Article Text

Abstract
Objective Recent evidence points to the gut microbiome’s involvement in postoperative outcomes, including after gastrectomy. Here, we investigated the influence of gastrectomy for gastric cancer on the gut microbiome and metabolome, and how it related to postgastrectomy conditions.
Design We performed shotgun metagenomics sequencing and capillary electrophoresis time-of-flight mass spectrometry-based metabolomics analyses on faecal samples collected from participants with a history of gastrectomy for gastric cancer (n=50) and compared them with control participants (n=56).
Results The gut microbiota in the gastrectomy group showed higher species diversity and richness (p<0.05), together with greater abundance of aerobes, facultative anaerobes and oral microbes. Moreover, bile acids such as genotoxic deoxycholic acid and branched-chain amino acids were differentially abundant between the two groups (linear discriminant analysis (LDA) effect size (LEfSe): p<0.05, q<0.1, LDA>2.0), as were also Kyoto Encyclopedia of Genes and Genomes modules involved in nutrient transport and organic compounds biosynthesis (LEfSe: p<0.05, q<0.1, LDA>2.0).
Conclusion Our results reveal alterations of gut microbiota after gastrectomy, suggesting its association with postoperative comorbidities. The multi-omic approach applied in this study could complement the follow-up of patients after gastrectomy.
- gastrectomy
- metagenome
- gutmicrobiome
- metabolome
- gastric cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Gastrectomy is a surgical treatment for gastric cancer and morbid obesity.
Gastrectomy alters physiological properties, such as oxygen availability, pH, food transit time, intestinal motility and hormonal conditions.
Case studies in obesity treatment have shown that faecal microbiome and metabolome alterations persist over time after surgery; in particular, these alterations correlate with metabolic improvements in patients who are morbidly obese.
Case studies in gastrectomy for gastric cancer using 16S rRNA amplicon sequencing have shown alterations of gut microbiome composition in patients with subtotal gastrectomy.
Increased risk of developing metachronous colorectal cancer has been reported in patients with gastric cancer.
What are the new findings?
The present shotgun metagenomic approach demonstrates overall microbiome community structure changes such as higher abundance of oral microbes, aerobes and facultative anaerobes in faecal samples, which can be related to reconstruction of the GI tract of patients with gastric cancer.
Gastrectomy-associated alterations in microbial functions, such as nutrient transport and biosynthesis of organic compounds, might relate to changes in postgastrectomy metabolism.
Several colorectal cancer-related bacteria displayed a similar pattern in postgastrectomy patients: Fusobacterium nucleatum, in particular, was significantly enriched (p<0.05) in the total gastrectomy compared with the control group.
The present metabolomic analysis shows enrichment of deoxycholic acid and branched-chain amino acids in postgastrectomy patients.
How might it impact on clinical practice in the foreseeable future?
The present study provides novel insights into microbiome/metabolome features underlying postgastrectomy comorbidities in patients with gastric cancer; in particular, previously reported occurrence of metachronous colorectal cancer following gastrectomy might be associated with alterations of the gut microbiome.
Introduction
Recent evidence indicates the involvement of the gut microbiome in disease onset and progression and in postoperative outcome.1–3 A study on the gut mucosal microbiota following ileocolonic resection for Crohn’s disease revealed that microbial community structure was associated with disease recurrence or maintenance of remission.4 Accordingly, microbiota structure might play a role in clinical outcomes.
Several studies have also highlighted gut microbiota alterations following gastrectomy, which represents the primary treatment for gastric cancer and recently for morbid obesity.5 6 Even though the two procedures have different aims, they share similar anatomical and technical features. Several obesity studies indicate that changes in microbial community and functional potential correlate with weight loss and persistent long-term effects on the gut microbiome.7–9 Nevertheless, Aron-Wisnewsky et al showed that the gut microbiome gene richness was partially restored 1 year after gastrectomy, in spite of weight loss and improved metabolism.10
Gastrectomy as a curative resection for gastric cancer aims to obtain complete histopathological clearance and involves radical resection of the primary site, as well as resection of affected lymph nodes and adjacent organs if necessary.6 Two studies using 16S rRNA sequencing have reported microbiome alterations after gastrectomy in patients with gastric cancer. Tseng et al revealed that subtotal gastrectomy altered the diversity, community composition and predicted gene functions of gastric microbiota, which associated closely with the altered gastric environment after surgery.11 Lin et al confirmed these findings by analysing the faecal microbiome over the long term after gastrectomy.12 They showed that patients with subtotal gastrectomy, and in particular Roux-en-Y gastrojejuno anastomosis, had subsequent lower occurrence of type II diabetes and metabolic syndrome than the controls. However, the possible consequences of microbiota alterations on the patients’ condition after gastrectomy, especially total gastrectomy, remain poorly understood.
Increasing evidence indicates a possible link between gut microbiota and postoperative outcome after gastrectomy. For example, patients with gastric cancer could be at an increased risk of developing metachronous cancer including colorectal cancer (CRC) after gastrectomy.13 14 Because the gut microbiota is known to be associated with CRC,15–17 it may also affect development of metachronous CRC in postgastrectomy patients. Here, we characterised the faecal microbiome of patients with a history of gastrectomy for gastric cancer and compared them with control participants. To provide a better understanding of microbial metabolism, we complemented these data with metabolome profiles. The study aims to comprehensively characterise the influence of gastrectomy for gastric cancer on the gut microbiome, with the intention of improving nutrition and follow-up examinations in postgastrectomy patients.
Materials and methods
Study participants and faecal sample collection
The samples and clinical information used in this study were obtained under conditions of informed consent and with the approval of the institutional review board of each participating institute. A total of 106 participants undergoing total colonoscopy at the National Cancer Center Hospital, Tokyo, Japan, were enrolled. Fifty participants had previously undergone gastrectomy for gastric cancer and did not show signs of gastric cancer recurrence. Those who showed abnormal findings, including precancerous lesions such as adenomatous polyps or carcinomas, were excluded. The remaining 56 participants who did not show any colorectal findings, nor had any history of gastroenterological surgery were included as controls (table 1 and online supplementary tables S1 and S2).
