Article Text

Abstract
Objective and design Human stem cell-derived hepatocyte-like cells (HLCs) have shown high potential as authentic model for dissection of the HCV life cycle and virus-induced pathogenesis. However, modest HCV replication, possibly due to robust innate immune responses, limits their broader use. To overcome these limitations and to dissect the mechanisms responsible for control of HCV, we analysed expression of key components of the interferon (IFN) system in HLCs, assessed permissiveness for different HCV strains and blocked innate immune signalling by pharmacological intervention.
Results Transcriptional profiling revealed that HLCs constitutively express messenger RNA of RLRs, and members of the IFN pathway. Moreover, HLCs upregulated IFNs and canonical interferon-regulated genes (IRGs) upon transfection with the double-stranded RNA mimic poly(I:C). Infection of HLCs with Jc1-HCVcc produced only limited viral progeny. In contrast, infection with p100, a Jc1-derived virus population with enhanced replication fitness and partial resistance to IFN, resulted in robust yet transient viraemia. Viral titres declined concomitant with a peak of IRG induction. Addition of ruxolitinib, a JAK/STAT inhibitor, permitted chronic infection and raised p100 infectious virus titres to 1×105 FFU/mL. IRGs expression profiling in infected HLCs revealed a landscape of HCV-dependent transcriptional changes similar to HCV-infected primary human hepatocytes, but distinct from Huh-7.5 cells. Withdrawal of ruxolitinib restored innate immune responses and resulted in HCV clearance.
Conclusion This authentic human cell model is well suited to examine acute and chronic host-HCV interactions, particularly IFN-triggered antiviral effector functions and mechanisms of innate immune control of HCV infection.
- stem cells
- hepatocyte
- HCV
- immune response
- interferon
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Stem cell-derived hepatocyte-like cells (HLCs) represent a relatively mature model of hepatocytes.
HLCs are permissive to HCV infection, but infection efficiency is low.
Primary human hepatocytes are susceptible to HCV infection, however, rapid de-differentiation and donor-to-donor variation complicate analyses.
What are the new findings?
Inoculation of HLCs with cell culture-adapted p100 HCV population allows complete and efficient replication of the virus.
HLCs clear acute HCV infection by a robust innate immune response, which is comparable to the one observed in primary adult hepatocytes.
Blockade of JAK/STAT signalling permits chronic HCV infection of HLCs, time-dependent experimental control of the innate immune response and thus analysis of effector functions mediating viral clearance.
How might it impact on clinical practice in the foreseeable future?
Our findings establish stem cell-derived HLCs and p100 infection as tractable and authentic model for studying of acute and chronic host-HCV interactions.
They open the way to personalised cell culture models of HCV infection, potentially providing critical insights for future personalised infection medicine.
Introduction
Although efficient direct-acting antivirals (DAAs) are available, HCV infection remains a global health problem, with ca. 70 million chronically infected patients.1 2 HCV vaccine development represents an important research goal, and benefits from a comprehensive understanding of the characteristics of protective immunity to HCV as well as the mechanisms of viral innate and adaptive immune evasion. These aspects have been heavily investigated using in vitro models for propagation of HCV relying mainly on the use of the Huh-7.5 hepatoma cell line. Notably, Huh-7.5 cells encode a defective retinoic acid inducible gene I (RIG-I), and therefore exhibit a blunted innate immunity,3 making them unsuitable for investigation of innate immune mechanisms associated with HCV clearance or persistence. Cultures of primary human hepatocytes (PHHs) are generally permissive to HCV in vitro infection4 and represent a more mature model to analyse innate immunity in response to HCV infection.5 6 However, PHHs present important limitations: limited availability, donor-specific differences in permissiveness, short duration of culture and de-differentiation on in vitro culture.
