Article Text

Abstract
Objective Although glial cell line-derived neurotrophic factor (GDNF) is a member of the transforming growth factor-β superfamily, its function in liver fibrosis has rarely been studied. Here, we investigated the role of GDNF in hepatic stellate cell (HSC) activation and liver fibrosis in humans and mice.
Design GDNF expression was examined in liver biopsies and sera from patients with liver fibrosis. The functional role of GDNF in liver fibrosis was examined in mice with adenoviral delivery of the GDNF gene, GDNF sgRNA CRISPR/Cas9 and the administration of GDNF-blocking antibodies. GDNF was examined on HSC activation using human and mouse primary HSCs. The binding of activin receptor-like kinase 5 (ALK5) to GDNF was determined using surface plasmon resonance (SPR), molecular docking, mutagenesis and co-immunoprecipitation.
Results GDNF mRNA and protein levels are significantly upregulated in patients with stage F4 fibrosis. Serum GDNF content correlates positively with α-smooth muscle actin (α-SMA) and Col1A1 mRNA in human fibrotic livers. Mice with overexpressed GDNF display aggravated liver fibrosis, while mice with silenced GDNF expression or signalling inhibition by GDNF-blocking antibodies have reduced fibrosis and HSC activation. GDNF is confined mainly to HSCs and contributes to HSC activation through ALK5 at His39 and Asp76 and through downstream signalling via Smad2/3, but not through GDNF family receptor alpha-1 (GFRα1). GDNF, ALK5 and α-SMA colocalise in human and mouse HSCs, as demonstrated by confocal microscopy.
Conclusions GDNF promotes HSC activation and liver fibrosis through ALK5/Smad signalling. Inhibition of GDNF could be a novel therapeutic strategy to combat liver fibrosis.
- hepatic fibrosis
- chronic liver disease
- hepatic stellate cell
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Glial cell line-derived neurotrophic factor (GDNF) is a member of the transforming growth factor-β superfamily and an important player in neuronal survival.
GDNF ligands canonically signal by binding to glycosyl-phosphatidylinositol (GPI)-anchored receptors, termed GDNF family receptor alpha-1 (GFRα1), in collaboration with signalling receptor subunits, such as the Ret tyrosine kinase.
What are the new findings?
GDNF in the serum increased significantly in human liver fibrosis compared with healthy controls.
Serum levels of GDNF are positively associated with hepatic stellate cell (HSC) activation and liver fibrosis progression.
GDNF promotes HSC activation through activin receptor-like kinase 5 at His39 and Asp76 and downstream signalling via Smad2/3, but not through GFRα1.
Mice with overexpression of GDNF display aggravated liver fibrosis, while mice with silenced GDNF expression or treated with GDNF-blocking antibodies have reduced fibrosis and HSC activation in carbon tetrachloride and bile duct ligation (BDL) disease models.
How might it impact on clinical practice in the foreseeable future?
GDNF upregulation presents as a new biomarker for HSC activation and liver fibrosis progression that is measurable in blood and thus has potential for clinical diagnosis of liver fibrosis; moreover, inhibition of GDNF can be developed as novel therapeutic approach for liver fibrosis.
Introduction
Liver fibrosis, which is characterised by excessive extracellular matrix (ECM) deposition, results from chronic liver injury of different aetiologies and represents a major health problem worldwide.1–3 The progression of liver fibrosis to cirrhosis gives rise to severe complications including portal hypertension, liver failure and hepatocellular carcinoma.4 Hepatic stellate cells (HSCs) reside in the space of Disse in the liver,5 and on liver injury, quiescent HSCs undergo a phenotypic and metabolic switch that leads to the acquisition of a myofibroblast (MFB)-like phenotype. Such HSC-derived MFBs are the predominant liver cell type that produces ECM in response to various damaging insults.6 7 Moreover, activated HSCs release cytokines and chemokines and express specific receptors, allowing them to communicate with other liver cells.8 Thus, inhibition of HSC activation is proposed as a therapeutic strategy for patients with liver fibrosis.9 10 The HSC activation process is induced by various stimulatory factors, including transforming growth factor beta 1 (TGFβ1).11 TGFβ1 is a potent profibrogenic cytokine due to its role in HSC activation. In this regard, TGFβ and its downstream targets have been implicated in both animal models and patients. However, interest in the use of anti-TGFβ biological agents has been significantly tempered by reports of serious adverse effects.12
Glial cell line-derived neurotrophic factor (GDNF), as a glycosylated, disulfide-bonded homodimer, is a member of the TGFβ superfamily.13 The contributions of GDNF have been described in renal morphogenesis14 and spermatogonial cell development.15 GDNF exerts a critical function in the peripheral taste system.16 GDNF is currently being evaluated for the treatment of Parkinson’s disease in humans.17 GDNF family signalling pathways ligands are involved in the pathogenesis of bone pain and could be targeted for pharmacological intervention.18 GDNF was identified as a key determinant of resistance to aromatase inhibitor therapy in oestrogen receptor-positive breast cancer.19 In the canonical signalling pathway, GDNF binds to a GDNF family receptor (GFRα) in conjunction with Ret tyrosine kinase.20–22 Inverse effects of GFR and Ret in the context of vitamin D stimulation have also been reported.23 Chen et al demonstrated that p75 interacts with Ret and GFRα1 on stimulation with GDNF.24 However, the molecular mechanisms underlying GDNF-mediated HSC activation and liver fibrosis remain unknown.
We hypothesised that GDNF induces HSC activation and liver fibrosis through the activin receptor-like kinase 5 (ALK5)/Smad pathway. Therefore, we examined the role of GDNF/Smad signalling in HSC activation and provide proof of concept of this pathway as a therapeutic target.
Materials and methods
Patient samples and clinical data information
The study conforms with the provisions of the Declaration of Helsinki.