Supplemental material
Supplemental material
Faecal samples were collected immediately at the first defaecation after starting the oral administration of bowel cleansing agents at the hospital on the day of colonoscopy.15 18 The samples were placed directly on dry ice and subsequently stored at −80°C for metagenomic and metabolomic analyses. The participants’ lifestyle data, including dietary habits and medical records, were acquired from questionnaires (475 questions, 25 pages), based on the example used in the Japan Public Health Center study19 (online supplementary figure S1A).
Supplemental material
Shotgun metagenomic sequencing and metabolomic quantification
DNA extraction and shotgun metagenomic sequencing of faecal samples (online supplementary methods) generated, on average, 57, 888, 536 reads per sample and read quality was assessed (online supplementary figure S1B and supplementary table S3). We quantified the metabolites in faecal samples from 44 gastrectomy and 54 control participants using capillary electrophoresis time-of-flight mass spectrometry20 (online supplementary methods).
Supplemental material
Supplemental material
Taxonomic profiling
Metagenomic sequencing results were subjected to taxonomic and functional profiling following read quality filtering (online supplementary figure S1B and supplementary methods). Taxonomic profiling using the mOTU pipeline annotated 606 species,21 whereas 403 species-level clades were obtained using the MetaPhlAn2 pipeline.22 We categorised the annotated species as oral microbes or others based on the expanded Human Oral Microbiome Database23 (online supplementary methods). High-level phenotypes were identified by BugBase24 (online supplementary methods).
Functional profiling
Functional profiles were generated using our in-house pipeline (online supplementary methods) and the Human Microbiome Project Unified Metabolic AnalysisNetwork 2 (HUMAnN2) pipeline.25 Annotation results were then summarised into Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology (KO) and KEGG modules26 profiles (online supplementary figure S1B and supplementary methods). We annotated 498 KEGG modules that collectively contained 6,108 KOs from our in-house pipeline. For the HUMAnN2 pipeline, we used UniRef90 as reference and acquired 437 KEGG modules containing 5,971 KOs.
Statistical analysis
Low-frequency and less abundant microbial features (species, functional modules and metabolites) were discarded (online supplementary methods). Principal coordinates analysis (PCoA) with Bray-Curtis distance was used to examine the separation of species and metabolites across samples. Permutational multivariate analysis of variance (PERMANOVA) (‘adonis’ function, vegan package in R) was applied to test microbial composition between groups. Chao1 richness and Shannon-Wiener alpha-diversity index were calculated to estimate microbial diversity between gastrectomy and control groups. Differences in relative abundance of the microbial features were determined by linear discriminant analysis (LDA) effect size (LEfSe)27 (online supplementary methods).
Associations between microbes, KEGG modules or metabolites and clinical parameters (eg, serum glucose, total cholesterol, body mass index (BMI)) and demographic information (eg, age, gender, smoking habits, alcohol consumption, medical history) were assessed using boosted additive generalised linear models available in the MaAsLin R package28 (online supplementary methods). Significantly different clinical parameters, demographic characteristics, medical history and diet were tested by a two-sided Mann-Whitney U (MWU) test for numerical data and Fisher exact test for categorical data.
We estimated genus and species association in each group using SparCC29 (bootstrap n=5000) on selected significantly different genera and species that overlapped between mOTU and MetaPhlAn2 annotations. Significant co-occurrence and co-excluding interaction (SparCC correlation scores ⍴<−0.2 or >0.2 with p<0.05) were visualised and analysed using igraph. To determine inter-omics (species and metabolome) correlations, we performed Procrustes analysis (online supplementary methods). Additionally, Spearman’s correlation analysis was performed on significantly different metabolites and genera to assess their putative correlation (online supplementary methods). Finally, we applied metabolic model-based integration of metabolite observations and species abundances (MIMOSA)30 to compare the predicted and measured metabolome trends (online supplementary methods).
Results
Participants’ characteristics and microbial community structure of gastrectomy and control groups
Participants’ characteristics did not show significant differences between the gastrectomy (n=50) and control (n=56) groups (table 1 and online supplementary table S1). BMI (two-sided MWU test: p=1.22×10−5) and total cholesterol (two-sided MWU test: p=0.0145) were significantly lower in the gastrectomy group, although average cholesterol content was within the normal range (128–219 mg/dL) in each group. Preoperational BMI data for 47 (94%) gastrectomy participants indicated lower BMI at the time of faecal sampling (paired sample t-test: p=7.00×10−5) than prior to surgery, suggesting that weight loss could be attributed to postoperative malnutrition, for example, as a result of dumping syndrome.31 Fifteen postgastrectomy patients (30%) experienced dumping syndrome.
Participants’ characteristics
We further examined the influence of gastrectomy on overall microbiome composition and metabolite profiles in faecal samples by performing PERMANOVA with Bray-Curtis distance (figure 1A and B and online supplementary table S4). Species composition differed significantly between the two groups (adonis: R2=0.0369, p=9.99×10−4; R2=0.0387, p=9.99×10−4, in mOTU and MetaPhlAn2, respectively). Within-group Bray-Curtis distance was significantly lower in the gastrectomy group than in the control group (two-sided MWU: p=5.53×10−7) (figure 1C). Metabolite profiles were also significantly different between groups (figure 1D) (adonis: R2=0.0647, p=9.99×10−4).
Supplemental material

Community structure of the faecal microbiome and metabolome in postgastrectomy and control participants. Principal coordinates analysis (PCoA) with Bray-Curtis distance was performed to assess the community structure of species’ relative abundance obtained by mOTU (A) and MetaPhlAn2 (B) in the gastrectomy group (n=50) (orange) and in the control group (n=56) (blue). The PCoA trend was confirmed by significantly lower microbial structure dissimilarity (Bray-Curtis) within groups (p=5.53×10−7) (C). PCoA was performed also on faecal metabolite concentrations in the gastrectomy group (n=44) (orange) and the control group (n=54) (blue) (D).
