mice (figure 6C). Consequently, TUNEL staining in liver tissue sections from Nec-1s treated Fmr1null mice was reduced when compared with Fmr1null mice (figure 6D). Consistently, when we administered Nec-1s during D-Gal/TNF injections, we observed reduction of liver enzyme activity including ALT, AST and LDH in the sera of treated Fmr1null mice when compared with untreated animals (figure 6E). These findings were also obtained using the inhibitor Nec-1 (online supplementary figure 6K). Taken together, the RIPK1 inhibitor Nec-1s can reduce the presence of proteins indicating apoptosis and necroptosis and consequently reduce liver damage following TNF/D-Gal treatment in Fmr1null mice.

Nec-1s treatment reduced TNF-induced liver damage in absence of FMRP. (A) Control and Fmr1null (Fmr1 KO) mice were treated with Flag-mTNFα (200 ng/mouse) with Nec-1s (6 µg/g) or without Nec-1s. RIPK1 was immunoprecipitated followed by immunoblotting of p-RIPK1 and RIPK1 (one representative of n=4 is shown). (B) p-MLKL and MLKL expression were detected in liver tissue lysates from Fmr1null mice and Nec-1s treated Fmr1null mice 5 hours post D-Gal/rTNF treatment. Lower panel shows quantification from n=6 mice. (C) Active-caspase-3 and (D) TUNEL staining of liver tissue sections from Fmr1null mice and Nec-1s treated Fmr1null mice 5-hour post D-Gal/rTNF treatment. Right panel indicates quantification (n=6, one representative picture is shown, scale bar=100 µm). (E) ALT, AST and LDH activities were measured in serum samples of Fmr1null mice and Nec-1s treated Fmr1null mice 5 hours post D-Gal/rTNF treatment (n=6). Error bars in all graphs indicate SEM. *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; FMRP, Fragile X mental retardation protein; LDH, lactate dehydrogenase; Nec-1s, necrostatin-1s; TNF, tumour necrosis factor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.
Expression of FMRP alleviates liver damage and disease during BDL
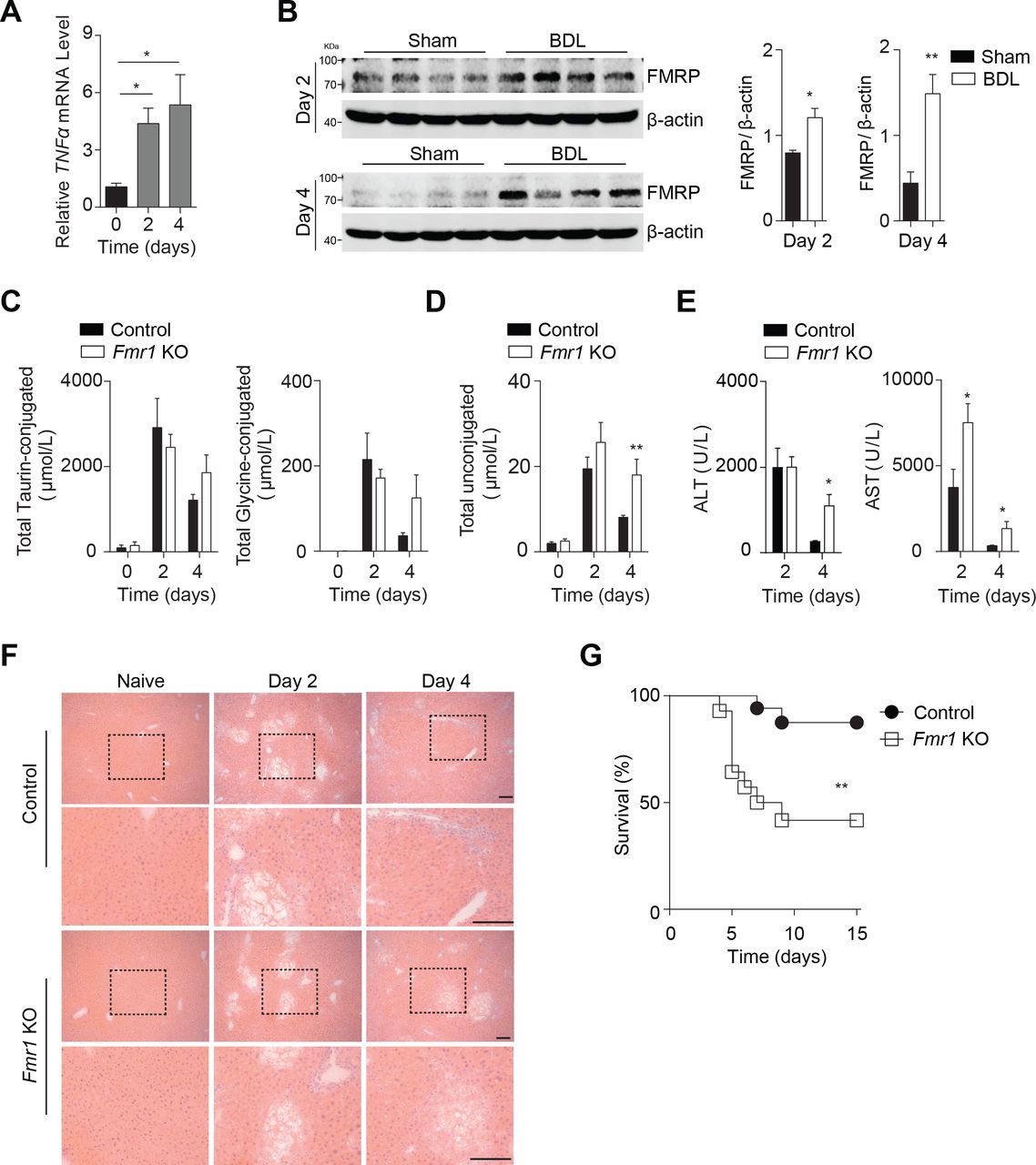