Serum samples collected from 157 healthy controls (HCs) at Putuo Hospital (December 2017 to April 2018) and from 239 patients with liver biopsy (F0=25, F1=48, F2=87, F3=51, F4=28; June 2011 to July 2018) were used for GDNF concentration measurement. GDNF mRNA expression was investigated in 165 patients (F0=13, F1=35, F2=61, F3=40, F4=16) who underwent liver biopsies between February 2013 and July 2018. GDNF protein expression was examined in 21 fibrotic tissue samples (F0=3, F1=6, F2=6, F3=6, F4=3). The aetiological background of patients was chronic hepatitis B. Fibrosis stages were defined based on Scheuer criteria.25
Liver biopsy samples of patients with non-alcoholic fatty liver disease (NAFLD) were collected from May 2014 to June 2018 in Shanghai General Hospital, Jiao Tong University School of Medicine. Pathologists examined the specimens and determined degree of fat, lobular/portal inflammation, hepatocyte ballooning and fibrosis stages. Controls (F0) were obtained from patients with NAFLD without fibrosis on biopsies. Paraffin liver sections (F0/1=7; F2=7; F3/4=5) were used for histology examination and GDNF staining, frozen liver samples (F0/1=11; F2/3=7) for GDNF mRNA analyses.
Serum samples of patients with alcoholic hepatitis (AH) or alcoholic cirrhosis (AC) were collected from October 2017 to July 2018 in Putuo Hospital, Shanghai University of Traditional Chinese Medicine, excluding patients with evidence of other liver diseases, based on standard clinical, laboratory and histological assessments. AC diagnosis was defined as chronic heavy alcohol consumption with portal hypertension, estimated by CT, causing variceal bleeding, ascites and hepatic encephalopathy.26 27 Patients were not biopsied but characterised as having histological stage F4 based on imaging. Written informed consent was obtained from all patients.
Surface plasmon resonance
To determine the binding of ALK5 to immobilised GDNF, we used a BIAcore T200 instrument (GE Healthcare) with a CM5 sensor chip (GE Healthcare). The activation, deactivation and preparation of the coupled flow cell as well as the ligand-binding assay were performed essentially as described previously.28 Briefly, GDNF in sodium acetate buffer (1 µg/mL, pH 5.5) was immobilised on a CM5 sensor chip, and ALK5 was analysed at a series of concentrations. The experiments were conducted with phosphate buffer saline, and the analyte was injected at a flow rate of 30 µL/min. The association time was 120 s, and the dissociation time was 420 s. The binding constant was obtained using a 1:1 Langmuir binding model via a BIAcore evaluation software program.
Other methods and statistical analyses are described in online supplementary materials.
Supplementary file 1
Results
Patient characteristics
As shown in table 1, F4 stage patients had higher alanine transaminase (ALT) and aspartate transaminase (AST) levels and lower platelet levels than HCs and F0 stage patients (p<0.05). F4 stage patients had significantly higher levels of hyaluronic acid and collagen IV than F0 stage patients (p<0.01). Other patient characteristics are summarised in table 1. NAFLD and alcoholic liver disease (ALD) patient information is included in online supplementary supporting table 1 and 2.
HBV patient characteristics
GDNF expression in patients with liver fibrosis and mouse models of liver fibrosis
GDNF concentrations were examined in 366 serum samples (127 HCs and 239 patients with liver fibrosis) using ELISA. Prominent increase in GDNF levels was evident in F4 patients compared with HC and F0–F2 patients (p<0.01; figure 1A). In addition, GDNF levels in human serum samples correlated significantly with the F stage (online supplementary supporting figure 1A, r=0.8137, p<0.0001).

Glial cell line-derived neurotrophic factor (GDNF) expression in liver fibrosis. (A) GDNF concentrations in 366 human serum samples measured by ELISA (healthy control (HC)=127; F0=25; F1=48; F2=87; F3=51; F4=28). (B) GDNF mRNA in 165 human liver specimens examined by real-time PCR. The levels of target mRNAs were normalised to that of 18S rRNA (F0=13; F1=35; F2=61; F3=40; F4=16). (C) Correlation analysis between GDNF concentrations in the serum and GDNF mRNA expression. Spearman’s correlation coefficients (r), p values and the number of patients are indicated. (D) Correlation analysis between GDNF and α-SMA mRNA expression. Spearman’s correlation coefficients (r), p values and the number of patients are indicated. (E) Immunohistochemistry of GDNF in frozen liver sections of 40 HBV patient samples (F0/1=10; F2=10; F3=10; F4=10). Upper original magnification x100, middle x600, lower x600, negative control; the red arrow indicates GDNF-positive staining. (F) Immunohistochemistry of GDNF in liver paraffin sections of non-alcoholic steatohepatitis (NASH) patient samples (F0/1=7; F2=7; F3/4=5). Upper original magnification x100, middle x600, lower x600, negative control; the red arrow indicates GDNF-positive staining. (G) Semiquantification of GDNF from figure 1F. (H) Hepatic GDNF mRNA expression in patients with NASH. (I) GDNF concentrations in serum samples from 30 HCs and 37 patients with alcoholic liver disease (ALD), as measured by ELISA (HC=30; alcoholic hepatitis (AH)=26; alcoholic cirrhosis (AC)=11). Bars indicate the mean±SD of three independent experiments; one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in A, B, G–I. A non-parametric correlation (Spearman’s) two-tailed test was used in C and D.