PERMANOVA analysis using different surgery types and reconstructions as predictors (online supplementary results and supplementary table S4) revealed no significantly different microbiome and metabolome distributions across different types of surgery (online supplementary figures S2) except for Fusobacterium nucleatum. F. nucleatum was highly enriched in total gastrectomy compared with control (LEfSe: p=1.53×10−5, q=0.00205, LDA=2.87; p=4.12×10−6, q=3.34×10−4, LDA=1.43, in mOTU and MetaPhlAn2, respectively) and subtotal gastrectomy group (LEfSe: p=5.58×10−5, q=0.0150, LDA=2.34; p=1.45×10−5, q=0.00366, LDA=2.14, in mOTU and MetaPhlAn2, respectively) (online supplementary results and supplementary table S5). Owing to an unbalanced sample size, statistical power may have been insufficient to detect microbiome and metabolome differences across different reconstructions; however, we could clearly observe a different distribution pattern of predominant species and metabolites (online supplementary results and supplementary figures S3 and S4).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
To examine the possible confounding effects of clinical parameters (eg, BMI, serum glucose, total cholesterol), demographic data (eg, age, gender) and medical history (eg, medication, diseases), we performed PERMANOVA and MaAsLin. PERMANOVA showed that the participants’ grouping explained the variance in species composition and metabolite profiles better than any of the other predictors (online supplementary results and supplementary rable S4). MaAsLin indicated that the association coefficients were not meaningfully affected by adjustment for possible confounders (online supplementary figure S5).
Supplemental material
Underlying comorbidities and concurrent medication of the participants may influence gut microbiota. The number of participants using gastric acid secretion inhibitors and diabetes therapeutic drugs varied significantly between the gastrectomy and control groups. Statistical analyses on subsets of the original participants that excluded such users confirmed the lack of any effect of the exclusion on originally reported gastrectomy-enriched microbiome and metabolome signatures (online supplementary results and supplementary tables S6 and S7). The microbial features that were significantly enriched (LEfSe: p<0.05, q<0.1, LDA>2.0) in control individuals who took gastric acid secretion inhibitors or diabetes therapeutic drugs are also reported (online supplementary results and supplementary tables S8 and S9).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Microbial diversity and richness are higher in gastrectomy patients
The gastrectomy group showed higher Chao1 index (mOTU: p=4.64×10−4; MetaPhlAn2: p=1.93×10−6) (figure 2A and B) and Shannon diversity index (mOTU: p=0.0954; MetaPhlAn2: p=0.0104) (figure 2C and D) for species composition than their control counterparts. Higher Shannon diversity indices were conserved across the three major phyla: Firmicutes (mOTU: p=8.98×10−7; MetaPhlAn2: p=2.91×10−4), Actinobacteria (mOTU: p=7.69×10−4; MetaPhlAn2: p=0.0419) and Bacteroidetes (mOTU: p=0.225; MetaPhlAn2: p=0.0283) (figure 2E and F). Our results were in accordance with previous studies, in that species diversity was higher in patients with postgastrectomy gastric cancer12 and obesity9 32 33 than in non-surgical participants.
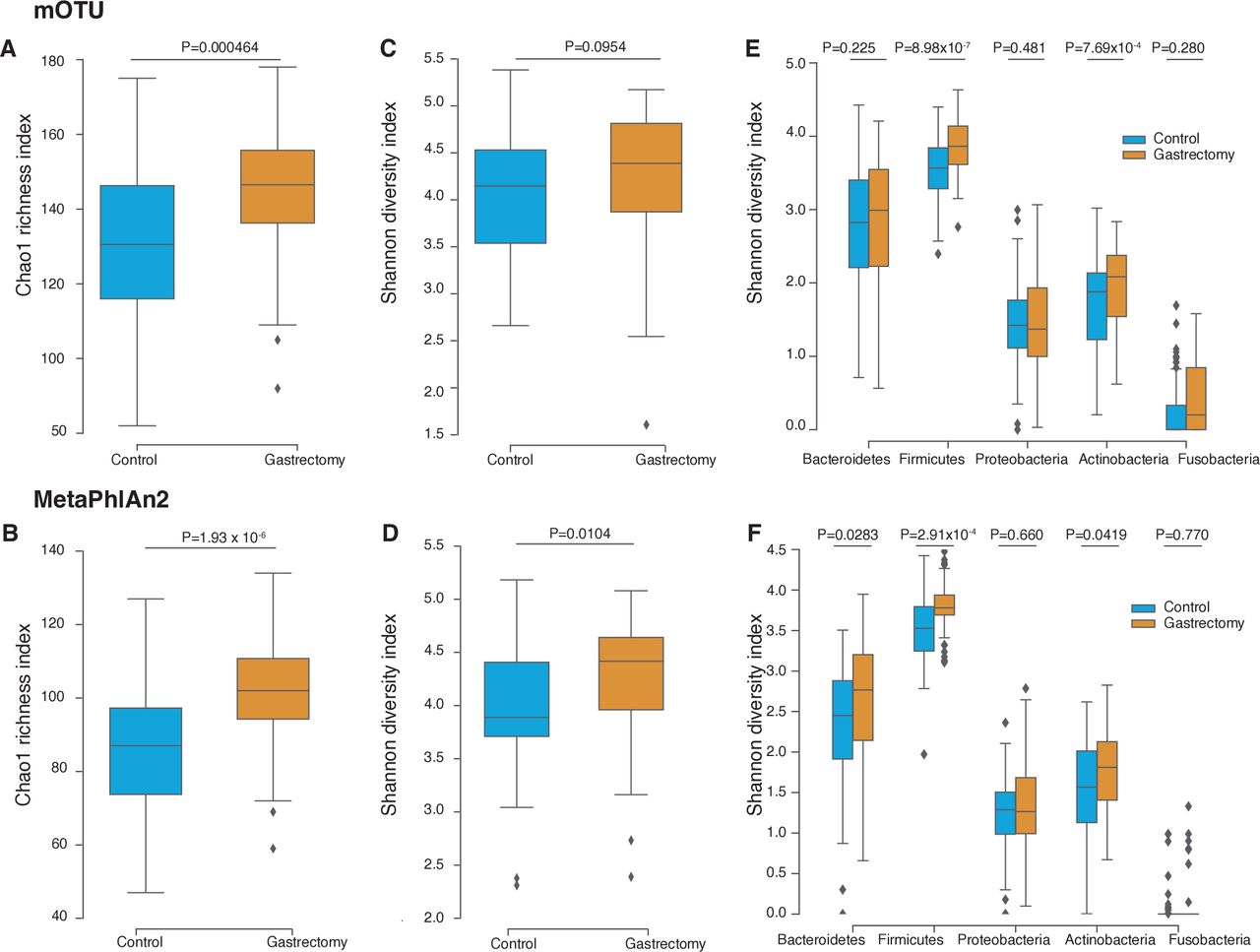
Microbiome diversity in gastrectomy and control groups. Species richness was measured using the Chao1 index calculated from the species annotated by mOTU (A) and MetaPhlAn2 (B). Species alpha-diversity was measured using the Shannon-Wiener index based on mOTU (C) and MetaPhlAn2 (D) annotation. Species alpha-diversity was measured for each major phylum using mOTU (E) and MetaPhlAn2 (F) annotation.