Human pluripotent stem cells (embryonic (hESCs) or induced (hiPSCs)) can be differentiated in vitro into cells reproducing key features of hepatocytes.7 These cells, termed hepatocyte-like cells (HLCs), are permissive to HBV,8 9 HCV10–13 and HEV14 infection in vitro. Upon inoculation with cell culture-derived HCV (HCVcc) containing the JFH1 (Gt2a) replication machinery,15 HLCs yield detectable intracellular HCV RNA replication, but only limited and transient production of infectious progeny viruses. It is generally assumed that active innate immunity is responsible for this acute HCV infection.16–18
The HCV genome replicates via the formation of a viral double-stranded (ds)RNA, a hallmark of viral replication, sensed by cellular pattern recognition receptors (PRRs). Among them are toll-like receptors (TLRs), particularly TLR3 detecting endosomal dsRNA,19 and the RIG-I-like receptors (RLRs) RIG-I20 21 and MDA520 22 23 detecting cytoplasmic dsRNA and signalling through the adaptor protein MAVS. Upon triggering, these PRRs induce interferon regulatory factor (IRF)3 translocation and interferon (IFN) production. Upon binding of type I or type III IFN to the cell surface receptors, the JAK/STAT signalling cascade activate transcription of numerous interferon-regulated genes (IRGs) controlling HCV infection.24 25 Among previously described IRGs with anti-HCV action are the PRRs MDA5 and RIG-I,26 ISG 1527 and MXA.28 Modulation of the JAK/STAT signalling pathway in HLCs has been previously described in order to improve HEV14 and HCV16–18 infection, but with conflicting results reported.17 Another approach to improve viral replication is to use HCVcc virus populations, selected for higher viral replication in Huh7-derived cell lines. Among them, a recently characterised Jc1-derived HCVcc viral population (termed p100) was isolated by long-term passage in Huh-7.5 cells; p100 exhibits higher replicative capabilities and partial resistance to IFN.29 30
Here, we infected HLCs with parental and adapted HCVcc and monitored innate immune responses during acute infection. We compared their innate immune response with the one in PHHs and Huh-7.5 cells, and investigated pharmacological intervention to modulate it, in order to make this model more tractable and valuable to decipher IFN-based antiviral effector functions and cellular mechanisms involved in viral clearance or persistence.
Results
HLCs possess an inducible, hepatocyte-like, innate immunity
HLCs were produced using standard protocols (online supplementary figures S1 and S2).12 31 They expressed key viral PRRs, IFN receptors and genes involved in the IFN pathway (online supplementary figure S3). HLCs can sense dsRNA through their RLRs, leading to IRF3 translocation, production of IFNs and subsequent induction of IRGs, making them a potential valuable model to analyse the innate immune response to HCV (online supplementary figure S4).
HCVcc p100 infection of HLCs and IRGs induction
HLCs were infected with the cell culture adapted p100 HCV virus population (figure 1A), resulting in enhanced viral replication when compared with the Jc1 parent virus (online supplementary figure S5). DAAs completely abrogated production of progeny p100 virus (online supplementary figures S5 and S6). Interestingly, treatment with ruxolitinib, a JAK/STAT pathway inhibitor (JSi), led to higher p100 viral replication, in HLCs (online supplementary figures S5 and S6) and PHHs (online supplementary figure S8) suggesting an important role of cellular innate immunity during acute p100 HCV infection of HLCs and PHHs.

Time and JAK/STAT-dependent control of stem cell-derived hepatocyte-like cell (HLC) infection by HCV p100. HLCs were left untreated (black) or inoculated with p100 (Multiplicity of Infection (MOI): 1) in presence of solvent (red), Telaprevir (direct-acting antiviral (DAA), blue) or ruxolitinib (JAK/STAT pathway inhibitor (JSi), green). (A) Kinetics of infectious virus production in supernatants of infected HLCs. (B) Time-dependent and treatment-dependent induction of selected interferon-regulated genes (IRGs), monitored by RT-qPCR, and expressed relative to uninfected cells. (C) Immunostaining for ISG15 and HCV NS5A in p100-inoculated HLCs. (D) Mean fluorescent intensity quantification of ISG15 staining in panel C. (E) HCV NS5A and interferon regulatory factor (IRF)3 co-staining on p100-infected HLCs, 7 dpi. (F) Type I and III interferon (IFN) induction on HCV p100 infection of HLCs. Means with SD of 3 independent experiments are given, statistical significance: *p<0.05; **p<0.01.