GDNF mRNA expression was significantly higher in F3 and F4 stages than in F0 patients (p<0.001), as determined in 165 patients (figure 1B). GDNF concentrations in serum positively correlated with GDNF mRNA levels in liver (figure 1C, r=0.7250, p<0.0001). A positive correlation was observed between GDNF mRNA expression and F stage in livers of patients (online supplementary supporting figure 1A, r=0.5490; p<0.0001). We also tested HSC activation and collagen production at mRNA levels (online supplementary supporting figure 1B). α-SMA and Col1A1 mRNA expression increased with disease progression (F3 and F4 compared with F0; p<0.001). Because GDNF mRNA levels coincided with α-SMA levels, we evaluated these values in each patient with liver fibrosis and found a positive correlation (figure 1D, r=0.7837, p<0.0001). GDNF levels in serum also significantly correlated with α-SMA (r=0.8409, p<0.0001) and Col1A1 (r=0.8345, p<0.0001) expression in liver (online supplementary supporting figure 1C).
Localisation of GDNF was determined by immunohistochemistry (figure 1E) and positive staining was evident in the hepatic sinusoids of patients with mild liver fibrosis. With increasing disease severity, GDNF-positive cells were also found in portal areas and adjacent to fibrotic septa (online supplementary supporting figure 1D).
Similar to our observations in viral hepatitis, non-alcoholic steatohepatitis (NASH)-induced liver fibrosis also presented with increased GDNF expression (figure 1F–H). Finally, ELISA data from patients with ALD show increased GDNF serum levels in patients with advanced cirrhosis compared with HC and patients with AH (p<0.01; figure 1I).
mRNA data were validated by immunoblotting for protein expression. Band intensities for GDNF and α-SMA are increasing with disease severity (online supplementary supporting figure 1E), and showed a positive correlation (r=0. 8478, p<0.0001). Increasing numbers of α-SMA-positive and GDNF-positive cells colocalised along with fibrosis progression (online supplementary supporting figure 2), as evidenced using sequential (serial sections) or dual immunohistochemistry.
We next investigated mouse models of liver fibrosis, namely carbon tetrachloride (CCl4) for 6 weeks, BDL for 2 weeks, methionine-choline-deficient diet (MCD) for 8 weeks, and high fat diet (HFD) for 14 weeks. GDNF was present throughout in non-parenchymal cells of fibrotic livers (figure 2A). GDNF mRNA (figure 2B) and protein levels were increased with fibrosis progression (online supplementary supporting figure 3A and B). GDNF serum concentration increased significantly in fibrotic mice compared with control mice (p<0.01; online supplementary supporting figure 3C).

Glial cell line-derived neurotrophic factor (GDNF) is upregulated in mouse liver fibrosis. (A) Representative images of GDNF staining in mouse liver fibrosis from carbon tetrachloride (CCl4), BDL, methionine-choline-sufficient (MCS), methionine-choline-deficient (MCD), normal chow diet (NCD), high fat diet (HFD) samples. Original magnification x100, the third line is x600, fourth line is the negative control showing a serial section of the third line x600, the red arrow indicates GDNF-positive staining, n=5 per group. (B) Gdnf mRNA expression in whole liver of CCl4, BDL, MCD and HFD mice. Bars indicate the mean±SD of three independent experiments; n=5 per group; the t-test with the non-parametric Mann-Whiney U test was used. (C) Frozen mouse liver sections on dual α-SMA and GDNF immunohistochemistry (original magnification x100), the lower part represents the boxed area with x600 magnification, the green arrow indicates α-SMA and GDNF dual positive staining, n=5 per group.
GDNF overexpression aggravates experimental liver fibrosis
To determine the functional impact of GDNF on liver fibrosis development, mice exposed to CCl4 were injected with an adenovirus expressing GDNF (Ad-GDNF) and a corresponding mock control. GDNF overexpression enhanced the architectural disturbance and collagen deposition in CCl4-treated mice (figure 3A and B). Ectopic GDNF expression clearly increased collagen deposition and α-SMA expression, as determined by Sirius red staining and immunohistochemistry (figure 3B-3D). Furthermore, ALT and AST levels in serum increased in control and CCl4-treated mice with ectopic GDNF expression, indicating GDNF-mediated hepatocyte damage (figure 3E). mRNA levels of pro-inflammatory markers and chemokines, for example, Cd68 and Ccl2, are increased in Ad-GDNF treatment groups (figure 3F). GDNF overexpression also increased gene expression of fibrogenic parameters α-Sma, Mmp2, Timp1 and Col1α1 (figure 3G) in CCl4-treated mice and controls.

Glial cell line-derived neurotrophic factor (GDNF) overexpression exacerbates carbon tetrachloride (CCl4)-induced liver fibrosis in mice. (A) GDNF, α-SMA and Col1 expression examined by immunoblotting and semiquantitative analysis. Liver samples were collected from CCl4-treated mice treated with Ad-GDNF or Ad-shuttle vectors (n=5 per group). Representative images of GDNF (B), Sirius red (C) and α-SMA staining and the negative control with the same location (D), with the red arrow denoting the α-SMA-positive staining (original magnification, x100). The larger box shows a x600 magnification of the small box (n=5 for each group). (E) Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in mouse serum samples (n=5 per group). mRNA expression of pro-inflammatory cytokines and chemokines (F), as well as fibrogenic parameters (G), examined by real-time PCR (n=5 per group). Bars indicate the mean±SD of three independent experiments; n=5 per group; one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in parts A, E–G.
Results from CCl4 mice were confirmed in BDL mice. Ad-GDNF infection augmented BDL-induced liver fibrosis, as evident from histopathology of H&E and Sirius red staining, and α-SMA immunohistochemistry (supplementary supporting figure 4A-D). Serum ALT and AST levels were slightly increased, Cd68, α-Sma mRNA and profibrogenic gene expression were upregulated (online supplementary supporting figure 4E-G).