Postgastrectomy patients exhibit a distinct faecal microbiota
Several taxa, mostly Bacilli, were significantly enriched in the gastrectomy group compared with controls (LEfSe: p<0.05, q<0.1, LDA>2.0) (figure 3A and B). Bacilli were previously reported to be enriched in the gut12 and gastric microbiota of patients after gastrectomy for gastric cancer.11 In obesity cases, Bacilli can be used to discriminate between postgastrectomy patients and non-surgical controls.32

Differential enrichment of microbes in gastrectomy and control groups. Cladogram of species annotated by mOTU (A) and MetaPhlAn2 (B). Each dot represents a taxonomic hierarchy. Dots are marked for significant (LEfSe: p<0.05, q<0.1) enrichment either in the gastrectomy group (n=50) (orange) or in the control group (n=56) (blue). Taxa that reached a linear discriminant analysis score (log10) >3.0 are highlighted and labelled accordingly. The summed relative abundances of oral microbes were compared between the gastrectomy (n=50) and control (n=56) groups based on the species annotated by mOTU (C) and MetaPhlAn2 (D). The summed relative abundances of aerobes (E) and facultative anaerobes (F) were compared between the two groups.
The mOTU annotation identified 89 species (online supplementary table S10), whereas MetaPhlAn2 annotation identified 76 species (online supplementary table S11) that differed significantly between gastrectomy and control groups (LEfSe: p<0.05, q<0.1, LDA>2.0), whereas 39 species overlapped between the two methods. The predominant species in postgastrectomy patients were Streptococcus (six species), followed by Prevotella (four species) and Veillonella and Lactobacillus (three species, each). Based on MaAsLin analysis of species composition with clinical parameters, enrichment of Roseburia hominis and Ruminococcus gnavus might be affected by BMI (online supplementary results and supplementary tables S10 and S11). None of the differentially enriched species exhibited significant associations with serum glucose or total cholesterol (MaAsLin: p>0.05, q>0.1).
Supplemental material
Supplemental material
Oral microbes (eg, Streptococcus, Veillonella, Prevotella) were relatively more abundant (two-sided MWU test: p<0.01) in the gastrectomy group (figure 3C and D), as did also aerobes and facultative anaerobes based on phenotype prediction (figure 3E and F, MWU: p=6.43×10−7, p=0.0337, respectively).
Gastrectomy alters microbial functional characteristics
To examine the functional consequences of microbial community changes, we annotated 73 KEGG modules (based on our in-house pipeline) and 119 KEGG modules (based on the HUMAnN2 pipeline) that were differentially abundant (LEfSe: p<0.05, q<0.1, LDA>2.0) between the gastrectomy and control groups (online supplementary table S12 and supplementary figure S6). Among the KEGG modules identified as differentially abundant by both pipelines were those for membrane transport, biosynthesis of organic compounds, multidrug resistance, two-component regulatory system (TCS) and others (figure 4A). In the membrane transport system, phosphate and several amino acid transporters were particularly enriched in the gastrectomy group, followed by manganese/iron and vitamin B12 transporters. Our results were in agreement with those reported in a previous obesity study.9 On the contrary, modules involved in raffinose/stachyose/melibiose transport, along with isoleucine and cobalamin (vitamin B12) biosynthesis, were enriched in the controls. We also observed significantly higher abundance of modules related to TCS and multidrug resistance in the gastrectomy group. KEGG modules’ association with the participants’ demographic characteristics revealed that control or gastrectomy group classification had a predominant impact on module composition (MaAsLin: p<0.05, q<0.1, online supplementary table S12).