Next, we wondered whether FMRP modulated liver cell death during another disease model such as acute cholestasis following BDL, a model system for liver fibrosis. TNF has been shown to contribute towards liver fibrosis following BDL.4 As expected, we found increased expression levels of Tnfa in mice following BDL (figure 7A). Consistently, we also identified increased expression of FMRP in liver tissue from BDL-operated animals, when compared with tissue from Sham-operated mice (figure 7B). Notably, we did not find a difference in circulating conjugated bile acids in control or Fmr1null mice following BDL (figure 7C). However, we identified increased levels of unconjugated bile acids 4 days after BDL (figure 7D). Furthermore, we found increased activity of liver enzymes in the blood stream in FMRP-deficient mice when compared with control mice, suggesting increased liver damage (figure 7E). Histological analyses showed increased disruption of the hepatic organisation in liver tissue harvested from Fmr1null mice compared with control mice following BDL (figure 7F). Notably, we did not find a major difference in granulocyte infiltration or expression of pro-inflammatory cytokines in this setting (online supplementary figure 7A-C). However, following BDL, FMRP-deficient mice showed decreased survival compared with control animals (figure 7G).

Lack of FMRP triggers increased susceptibility towards BDL-induced liver damage. (A) Tnfα gene expression was quantified in liver tissue at indicated time points post-BDL (n=3–4). (B) Liver lysates were probed for FMRP at the indicated time points from Sham and BDL mice. Right panels indicate quantification of n=4 mice. (C) Total taurin-conjugated and glycine-conjugated bile acids, and (D) unconjugated bile acids were measured in serum samples of control and Fmr1null (Fmr1 KO) mice at indicated time points post-BDL (n=4–10). (E) ALT and AST activities were measured in serum samples of control and Fmr1null mice at indicated time points post-BDL (n=5–7). (F) Haematoxylin and eosin staining of liver tissue sections of control and Fmr1null mice at indicated time points post-BDL (one representative set of n=5–7 is shown, scale bar=50 µm). (G) Survival of control and Fmr1null mice after BDL was monitored (n=14–17). *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; BDL, bile duct ligation; FMRP, Fragile X mental retardation protein; KO, knockout; TNF, tumour necrosis factor.