GDNF knockdown ameliorates experimental liver fibrosis
To further investigate the role of endogenous GDNF in hepatic fibrosis, CCl4 intoxication and BDL mouse models were subjected to Gdnf sgRNA CRISPR/Cas9 adenovirus delivery to deplete GDNF expression. GDNF expression in fibrotic livers was examined by immunoblotting and immunohistochemistry (online supplementary supporting figure 5A, B, 6A and B). Sirius red and α-SMA staining revealed attenuation of fibrosis and collagen deposition in CCl4 mice on Gdnf depletion, as compared with scrambled sgRNA treatment (online supplementary supporting figure 5B-D and 6B-D). Moreover, mRNA levels of α-Sma and Col1α1 were significantly downregulated in Gdnf sgRNA/CCl4 mice in comparison to control CCl4 mice.
GDNF is profibrogenic and promotes HSC activation
To identify the cell type responsible for GDNF expression in liver fibrosis, we isolated the different cell types from human livers. GDNF mRNA was expressed at higher levels in HSCs than hepatocytes, Kupffer cells (KCs) or liver sinusoid endothelial cells (LSECs; figure 4A). GDNF mRNA expression was also higher in LX2 than HepG2, Huh7 or HUVEC cells (online supplementary supporting figure 7A). We also examined the expression of Gdnf in HSCs, KCs and hepatocytes from fibrotic mouse livers, showing upregulation in in vivo-activated HSCs from CCl4 and BDL mice, but not in KCs and hepatocytes (figure 4B and online supplementary supporting figure 7B). In a comparative investigation, we showed low Gdnf expression in all cell populations, except for activated HSCs (figure 4C).

Glial cell line-derived neurotrophic factor (GDNF) induces hepatic stellate cell (HSC) activation in human HSCs. Gdnf mRNA expression as determined by real-time PCR. (A) The different types of liver cells were extracted from human liver samples. (B) Gdnf mRNA expression in primary HSCs, hepatocyte (HEPs), KCs and liver sinusoidal endothelial cell (LSECs) isolated from olive oil or carbon tetrachloride (CCl4)-treated mice aged 6 weeks. N.S., not significant. (C) Gdnf mRNA expression in 7-day-culture-activated mouse HSC compared with HEPs, KCs, LSECs and peripheral blood mononuclear cells (PBMC) (n=3 per group). (D) Gdnf mRNA expression in cultured human and mouse HSCs at the indicated time points. (E) Real-time PCR and western blot analysis for α-SMA and Col1A1. Human and mouse primary HSCs were treated with GDNF (10 ng/mL) for the indicated time intervals. (F) Effect of GDNF on fully activated HSCs. GFP-Col-HSCs were plated and cultured in 6-well plates for 2 hours and then treated with GDNF (10 ng/mL) for an additional 48 hours; the cells were analysed by fluorescence microscopy (original magnification, x200, n=3) and positive cells were counted (%). (G) Western blot analysis for α-SMA and Col1A1. Primary human and mouse HSCs were plated and cultured in 6-well plates overnight, followed by si-GDNF or sgGdnf RNA (2 multiple of infection (MOI)) for 48 hours. Bars indicate the mean±SD of three independent experiments; n=3 per group; one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in parts A, C and E, and the t-test with the non-parametric Mann-Whiney U test was used in part B.
Gdnf mRNA expression increased over time in primary human and mouse HSCs during culture activation (figure 4D). To investigate the response of HSCs to GDNF stimulation, we treated both hHSCs and mHSCs with recombinant GDNF (10 ng/mL), which significantly increased α-SMA and Col1A1 mRNA and protein levels (figure 4E and F). In contrast, α-SMA and Col1 levels decreased on Gdnf sgRNA treatment (figure 4G).
Ret, but not GFRα1, via AKT and Smad2/3 downstream signalling participate in GDNF-induced culture activation of HSC
Mitogen-activated protein kinases participate in GDNF classical signalling.21 22 By immunoblotting, we demonstrated with that AKT but not ERK was phosphorylated in response to GDNF in hHSCs (figure 5A), mHSCs (figure 5B) and LX2 cells (online supplementary supporting figure 8A). α-SMA and Col1 protein levels were not increased following GDNF stimulation in hHSCs and LX2 cells when AKT expression was depleted with small interfering RNA (siRNA) or AKT signalling was blunted with a specific inhibitor; these results indicate that AKT participates in fibrogenic GDNF signalling (figure 5C, D and online supplementary supporting figure 8B, C).