Supplemental material
Supplemental material

Different trends of functional modules and metabolites from the faecal microbiomes of gastrectomy and control groups. Relative abundance and linear discriminant analysis (LDA) score (log10) of Kyoto Encyclopedia of Genes and Genomes (KEGG) modules annotated by the Human Microbiome Project Unified Metabolic Analysis Network 2 (HUMAnN2) and overlapping with those annotated by our in-house pipeline (linear discriminant analysis effect size (LEfSe): p<0.05, q<0.1, LDA>2.0) (A). Richness (Chao1) and alpha-diversity (Shannon-Wiener) of contributor species were estimated. Three modules (marked by an asterisk (*) in A) contributed by significantly more diverse and richer microbes (two-sided Mann-Whitney U test (MWU): p<0.001) in the gastrectomy group, in spite of their enrichment in the control group (B). The modules were M00196, M00535 and M00122 (two-sided MWU: p=5.31×10−4, 3.17×10−5, 7.14×10−6, for richness, respectively, and p=1.12×10−4, 2.22×10−4, 6.10×10−4, for alpha-diversity, respectively). KEGG modules’ relative abundance is represented by the top value of each stack of bars. Samples were subsequently sorted according to the dominant contributor to a module and then grouped as either gastrectomy or control (sample order differs between panels). Different trends in metabolites were also observed between the two groups (LEfSe: p<0.05, q<0.1, LDA>3.0) (C). Bile acid reaction consisted of deconjugation of conjugated bile acids (glycocholate and taurocholate) into their primary form (cholate) and amino acids (glycine and taurine) followed by 7-α/β-dehydroxylation to form secondary bile acids (deoxycholic acid (DCA)). The colours highlight enrichment in the control (blue) and gastrectomy (orange) groups (D).
To compare module composition between gastrectomy and control groups, we estimated species richness (Chao1) and alpha-diversity (Shannon-Wiener) of each module contributor previously confirmed by our in-house and HUMAnN2 pipelines based on the HUMAnN2 output (online supplementary table S13). The three most abundant modules in the control group, raffinose/stachyose/melibiose transport system (M00196), isoleucine biosynthesis (M00535) and cobalamin biosynthesis (M00122), had significantly higher species diversity and richness in the gastrectomy group (figure 4B and online supplementary table S13). Whereas the dominant species were identical among the two groups, their proportions were different. For example, Bacteroides vulgatus contributed predominantly to cobalamin biosynthesis in both groups; however, Escherichia coli was a major module contributor in the gastrectomy group (figure 4B). We also detected three different microbes’ groups that contributed to the enrichment of three different KEGG modules involved in manganese/iron/zinc/copper transport (online supplementary figure S7). Overall, these findings indicate a microbial community-level shift followed by an alteration in functional potential.
Supplemental material
Supplemental material
Metabolome profiles change in postgastrectomy patients
We observed different enrichment of metabolites between control (n=54) and gastrectomy (n=44) participants (figure 4C) (52 vs 46 metabolites, respectively; LEfSe: p<0.05, q<0.1, LDA>2.0, online supplementary table S14). Specifically, primary (cholate) and conjugated forms of bile acids (taurocholate and glycocholate) were more abundant in the control group, whereas the secondary form (deoxycholic acid (DCA)) was significantly more abundant in the gastrectomy group (figure 4C and D). Twelve amino acids, including all branched-chain amino acids and two aromatic amino acids (Ile, Leu, Val, Tyr, Phe), were significantly enriched in the gastrectomy group. Metabolites’ association with the participants’ demographic parameters by MaAsLin showed that cholate enrichment in the control group might be affected by BMI, whereas phenyl-lactate and arginine enrichment might be affected by total cholesterol (online supplementary results and supplementary table S14).
Supplemental material
Putative microbes-microbes and microbes-metabolites correlations in each group
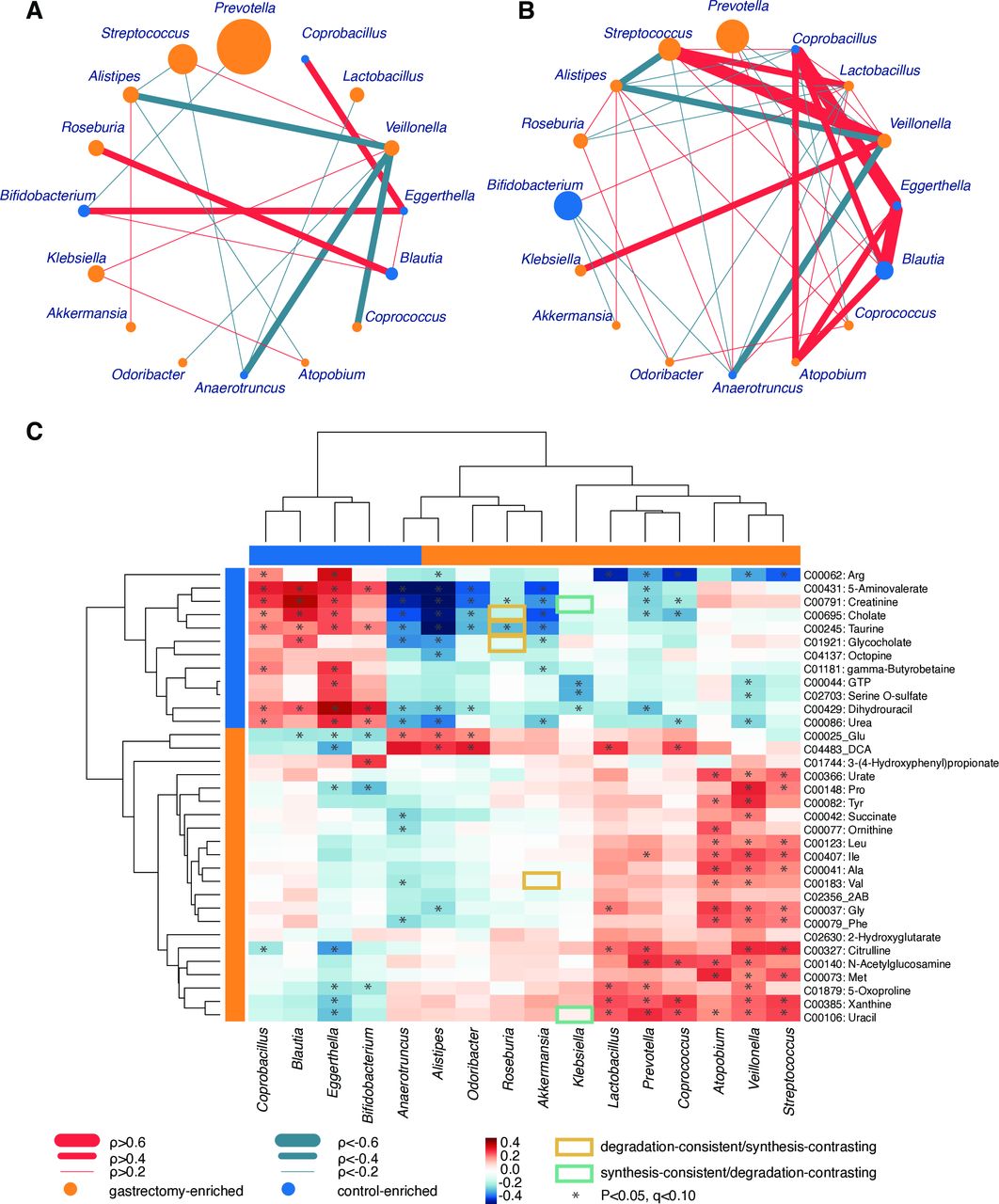