Infectious titres and production of IFNs and selected IRGs were concomitantly monitored during acute infection of HLCs by p100 HCV, in the presence or absence of ruxolitinib. During the initial days post infection (pi), infectious titres increased gradually to reach 104 FFU/mL in both ruxolitinib-treated and ruxolitinib-untreated infected HLCs (figure 1). However, in the following days, the titres decreased and infectivity was sometimes not detectable anymore at d11pi in non-treated cells, while in ruxolitinib-treated HLCs, infectious virions production did not decline, and titres reached up to 105 FFU/mL at d11pi. In infected HLCs, IRG messenger RNA (mRNA) induction (figures 1 and 2) and protein expression (figure 1C–D and online supplementary figure S6C) remained low during the first 4 days pi, followed by a rapid burst of induction, correlating with >100-fold drop of infectious virus titres. IRF3 nuclear translocation could sometimes be observed in HCV NS5A-positive HLCs (figure 1E), and type I and III IFNs upregulation was detectable concomitant with upregulation of IRGs and HCV clearance (figure 1F). Notably, very little, if any induction of IFN mRNAs was observed when infected HLCs were treated with ruxolitinib, suggesting that JAK/STAT-dependent feedback is necessary for full induction of type I and III IFN mRNA in this system. Importantly, ruxolitinib treatment did not affect levels of hepatic maturation or the expression of HCV-associated host factors in HLCs (online supplementary figure S7). Similar data were obtained when PHHs were infected with HCV p100 and treated with ruxolitinib (online supplementary figure S8).

Modulation of p100 HCV replication in stem cell-derived hepatocyte-like cells (HLCs). Production of infectious progeny in p100 HCV-infected HLCs (A) transfected with small interfering RNA (siRNA) against STAT1 and STAT2, (B) pre-incubated with anti-IFN-alpha receptor (anti IFNAR) antibodies, (C) treated with the PKR inhibitor C16, compared to non treated or Ruxolitinib (JSi)-treated HLCs. (D) Visualisation of infected HLCs, treated or not with C16 and/or JSi, 9 days post inoculation. (E) Interferon-regulated genes (IRGs) induction in HLCs treated or not with C16 or JSi. Means with SD of 2 (A, B) or 4 (C) independent experiments are given, statistical significance: *p<0.05; **p<0.01.
Transfection of HLCs with siRNA against STAT1, but not STAT2 (figure 2A), and blocking of the IFN receptor complex using anti-IFN-alpha receptor (IFNAR) antibodies (figure 2B) increased production of infectious progeny viruses, confirming that inhibition of the IFNAR signalling enhances HCV p100 infection of HLCs.
We previously reported that p100 HCV infection causes enhanced PKR activation compared with Jc1 infection.29 Moreover, other groups postulated that activation of PKR may be proviral by inhibiting translation of IFNs and IRGs.32–34 Therefore, we investigated the influence of PKR activation on p100 infection of HLCs by using oxindole-imidazole C16, a PKR inhibitor.35 C16 treatment decreased p100 replication on day 4 post inoculation, suggesting a proviral effect of PKR (figure 2C–D). However, at later time points, concomitant with massive induction of IRGs (figure 2E), C16 no longer inhibited infection. These results suggest a time-dependent role of PKR activation for control of p100 HCV infection of HLCs.
RNA-Seq analysis of HLCs infected with p100
To globally assess HCV-mediated innate immune activation, we performed RNA-Seq analyses of infected HLCs, with or without ruxolitinib or telaprevir, and compared them with uninfected HLCs. Expression levels of cellular mRNAs were quantified by mapping reads to the hg19 reference genome and normalisation performed to allow comparison between experiments. Normalised expression values for individual genes are presented as the number of Reads Per Kilobase of transcript, per Million mapped reads (RPKM). Analyses were performed at time points when infectious titre in infected, non-JSI-treated cells were dropping (online supplementary table S1, GEO accession GSE132606).
Pathway analyses showed that IFN response and other innate immune pathways are the most prominently dysregulated pathways on HCV infection of HLCs (figure 3A) (online supplementary figure 9A-C)(online supplementary tables S2 and S3). Upon ruxolitinib or DAA treatment, no immune-related pathways were enriched, indicating the dominance of HCV-triggered innate immune response pathways and highlighting replication-dependent triggering of these pathways.