Glial cell line-derived neurotrophic factor (GDNF) induces hepatic stellate cell (HSC) activation via the activin receptor-like kinase 5 (ALK5)/Smad pathway. Western blot analysis for (p-)AKT and (p-)Erk and semiquantification. Human (A) and mouse (B) HSCs were treated with GDNF (10 ng/mL) for the indicated time. (C) Western blot analysis for α-SMA and Col1A1. AKT small interfering RNA (siRNA) was transfected into human HSCs for 12 hours and then the cells were treated with 10 ng/mL GDNF for an additional 48 hours. (D) Western blot analysis for α-SMA and Col1A1. Human HSCs were treated with MK2206 2HCL (0.5 µM) for 1 hour, followed by 10 ng/mL GDNF for an additional 48 hours. (E) Western blot analysis for (p-)AKT and (p-)Smad2. Human HSCs were transfected with GFRα1 siRNA for 48 hours and subsequently treated with 10 ng/mL GDNF for an additional 2 hours. (F) Western blot analysis for (p-)AKT and (p-)Smad2. Human HSCs were treated with RPI-1 (60 µM) for 1 hour, followed by treatment with 10 ng/mL GDNF for additional 2 hours. (G, H) GFRα1 and Ret mRNA expression in 165 human liver specimens. The levels of target mRNAs were normalised to that of 18S rRNA (F0=13; F1=35; F2=61; F3=40; F4=16). (I, J) Western blot analysis for (p-)Smad2 and (p-)Smad3 and semiquantification. Human (I) and mouse (J) HSCs were treated with GDNF (10 ng/mL) as indicated. (K) Western blot analysis for Smad2 and Smad3. Human HSCs were treated with GDNF (10 ng/mL) for 2 hours, and nuclear and cytoplasmic extracts were tested for Smad2/3 levels. (L, M) Western blot analysis for α-SMA and Col1A1. Human HSCs were transfected with siRNA for Smad2 or Smad3 for 12 hours and then treated with GDNF (10 ng/mL) for additional 48 hours. (N) Western blot analysis for α-SMA and Col1A1. Human HSCs were treated with LY2157299 (0.5 µM) for 1 hour and then GDNF (10 ng/mL) for an additional 48 hours. (O) Western blot analysis for (p-)AKT and (p-)Smad2. Human HSCs were treated with SB431542 (10 µM) for 1 hour and then GDNF (10 ng/mL) for an additional 2 hours. (P) Western blot analysis for (p-)Smad2 and (p-)Smad3. Human HSCs were transfected with ALK5 siRNA for 12 hours and then GDNF (10 ng/mL) for an additional 48 hours. (Q, R) mRNA expression of ALK5 in 165 human liver specimens. The level of ALK mRNAs was normalised to that of 18S rRNA (F0=13; F1=35; F2=61; F3=40; F4=16). Correlation analysis between ALK5 and GDNF mRNA expression in patients with liver fibrosis. Spearman’s correlation coefficients (r), p values and the number of patients are indicated. Bars indicate the mean±SD of three independent experiments; n=3 per group; one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in parts A, B, G–I, J and K, and the non-parametric correlation (Spearman’s) two-tailed test was used in part R.
Canonical GDNF signalling is initiated by binding to GFRα1. This complex then binds to and activates the transmembrane receptor Ret.15 Therefore, we wanted to determine whether GFRα1 and Ret are the receptors required for GDNF-induced HSC activation. To validate this proposition, we selectively inhibited GFRα1 and Ret, using GFRα1-specific siRNA and RPI-1, a compound Ret inhibitor and measured the impact on activation in hHSC and LX2 cells. Notably, pAKT and pSmad2 protein levels were still increased by GDNF treatment in GFRα1-silenced hHSCs and LX2 cells (figure 5E, F and supplementary supporting figure 8D, E), whereas RPI-1-pretreated, GDNF-stimulated HSCs had pAKT protein levels similar to those of untreated cells. Furthermore, hepatic GFRα1 mRNA levels were unchanged in patients with liver fibrosis (figure 5G), whereas Ret mRNA expression was significantly higher in F3 and F4 stage patients than in F0 patients (figure 5H). These results indicate that Ret, but not GFRα1, contributes to GDNF-induced HSC activation in vitro.
Because Smad signalling is the predominant profibrogenic pathway in HSC, and GDNF is a member of the TGFβ family, we examined whether GDNF could activate Smad signalling in HSCs. Indeed, both pSmad2 and pSmad3 were significantly increased between 1 and 4 hours after GDNF stimulation in hHSCs (figure 5I), mHSCs (figure 5J) and LX2 cells (online supplementary supporting figure 8F). Using cytoplasmic and nuclear cell extraction, we also found that nuclear presence of Smad2 and Smad3 increased significantly after GDNF stimulation in hHSCs (figure 5K) and LX2 cells (online supplementary supporting figure 8G). Finally, GDNF stimulation did not induce α-SMA and Col1A1 at the protein level in HSCs, when Smad2 and Smad3 were knocked down by RNAi (figure 5L and M).
ALK5 is required for GDNF-mediated HSC activation
ALK5 is the R-Smad that activates the receptor of TGFβ.29 Since we found, for the first time, that GDNF induces Smad2 and Smad3 phosphorylation, and since GFRα1 is not involved in this process, we wanted to validate whether ALK5 contributes to the GDNF signalling complex at the cell surface to transduce profibrogenic signalling. We pretreated the cells with the ALK5 inhibitor LY2157299 before GDNF stimulation. In this setting, induction of α-SMA and Col1A1 protein expression was blunted (figure 5N). In line with this finding, induction of pAKT and pSmad2 was abrogated in GDNF-treated LX2 cells and hHSCs when we inhibited ALK5-dependent signalling by SB431542 (another established ALK5 inhibitor) or when we decreased ALK5 expression with siRNA (figure 5O, P and online supplementary supporting figure 8H, I). These results suggest that ALK5 functions as a signalling receptor in GDNF-mediated HSC activation. Accordingly, ALK5 mRNA expression was significantly elevated in fibrotic liver of F3 and F4 stage patients than F0 patients (p<0.01; figure 5Q), and we could identify a positive correlation between ALK5 and GDNF (r=0.8591; p<0.0001) in patients with liver fibrosis (figure 5R).
To gain further insight into signalling receptor complex formation, we used surface plasmon resonance (SPR) to investigate the binding affinity of GDNF to the ALK5 receptor. As shown in figure 6A, binding of ALK5 to GDNF was dose-dependent, exhibiting a fast association-dissociation process. The response units at equilibrium were plotted against ALK5 concentrations (figure 6B), and the dissociation constant (KD) was calculated by non-linear regression suggesting that GDNF binds to ALK5 with a KD value of 85.47 nM.