Given the notably distinct microbiome composition between gastrectomy and control groups, we compared the topology of genus (figure 5A and B) and species (online supplementary figure S8) co-occurrence and co-excluding networks. In the genus network, the number of edges was higher in the control group (co-occurrence, 25; co-excluding, 15) compared with the gastrectomy group (co-occurrence, 9; co-excluding, 9). Although the edges do not necessarily represent ecological interactions (eg, mutualism, competition), the microbial network can show which organisms form the hub of community composition.34 Most close relationships (⍴>0.6) in the control group (eg, Coprobacillus, Eggerthella) did not appear or showed much lower values (0.2<⍴<0.4) in the gastrectomy group. In this group, Veillonella contributed to most connections (six genera), but co-excluded (⍴<−0.3) with four genera including Coprococcus (⍴=−0.569, p=4.00×10−4) and Odoribacter (⍴=−0.384, p=0.0160) (online supplementary table S10e). That might reflect the role of Veillonella as a network-hub in the gastrectomy group and its importance in microbial community alteration.34 Results were similar at species level, whereby co-occurrence networks were more abundant in the control (co-occurrence, 31; co-excluding, 8) compared with the gastrectomy group (co-occurrence, 25; co-excluding, 1). Several co-occurrence and co-excluding patterns were shared among genus-level and species-level networks (online supplementary results and supplementary figure S8).
Supplemental material
Supplemental material

Genus-genus and genus-metabolite correlations. Co-occurrence (red) and co-excluding (green) relationships between genera (SparCC: −0.2<⍴<0.2, p<0.05) in gastrectomy (n=50) (A) and control (n=56) (B) groups. The edge width corresponds to SparCC correlation coefficients. The nodes’ size is scaled based on the genus relative abundance averaged over participants within each group. Nodes’ colour represents enrichment of the genus in gastrectomy (orange) and control (blue) participants. Spearman’s correlation analysis was performed between differentially abundant genera (linear discriminant analysis effect size (LEfSe): p<0.05, q<0.1, linear discriminant analysis (LDA)>2.0) and metabolites (LEfSe: p<0.05, q<0.1, LDA>3.0) from gastrectomy (n=44) and control (n=54) participants (C). The matrices were derived from Euclidean distance-based bi-clustering of Spearman’s RANK correlation matrices. Correlation coefficients in each square represent positive (red) and negative (blue) relationships. Colours are proportional to the absolute value of Spearman’s RANK correlations (see legend in the figure). Statistically significant correlations (p<0.05, q<0.1) are marked with asterisks (*). Correlations confirmed by Model-based Integration of Metabolite Observations and Species Abundances are marked by a rectangle (see legend in the figure).
Next, we examined microbiome and metabolite correlations. Procrustes analysis showed an overall significant inter-omics relationship between metabolites and species (mOTU: r=0.279, p=0.003, online supplementary figure S9D; MetaPhlAn2: r=0.292, p=0.002, online supplementary figure S9E). Bi-clustering of correlations between significantly different genera (LEfSe: p<0.05, q<0.1, LDA>2.0, by mOTU and MetaPhlAn2 annotation) and metabolites (LEfSe: p<0.05, q<0.1, LDA>3.0) revealed distinct clusters (figure 5C). Positive correlation clusters were observed in gastrectomy-enriched metabolites and genera, as well as in control-enriched metabolites and genera. Control-enriched clusters included positive correlations between primary and conjugated bile acids (eg, cholate, taurine, glycocholate) and Coprobacillus, Blautia, Eggerthella and Bifidobacterium. In contrast, DCA exhibited a significantly positive correlation with Alistipes, Odoribacter, Lactobacillus and Coprococcus, which were mutually enriched in gastrectomy patients.
Supplemental material
The limited fraction of species contributing to metabolome data (134 out of 326 detected metabolomes) was predicted using MIMOSA based on the KEGG reactions information (online supplementary table S15, supplementary figure S10). In particular, the genus Roseburia contributed to the metabolism of bile acids-related metabolites (eg, glycocholate, taurine, cholate) (figure 5C). Roseburia was enriched in the gastrectomy group and showed a negative correlation with bile acid metabolites that were enriched in the control group. The prediction indicated a consistent contribution of Roseburia to the degradation of those metabolites.
Supplemental material
Four (Leu, Ile, Ala, Val) out of nine amino acids formed a cluster and exhibited significant positive correlation with Atopobium, Veillonella and Streptococcus, which were mutually enriched in postgastrectomy patients. Streptococcus and Veillonella have been reported to ferment amino acids.35 We also observed the contribution of several gastrectomy-enriched species to amino acids synthesis (eg, Bacteroides fragilis) or degradation (eg, Akkermansia muciniphila) (online supplementary table S15). Accordingly, a gastrectomy-associated increase in the availability of amino acids might lead to more amino acid fermentators.