Next, we wondered by which mechanism FMRP-deficient mice exhibited high susceptibility towards acute cholestasis. We did not observe any significant differences in the expression levels of Cola1, Cola3 and Acta2 between control and Fmr1null mice (online supplementary figure 8A). Consistently, we did not find any difference in hepatic αSMA expression between Fmr1null and control mice at these early time points (online supplementary figure 8B). Notably, we found a transient increase in TUNEL+ cells in absence of FMRP (figure 8A). Furthermore, histological analyses of the proliferation marker Ki67 showed reduced expression in liver tissue harvested from Fmr1null mice when compared with control animals (figure 8B). Consistently, RNA expression levels of Ki67 were reduced in FMRP-deficient mice (figure 8C). These findings were further supported by the reduced expression of PCNA, which is associated with hepatocyte proliferation following liver damage (figure 8D). Notably, we did not observe increased presence of cleaved Casp3 or cleaved Casp8 in Fmr1null mice following BDL (online supplementary figure 8C, D). In contrast, LCMV infection resulted in increased presence of cleaved Casp3 in liver tissue of Fmr1null mice compared with control mice (online supplementary figure 9A, B). Consistent with the increased cell death and reduced proliferation during BDL, we found increased presence of RIPK1 in FMRP-deficient mice when compared with control animals following BDL (figure 8E). RIPK1- dependent recruitment of RIPK3 is a critical step in TNF-mediated necroptosis.10–12 We observed increased RIPK3 expression in absence of FMRP when compared with FMRP competent mice (figure 8F). RIPK3 can phosphorylate the pseudokinase MLKL, which mediates necroptosis.43 Consistently, we detected increased presence of p-MLKL in Fmr1null liver tissue when compared with control tissue (figure 8G). Furthermore, we observed increased presence of RIPK1, RIPK3 and p-MLKL following LCMV infection (online supplementary figure 9C-E). Treatment of Fmr1null animals with Nec-1s could improve the pathology observed following BDL (figure 8H). Taken together, absence of FMRP results in increased expression of RIPK1 and accordingly increased pathology following acute cholestasis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Increased expression of RIPK1 triggers BDL-induced pathology in absence of FMRP. (A) TUNEL staining of liver tissue sections harvested from control and Fmr1null (Fmr1 KO) mice at indicated time points after BDL. Right panel indicates quantification (n=5–7, one representative set of pictures is shown, scale bar=50 µm). (B) Sections of liver tissue harvested from control and Fmr1null mice were stained for Ki67 at indicated time points after BDL (n=5–7, scale bar=50 µm). Representative images are shown. (C) Ki67 gene expression was quantified in liver tissue harvested from control and Fmr1null mice at indicated time points post-BDL (n=5–7). (D) PCNA expression was detected at the indicated time points from BDL control and FMRP-deficient mice (one representative set of immunoblots of n=4 is shown). Bottom panels indicate quantification. (E–G) RIPK1 (E), RIPK3 (F), p-MLKL and MLKL (G) expression were detected in liver tissue lysates from control and Fmr1null mice at the indicated time points (one representative set of immunoblots of n=3–4 is shown). Bottom panels indicate quantification of day 2. (H) Survival of Fmr1null and Nec-1s treated Fmr1null mice after BDL was monitored (n=7–8). *p<0.05, **p<0.01. BDL, bile duct ligation; FMRP, Fragile X mental retardation protein; KO, knockout; PCNA, proliferating cell nuclear antigen; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.
Discussion
This study shows that lack of FMRP results in prolonged presence of RIP1K affecting TNFR signalling and consequently leading to increased susceptibility towards TNF-mediated liver cell death. Hence, Fmr1null mice exhibited increased TNF-mediated liver damage, septic shock, LCMV-induced hepatitis and pathology during BDL. Application of the RIP1K inhibitor Nec-1s could alleviate liver damage following TNF challenge or pathology during BDL in Fmr1null mice.
Despite its ubiquitous expression, little is known about the FMRP function outside the CNS. Fmr1null mice exhibit macroorchidism and increased ovarian weight.23 24 Moreover, increased expression of Fmr1 correlates with aggressive cancer growth.27 Furthermore, in hepatocellular carcinoma tissue, FMR1 expression levels were increased when compared with tumour-free tissue.45 Considering our data, increased presence of FMRP could affect RIPK1 expression and accordingly prevent susceptibility of cancer cells towards TNF-mediated cell death. Hence, strategies to inhibit FMRP might increase the susceptibility of cancer cells towards therapies involving TNF including immunotherapies. Whether patients suffering from Fragile X syndrome exhibit increased liver damage is not sufficiently studied. Notably, we did not observe increased liver enzymes or gross defects in liver tissue in naïve mice. Only following stimulation with TNF-dependent disease models Fmr1null mice exhibited increased liver damage and disease. Patients carrying CGG repeats in the locus Xq27.3 might also be asymptomatic and would not exhibit gross liver damage. However, during infections or intoxication TNF-mediated liver damage might be increased. Future studies on patient cohorts are needed to address the role of FMRP during liver damage and disease.