Determination of two critical sites of activin receptor-like kinase 5 (ALK5) for glial cell line-derived neurotrophic factor (GDNF) binding. (A) The binding affinity of GDNF to ALK5 was determined using surface plasmon resonance (SPR). (B) The dissociation constant (KD) of GDNF and ALK5 was calculated by non-linear regression analysis. GDNF bound to ALK5 with a KD value of 85.47 nM. (C) The complex of ALK5 and GDNF was generated by in silico estimation using a rigid-body docking approach with PatchDock software (http://bioinfo3d.cs.tau.ac.il/PatchDock/). (D) Expression plasmid sequences of ALK5-wt and ALK5-mut were validated. (E) ALK5-wt and ALK5-mut amino acid sequences. (F) co-immunoprecipitation (co-IP) analysis of ALK5/GDNF interaction comparing wt and His39 and Asp76 mutated ALK5. LX2 cells were transfected with pcDNA3-ALK5 plasmids for 12 hours and subsequently treated with Ad-GDNF for an additional 48 hours. (G) Direct interaction of ALK5 with Ret after GDNF stimulation. After IP with an anti-ALK5 antibody, immunoblotting for Ret was performed. (H) Direct interaction of ALK5 with p-Smad2 after GDNF stimulation. After IP with an anti-ALK5 antibody, immunoblotting for p-Smad2 was performed.
To define how GDNF binds to ALK5, we constructed a virtual GDNF-ALK5 structure model (figure 6C), suggesting an electro bond interaction between Glu153 of GDNF and His39 of ALK5, as well as a hydrogen bond interaction between Tyr156 of GDNF and Asp76 of ALK5. Guided by the model, we mutated His39 and Asp76 of ALK5 to Ala (figure 6D and E) and subsequently examined the GDNF/ALK5 interaction by co-immunoprecipitation (co-IP). The interaction of GDNF with ALK5 could be demonstrated in LX2 cells with ectopic GDNF and ALK5 expression, and was abrogated by expressing a mutated ALK5 receptor (figure 6F).
Finally, we confirmed the ALK5, Ret and p-Smad2 interactions and participation in GDNF-induced complex formation by co-IP experiments (figure 6G). Taken together, data suggest that ALK5 is the coreceptor of Ret for GDNF-induced HSC activation.
GDNF, ALK5 and α-SMA colocalise in liver fibrosis in humans and mice
To investigate the interaction between GDNF and ALK5 during HSC activation in human liver fibrosis, we analysed colocalisation experiments with confocal microscopy (online supplementary supporting figure 9). GDNF, ALK5 and α-SMA colocalised in liver sinusoids of F0 and F1 patients (online supplementary supporting figure 9). During fibrosis progression, F2–F4 stage patients displayed a significant increase in numbers of α-SMA-positive cells in hepatic lobules and fibrotic septa. GDNF-positive and ALK5-positive cell numbers also increased in parallel. This result in patients with liver fibrosis was consistent with the increased GDNF and ALK5 expression in mice with liver fibrosis (online supplementary supporting figure 10).
GDNF targeting antibody administration inhibits liver fibrosis in mice
To test whether targeting GDNF could mitigate the development of liver fibrosis, CCl4 mice were treated with anti-GDNF blocking antibodies or isotype IgG, starting after 3 weeks, and sacrificed the mice 1 week later (figure 7). Administration of either antibody no. 1 (Ab1; 400 ng/g BW; Abcam; cat. ab18956), Ab2 (400 ng/g BW; Santa Cruz; cat.sc-13147) inhibited fibrosis and HSC activation (figure 7). These results suggest that targeting GDNF could be an attractive therapeutic strategy for patients with liver fibrosis.
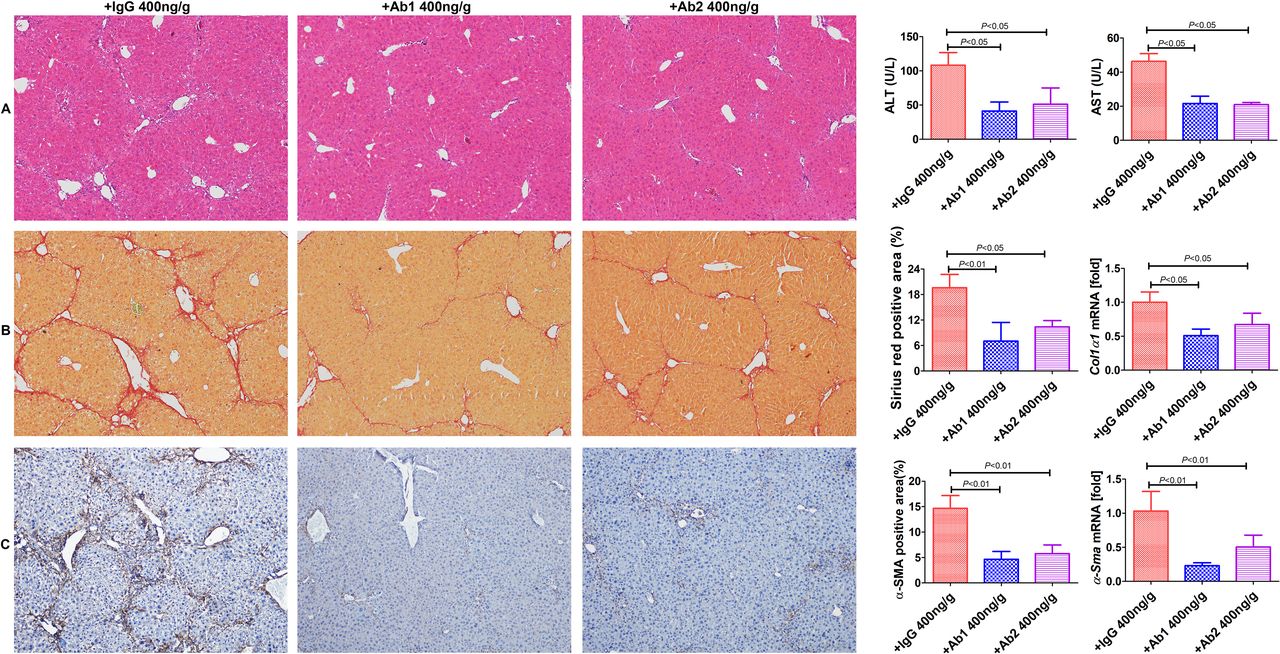
Administration of anti-glial cell line-derived neurotrophic factor (anti-GDNF) antibodies reduces liver fibrosis development in mice. (A) Anti-GDNF-blocking antibodies blunted carbon tetrachloride (CCl4)-induced liver injury (intraperitoneal injections; Ab1: 400 ng/g BW; Ab2: 400 ng/g BW administered for 3 weeks (once a week), starting from the second week after CCl4 treatment); controls were treated with IgG (400 ng/g BW). (B, C) Sirius red and α-SMA staining (original magnification, ×100). The positive stained area was calculated as a percentage. Col1α1 and α-Sma mRNA expression levels are shown as the fold downregulation in comparison to untreated animals. Bars indicate the mean±SD of three independent experiments; n=6 per group; one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in parts A–C.