Supplemental material
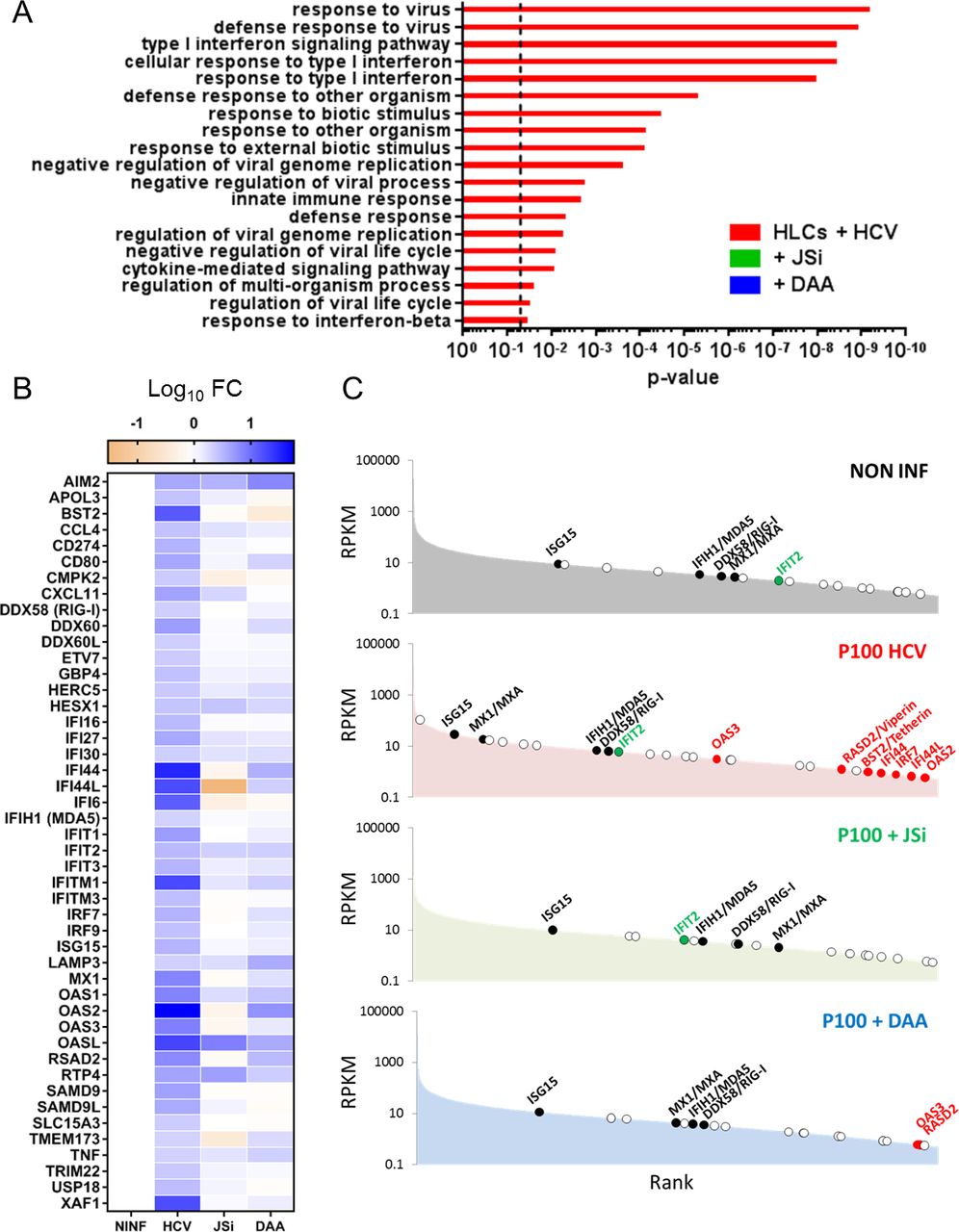
RNA-Seq analyses of p100-infected stem cell-derived hepatocyte-like cells (HLCs) (HCV), treated or not with 10 µM telaprevir (direct-acting antiviral (DAA)) or ruxolitinib (JAK/STATpathway inhibitor (JSi)), compared with non-infected HLCs. (A) Most upregulated pathways according to the Gene Ontology (GO) Enrichment Analysis tool. Histograms show p value calculated with Bonferroni correction. Dotted line indicates p value of 0.05. (B) Induction of selected interferon-regulated genes (IRGs) in each condition, expressed as log10 fold change of ‘Reads Per Kilobase of transcript, per Million mapped reads’ (RPKM) values compared with uninfected HLCs. Total list of studied IRGs can be found in online supplementary figure S10A. (C) RPKM ranking of selected IRGs in non-infected HLCs, p100-infected HLCs and infected HLCs treated with JJSi or telaprevir (DAA).