Absence of FMRP triggers TNF-mediated apoptosis and necroptosis in liver tissue. Positive TUNEL staining and caspase activation cannot distinguish between apoptosis or necroptosis.6 Notably, the deubiquitinase CYLD was increased in Fmr1null mice compared with controls. Presence of CYLD triggers deubiquitination of RIPK1 resulting in presence of non-ubiquitinated RIPK1 and preventing its degradation.7 9 Hence, we observed increased expression of RIPK1, a central switch towards necroptosis.6 Moreover, FLIPS expression was increased in the absence of FMRP, which triggers formation of the riptoptosome, promoting recruitment of RIPK3 and accordingly induction of necroptosis.14 15 Consistently, we observed increased presence of p-MLKL during TNF/D-Gal treatment, LCMV infection and BDL in Fmr1null mice when compared with control animals. During TNF/D-Gal treatment and LCMV infection, we also observed increased cleaved Casp3, which we did not observe during BDL. Hence, we speculate that FMRP can affect both apoptosis and necroptosis, which is likely context specific. Since treatment with Nec-1s can alleviate pathology during TNF/D-Gal stimulation and BDL in Fmr1null mice, RIPK1 is likely involved.
The function of TNF during FMRP deficiency might reach beyond liver pathology. Necroptosis and activation of RIPK1 are associated with a variety of neurological diseases including Alzheimers disease, amyotrophic lateralsclerosis (ALS) and multiple sclerosis.46 Hence, TNFR1-deficient animals exhibit reduced amyloid β generation and disease symptoms during Alzheimer model systems.47 RIPK1 mediated the microglial response, which is associated with disease.48 Moreover, activation of necroptosis can be observed in patient cohorts suffering from Alzheimer disease.49 Consistently, RIPK1 mediates axonal degradation during ALS.50 Accordingly, suppression of RIPK1 is associated with later onset of ALS.28 Furthermore, pathological TNF levels cause synaptic alterations resulting in memory impairment, which is dependent on TNFR1 on astrocytes.51 In line with this, TNF exacerbates neurotoxic effects during liver disease and acute ammonia intoxication.52 It is tempting to speculate that increased effects of TNF on neurons contribute to the phenotypical changes of neurons in absence of FMRP. Considering our data, absence of FMRP in neurons might trigger increased degradation during infection or intoxication. Nec-1 or other inhibitors of RIPK1 might be beneficial, not only in preventing liver pathology but also to improve neurological defects. While inhibition of RIPK1 could prevent neuronal degradation during infection, it remains unclear whether a long-term treatment would be possible and/or beneficial. However, the function of FMRP might also be cell type specific and our observations unrelated to neurological defects.
Taken together, we identified a protective role of FMRP in TNF-mediated liver damage and during liver disease.
References
Footnotes
KSL and PAL contributed equally.
Contributors YZ designed and conducted the experiments and analysed the data. HCX, PVS, JW, JV, BS, KB, JH, JIH, AB and KP conducted the experiment and analysed the data. MST, DH, EM, DB, MRA, VK, DW, DH, AAP and KSL analysed the data. AAP and KSL supervised the study and edited the manuscript. PAL designed the whole project, supervised the study and wrote the manuscript.
Funding This study was supported by the German Research Council (SFB974, KFO217, LA-2558/5-1). Furthermore, this study was supported by the Jürgen Manchot Graduate School MOI III and the Research Committee of the Medical Faculty of the Heinrich-Heine University Düsseldorf (grant number: 9772690), DB is supported by the FNR-ATTRACT program (A14/BM/7632103).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV), North Rhine-Westphalia, Germany. State Office for Nature, Environment and Consumer Protection.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.