TGFβ and GDNF activate the GDNF promoter and GDNF mRNA expression via Smad2/3
To estimate how GDNF mRNA expression is regulated in HSCs, we tested its transcriptional regulation on treatment with TGFβ. Indeed, TGFβ induced GDNF mRNA expression in HSCs (figure 8A). We also found that Tgfβ and Gdnf mRNA were both increased in CCl4-induced liver fibrosis, suggesting that TGFβ might be the initial inducer of GDNF expression in liver fibrosis (figure 8B). GDNF mRNA was not induced in response to TGFβ or GDNF stimulation, when Smad2/3 were knocked down by siRNA (figure 8C). These results indicate that Smad2/3 is required for GDNF mRNA upregulation during HSCs activation. Bioinformatics analysis of the human GDNF promoter revealed that the −60 to −46 site contained a Smad2/3 binding sequence. We experimentally confirmed Smad2/3 binding by chromatin immunoprecipitation (ChIP) assays (figure 8D). Furthermore, Smad2 and Smad3 expression levels were upregulated in fibrotic liver of patients (figure 8E, F), and were correlated with GDNF expression. Taken together, our data strongly suggest that GDNF facilitates liver fibrosis progression via Ret and ALK5 receptors and Akt/Smad2/3 pathways (figure 8G).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The glial cell line-derived neurotrophic factor (GDNF)/activin receptor-like kinase 5 (ALK5)/Ret/Smad2/3 circuit promotes hepatic stellate cell (HSC) activation. (A) Real-time PCR Gdnf mRNA detection after transforming growth factor-β (TGFβ) treatment in human and mouse HSCs. (B) Real-time PCR for Tgfβ and Gdnf mRNA in carbon tetrachloride (CCl4)-induced liver fibrosis. (C) GDNF mRNA was not induced in response to TGFβ and GDNF stimulation, when Smad2/3 were depleted using specific small interfering RNAs in LX2 cells. (D) Schematic diagram of the putative Smad2/3 binding site within the human GDNF promoter. LX2 cells were subjected to ChIP assays with anti-Smad2/3 or IgG antibodies. Representative results from three independent experiments are shown. mRNA expression of Smad2 (E) and Smad3 (F) in 165 human liver specimens. The levels of Smad2/3 mRNAs were normalised to that of 18S rRNA (F0=13; F1=35; F2=61; F3=40; F4=16). Correlation analysis between Smad2/3 and GDNF mRNA expression in patients with liver fibrosis. Spearman’s correlation coefficients (r), p values and the number of patients are indicated. Bars indicate the mean±SD of three independent experiments; n=3 per group; the t-test with the non-parametric Mann-Whiney U test was used in part A, one-way analysis of variance with the non-parametric Kruskal-Wallis test was used in parts C and E, and the non-parametric correlation (Spearman’s) two-tailed test was used in parts E and F. (G) Graphical summary of GDNF fibrogenic signalling in HSCs. HSCs express the GDNF receptors GFRα1 and Ret, whereas only Ret is upregulated in patients with liver fibrosis. GDNF induces AKT and Smad2/3 phosphorylation as a short-term response. As downstream events, GDNF induces GDNF transcription as an autocrine feedback stimulation and fibrogenic gene expression, as exemplified by α-SMA and Col1. ALK5 and Ret, but not GFRα1, are required for AKT/Smad2/3 activation and subsequent GDNF and α-SMA/Col1 expression. ALD, alcoholic liver disease; NASH, non-alcoholic steatohepatitis.
Discussion
Liver fibrosis involves several key factors, including HSC activation and profibrogenic signal transduction.30 31 In this study, we show that GDNF levels increase in chronic liver diseases in human and mouse. Our data reveal a novel mechanism of GDNF-induced HSC activation via the TGFβ receptor ALK5. We also discovered colocalisation of GDNF, ALK5 and α-SMA in liver of patients and mice with fibrosis. Administration of GDNF-blocking antibodies attenuated CCl4-induced liver fibrosis in mice, suggesting that GDNF could serve as an attractive target for antifibrotic therapy.
GDNF is well known as a trophic factor in neural cells (see online supplementary supporting discussion). In addition to the brain, the function of GDNF has been studied in other diseases. Currently, there is limited knowledge about its role in liver injury. It was recently reported that GDNF is protective in HFD-induced insulin resistance, hyperlipidemia and obesity in mice.32 33 In their experiments, Mwangi et al used transgenic GDNF expression in mice, and investigated GDNF responses of hepatocytes in experimental NAFLD. They convincingly showed that in the setting of hepatocyte steatosis, GDNF is protective through the downregulation of peroxisome proliferator-activated receptor-γ (PPAR-γ) expression. In HSCs, however, downregulation of PPAR-γ is required for activation, and thus is in line with a profibrogenic role of GDNF.34 In total, we demonstrated that serum GDNF concentrations were significantly higher in 239 patients with liver disease as compared with 127 HCs with a positive correlation to the fibrotic stage. GDNF expression in liver was remarkably higher in F3 and F4 stage than in F0 stage patients. We further showed that the liver itself was producing GDNF in advanced liver fibrosis, mainly in HSCs and demonstrated that hepatic GDNF mRNA and protein levels were increased in F2/F3/F4 stage patients with NASH. Human data could also be validated in mouse MCD and HFD NAFLD models. With that, we are first to determine HSCs as responsible cell type expressing GDNF in liver.