Discussion
Gastrectomy followed by reconstruction of the GI tract radically alters oxygen availability, gut pH, food transit time, intestinal motility and hormonal conditions.36 37 We hereby show that gastrectomy for gastric cancer affects also the faecal microbiome and metabolome. Their links to physiological alterations are summarised as a schematic hypothesis (figure 6). Community PCoA highlighted species-level differences in microbiome composition and metabolite profiles between gastrectomy and control groups. The dissimilarity index within the gastrectomy group was significantly lower than in the control counterpart, demonstrating greater species similarity among postgastrectomy individuals (figure 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Data integration-derived schematic hypothesis. Schematic hypothesis of gut microbiome and metabolite alterations after gastrectomy. The scheme is divided into three parts denoting physiological changes (grey), findings from our study (blue) and the possible consequences (red). Solid lines show the links confirmed by previous studies. Dashed lines show possible connections that can be inferred from our findings. The higher or lower levels observed in postgastrectomy patients are shown in comparison with those of control participants. BCAA, branched-chain amino acids.
Higher species richness and diversity in postgastrectomy patients (figure 2) confirms previous reports on both gastric cancer12 and obesity.9 32 33 This trend might be explained by a combination of major alterations in the gut environment that could support growth of several microbes. One such change is the presence of more oxygen in the gut after gastrectomy,38 which may provide a preferable niche for aerobic and facultative anaerobic microbes.7 9 32 Indeed, relative abundance of aerobes (Streptococcus and Enterococcus) and facultative anaerobes (Escherichia, Enterobacter and Streptococcus) was higher in postgastrectomy patients compared with control participants (figure 3E and F).
Another possibility might be the migration of oral microbes into the gut. Several microbes frequently detected in the oral ecosystem23 were significantly more abundant in postgastrectomy patients (figure 3C and D). They included several species of the genera Streptococcus, Veillonella and Prevotella (online supplementary tables S10 and S11). Veillonella, in particular, formed a network-hub in the gastrectomy group, suggesting an important role in microbial community alteration (figure 5A and B, and online supplementary figure S8). Interestingly, F. nucleatum was significantly enriched in the total gastrectomy group (n=12), compared with control (n=56) and subtotal gastrectomy (n=38) counterparts. The intestinal tract sustains lower acidity due to reduced gastric acid secretion following gastrectomy.23 32 These results indicate that gastrectomy promotes higher transport and survival, as well as growth of aerobes, facultative anaerobes and oral microbes in the distal GI tract.9 32 33
Previous studies indicated that stomach reconstruction affected microbial functions in the gut.9–11 32 Metagenome-based functional analysis revealed gastrectomy-associated enrichment of KEGG modules related to nutrient transport system, including manganese/iron/zinc/copper and vitamin B12. The obesity study suggested that this feature could reflect the increased availability of those substances to microbes.9 Here, no significant difference in dietary consumption was observed between the control and gastrectomy groups (online supplementary table S16).
Supplemental material
Regarding postoperative metabolism, several studies have reported nutrient intolerance (eg, dumping syndrome) and nutrient deficiency (eg, anaemia) in gastrectomy patients, which could be attributed to malabsorption, impaired food intake and altered transit time, and result in weight loss.31 39 The observed postoperative reduction in BMI might be associated with these metabolic deficiencies; however, we did not observe any microbiome or metabolites associated with patients having dumping syndrome (online supplementary results and online supplementary table S17). Furthermore, gastrectomy-associated enrichment of microbial nutrient transport might be associated with the host metabolic functions in two ways. First, postsurgical metabolic deficiencies are at least partially attributed to alterations in gut microbial function (figure 4B and online supplementary figure S7). Second, these changes might be associated with the impaired metabolism of the host. For example, enrichment of the vitamin B12 transporter (M00241) in the gastrectomy (online supplementary table S12) and particularly total gastrectomy group (online supplementary table S5) might be related to the malabsorption of vitamin B12. This may follow from inadequate secretion of intrinsic factors and reduced gastric acidity in the stomach, which causes a lack of absorption in the terminal ileum following gastrectomy.40 Consequently, vitamin B12 that remain unabsorbed in the colon and lead to more microbes with high vitamin B12 intake capacity.41 In the present study, we observed that the vitamin B12 biosynthesis module (M00122) was enriched in the control group. Further comprehensive analysis including the effect of preoperative and postoperative supplemental diet could help decipher these mechanisms.
Supplemental material
The present faecal metabolomic analysis also showed that DCA, one of the secondary bile acids, was enriched in postgastrectomy patients, while other forms, such as conjugated and primary bile acids, were enriched in the control counterparts (figure 4C and D). This might be explained by alterations in bile flow after gastrectomy stimulating the growth of bile acid-transforming bacteria. These bacteria play a key role in the deconjugation or transformation of bile acid to secondary bile acids.32 42 The process is typically mediated by a 7-α/β-dehydroxylation enzyme, expressed by specific members of Clostridium and Eubacterium in the human large intestine.42 43 Although not significant, we confirmed that Clostridium and Eubacterium genera were comparatively more abundant in postgastrectomy patients (online supplementary tables S10 and S11). DCA is a microbiome-produced carcinogen in liver cancer44 and CRC.15 16 45 Obesity studies have raised concerns regarding microbiome changes and secondary bile acids increment following gastrectomy, as well as their link to CRC occurrence.46 47 Patients with gastric cancer were also reported to be at an increased risk of developing metachronous CRC.13 14
Although the mechanism for metachronous CRC onset after gastrectomy may not be similar to sporadic cancer, we observed higher abundances of several CRC-enriched microbes (eg, F. nucleatum and Atopobium parvulum) in the gastrectomy group. F. nucleatum has been suggested to mediate the early steps of carcinogenesis,15 through FadA adhesion to the epithelium, activation of β-catenin signalling48 and infiltration of myeloid cells into the tumour microenvironment.49 Thus, enrichment of F. nucleatum in the total gastrectomy group is worth noting. In addition, A. parvulum has been associated with multiple polypoid adenomas and intramucosal carcinoma.15 50 Enrichment of these species suggests a form of dysbiosis, which can lead to CRC development after gastrectomy. In contrast, Parvimonas micra and Peptostreptococcus stomatis, which are also associated with CRC,51 were not significantly enriched in postgastrectomy patients (online supplementary tables S10 and S11). This may be partly explained by their relative abundances being high only in the presence of carcinomas.15
Of note is also our observation on higher branched-chain amino acids levels and species richness after gastrectomy. Branched-chain amino acids are elevated in plasma and tissue of gastric cancer52 and CRC.53 Additionally, increased species richness in CRC has been partly attributed to a greater abundance of oral microbes.17 Our findings question the supposed association between decreased species richness and intestinal dysbiosis.54 Altogether, they indicate that although the mechanism of metachronous CRC in postgastrectomy patients may not be exactly the same as that of non-gastrectomy CRC, the similar microbiome contribution can be speculated in the cancer development.