HSCs have been frequently described as relatives of glial or neuronal cells, since they express several respective markers, for example, glial fibrillary acidic protein (GFAP),35 neurotrophins and their receptors, including nerve growth factor (NGF), brain-derived neurotrophic factor, neurotrophin 4/5 and NGF receptor p75.36 Given the findings of the present study, we can now also add GDNF to this list as a neuronal marker protein expressed by stellate cells. Moreover, in response to this cytokine, GDNF increases α-SMA and Col1A1 expression in HSCs and, therewith, mediates HSC activation.
Despite the physiological and clinical significance of GDNF, the signalling pathway activated by GDNF has not yet been fully elucidated. Biochemical and cell culture studies suggest that GDNF binds to GFRα1 requiring the transmembrane tyrosine kinase Ret14 to form a functional receptor complex. Both proteins are coexpressed in the developing nephron, midbrain and motor neurons and mice carrying a disruption of either Ret or GFRα1 are deficient in renal and neuronal development and suffer from early postnatal death.15 GDNF/Ret signalling through the phosphatidylinositol 3-kinase/nuclear factor kappa B pathway is required for mitochondrial function and morphology,37 and seems also relevant for dopamine system maintenance to prevent Parkinson’s and Hirschsprung diseases.22 Besides, it has been reported that p140NCAM may interact with GFRα1 to mediate GDNF signalling independently from Ret in Schwann cell migration.38 Another study also describes that GDNF can regulate migration of GABAergic cells in the medial ganglionic eminence and cortex via GFRα1, but not Ret or NCAM.20 Finally, an inverse relationship between GFRα1 and Ret has been reported in dopaminergic neurons, where knocking down Ret leads to increased GFRα1 expression.23
In HSC, GDNF induces activation through Smad2/3 and AKT signalling. Our data showed that only Ret but not GFRα1 was required for GDNF-induced HSC activation. In line with these in vitro data, Ret mRNA levels were significantly elevated in F3 and F4 stage compared with F0 stage patients, where GFRα1 levels were not significantly changed. Because GFRα1 was not involved in the identified activation-related GDNF signalling in HSC, we wanted to identify a coreceptor of Ret in this cell type. GDNF is a member of the TGFβ superfamily and we found that it could induce HSC activation. TGFβ is mediating its multifunctional effects on liver cells by binding to ALK539 40 followed by subsequent downstream signalling via phosphorylation and nuclear translocation of Smad2/3 to regulate target gene expression in a context-dependent manner.41 Thus, we hypothesised that GDNF might involve ALK5 to induce HSC activation. Indeed, HSC activation in response to GDNF was suppressed by the ALK5 inhibitors SB451342 and LY2157299. Furthermore, SPR analysis, the gold standard method for studying protein-protein interactions, confirmed that GDNF bound to ALK5. After mutating Asp76 and His39 to Ala in recombinant ALK5 protein, which are the obvious critical binding sites as determined by computer simulation, the binding of GDNF and ALK5 was blunted, as measured by co-IP analysis. Considering these results, we concluded that ALK5 participates in GDNF-induced HSC activation. To the best of our knowledge, this is the first report to show that GDNF can induce HSC activation via the ALK5/Smad pathway. Finally, we confirmed the correlation between GDNF, ALK5 and α-SMA in liver fibrosis by colocalisation studies in humans with tricolour laser confocal microscopy.
In conclusion, our results show that GDNF is significantly upregulated in human liver fibrosis and is produced by HSCs. GDNF can induce HSC activation via Ret/ALK5 receptors and the AKT and Smad pathways. Blocking GDNF can inhibit fibrosis progression. Based on these findings, GDNF could be an attractive therapeutic target for the treatment of liver fibrosis in the future.
Acknowledgments
The authors would like to thank Dr Isaac M Chiu (Department of Microbiology and Immunobiology, Division of Immunology, Harvard Medical School) for his suggestions on GDNF and ALK5 interaction. The authors would like to thank Dr Xiaoling Song (Institute of Immunology, Shanghai Technology University) for her assistance in mutagenesis experiments.
References
Footnotes
LT, WM, LW and MX contributed equally.
Contributors CL, SD, PL and ES conceived and designed the experiments. LT, WM, LW, WS, MX, HL, RF, WZ and JX carried out experiments. YY, DX, JZ, MX and helped with the human liver biopsy specimen. ES assisted with data analysis. CL and SD wrote the manuscript, which was read, edited and approved by all authors.
Funding This work was supported by The National Natural Science Foundation of China (no. 81673788 (to CL), 81873136 (to CL), 81530101 (to PL), 81803232 (to WM), 81803898 (to LT), 81570547 (to MX), 81770597 (to MX), 81704027 (to WS), 81873109 (to HL)). The National Institutes of Health Grant (no. R01DK085252, R01AA027036 (to ES)) and Winnick Research award from Cedars-Sinai Medical Center (to ES). The Federal Ministry of Education and Research grant ‘LiSyM’ (to SD).
Competing interests None declared.
Ethics approval Human samples and study protocol were approved by the Clinical Ethics Committee of Putuo Hospital, Shanghai University of Traditional Chinese Medicine and Shanghai General Hospital, Jiao Tong University School of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. Figure 8 has been corrected.
Patient consent for publication Obtained.