We acknowledge that our study presents also some limitations. Even though our analysis was data-driven, it requires further validation. Future longitudinal or prospective studies with comprehensive medical records including health-related quality of life assessment (eg, based on Gastroenterology Quality of Life Index or Life After Gastric Surgery),55 intestinal inflammation under colonoscopy, intestinal permeability and nutrition assessment may provide further evidence to support our hypothesis. Additionally, most of our postgastrectomy patients underwent Roux-en-Y reconstruction, so our findings might be representative of this specific reconstruction (online supplementary results and online supplementary table S18).
Supplemental material
Our schematic hypothesis (figure 6) could be used as a framework for proof-of-concept studies that link the impact of microbiome alterations with patients’ outcome after gastrectomy. Our findings are in many ways consistent with results from other gastrectomy studies. We also identified several microbial functions and metabolites that might correlate with postsurgical metabolism. To determine if the microbiome is indeed involved in CRC occurrence following gastrectomy, further follow-up prospective studies are required. Our analysis also underlines the importance for gastrectomy patients to undergo intensified surveillance such as colonoscopy for early detection of possible metachronous CRC occurrence. To our knowledge, this is the first report on microbiome alterations using metagenomics and metabolomics data analysis after gastrectomy, especially total gastrectomy, for gastric cancer treatment. The present findings may be used to complement a non-invasive method for postsurgical prognosis assessment.
Acknowledgments
The authors would like to thank all participants, and their families, who participated in this study. The authors would like to thank Ms. Natsumi Sezawa and Ms. Manami Iwahara (National Cancer Center Research Institute, Japan), Ms. Yuka Ohara, Ms. Noriko Kagata and Ms. Kaori Igarashi (Institute for Advanced Biosciences, Keio University, Japan) for expert technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
- Data supplement 14
- Data supplement 15
- Data supplement 16
- Data supplement 17
- Data supplement 18
- Data supplement 19
- Data supplement 20
- Data supplement 21
- Data supplement 22
- Data supplement 23
- Data supplement 24
- Data supplement 25
- Data supplement 26
- Data supplement 27
- Data supplement 28
- Data supplement 29
- Data supplement 30
Footnotes
Twitter @erawijantaript
Contributors PPE, SM, SY and TY contributed to study concept and design. SY, SS, TN, TS and YS collected clinical samples and information. SF performed metabolome quantification. PPE, SM, HS and TY performed bioinformatics analyses on metagenomic and metabolomic data. PPE, SM, SY and TY wrote the manuscript. SY, SS and TY gave critical revision of the manuscript for important intellectual content. SY and TY supervised the study. All authors read and approved the final manuscript.
Funding This work was supported by grants from the Japan Agency for Medical Research and Development (AMED) (JP18ek0109187 to SF, SY and TY; JP19gm1010009 to SF and JP19cm0106464 to SY and TY); the National Cancer Center Research and Development Fund (25-A-4, 28-A-4, 29-A-13 and 29-A-6 to YS, SF, SY and TY); JST (Japan Science and Technology Agency)-PRESTO (JPMJPR1537 to SF and JPMJPRI507 to TY); JST-ERATO (JPMJER1902 to SF); JST-AIP Acceleration Research (JPMJCR19U3 to SY and TY); JSPS (Japan Society for the Promotion of Science) KAKENHI (142558 and 221S0002 to TY; 16H04901, 17H05654 and 18H04805 to SF); the Food Science Institute Foundation (to SF); the Program for the Advancement of Research in Core Projects under Keio University’s Longevity Initiative (to SF); Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University (SY); Joint Research Project of the Institute of Medical Science, the University of Tokyo (to SY); the Takeda Science Foundation (to SY and SF); the Suzuken Memorial Foundation (to SY); the Yasuda Memorial Medical Foundation (to SY) and Yakult Bio-Science Foundation (to SY).
Competing interests SF and TY are founders of Metabologenomics. The company is focused on the design and control of the gut environment for human health. The company had no control over the interpretation, writing or publication of this work. The terms of these arrangements are being managed by Keio University and Tokyo Institute of Technology in accordance with its conflict of interest policies.
Patient consent for publication Not required.
Ethics approval The samples and clinical information used in this study were obtained under conditions of informed consent and with approval by the institutional review boards of each participating institute (National Cancer Center, 2013-244; Tokyo Institute of Technology, 2014018 and Keio University, Shonan Fujisawa Campus, 78).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The raw sequencing data reported in this paper have been deposited at the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA), Tokyo, Japan under accession numbers DRA007281, DRA008243, DRA006684 and DRA008156. The source codes describing all of the analysis performed for this paper are available online (https://github.com/yamada-lab/GastrectomyGC_Microbiome).