Our dataset was then analysed for 417 IRGs, including 112 IRGs associated with HCV and other flaviviruses infection.24–26 The total list of analysed IRGs and their differential expression on HCV infection can be found in online supplementary figure S10A and online supplementary table S4. In the infected HLCs, 46 of the 417 analysed IRGs were upregulated more than two fold (figure 3B, ‘HCV’), when compared with control cells (figure 3B, ‘NINF’). Among them are the IRF7 and IRF9, and IRGs previously described as restricting HCV infection (IFI27, IFI44L, IFITM1, IFITM3, ISG15, MXA, the oligoadenylate synthetase system components OAS1, OAS2 and OAS3, OASL, RSAD2 (viperin) and BST2 (tetherin)). IRG upregulation was strongly inhibited in infected cells treated with telaprevir (figure 3B, ‘DAA’) or ruxolitinib (figure 3B, ‘JSi’), confirming that on sensing of the replicating viral RNA, the JAK/STAT and IFN pathways are critical for induction of IRGs and their subsequent antiviral effect.
A rank ordered mRNA expression analysis confirms that upon HCV infection, HLCs upregulate many IRGs (figure 3C), particularly IRGs with described anti-HCV properties expressed only at very low level (RPKM <0.05) in control cells (figure 3C, red dots). HCV infection induced expression of some IRGs, which are not, or only partially dependent on JAK/STAT signalling, as ruxolitinib treatment of p100-inoculated HLCs did not totally ablate their induction. This group of mRNAs includes OASL, RTP4 and IFIT2 (online supplementary figure S10 and figure 3C, green dots). Moreover, induction of the RLRs RIG-I and MDA5 depended on JAK/STAT signalling offering a possible explanation why ruxolitinib treatment affected HCV-dependent induction of type I and III IFN mRNAs (figure 1F) (online supplementary table S4B, C).
HLCs innate immunity is comparable to the one of PHHs
We then compared expression and induction of IRGs in our HLCs with the one in Huh-7.5 cells and in PHHs (online supplementary figure S9D–F), using comprehensive RNA-Seq datasets reported elsewhere (Tegtmeyer B and Vieyres G et al, manuscript in revision; GEO accession: GSE132548). As described above, upon HCV infection, HLCs upregulate 46 IRGs, while PHHs and Huh-7.5 cells upregulate 159 and 53 IRGs, respectively (figure 4A) (online supplementary table S4 and S5). Forty-three of the 46 IRGs upregulated in HLCs were also upregulated in PHHs (figure 4A–C) while IRGs upregulated in infected Huh-7.5 cells were mostly distinct from the one upregulated in HLCs and PHHs (figure 4B,C). Ten IRGs were upregulated in all three models, among them were the PRR MDA5 (IFIH1), MX1 and ISG15. The pattern and intensity of IRGs upregulation upon HCV infection in HLCs correlated better with the one observed in PHHs (r=0.3124) than with the one in Huh-7.5 cells (r=0.0562) (figure 4D). Huh-7.5 IRG upregulation was also poorly correlated with the response in PHHs (r=0.0133). Importantly, both basal and induced levels of expression of IRGs in Huh-7.5 cells were very low compared with PHHs and HLCs (figure 4E). Taken together, these results confirm that Huh-7.5 cells represent a poor model to analyse innate immune response to HCV. HLCs on the other hand are a good substitute model to the innate immune activation of the native host cell of HCV, the PHH.
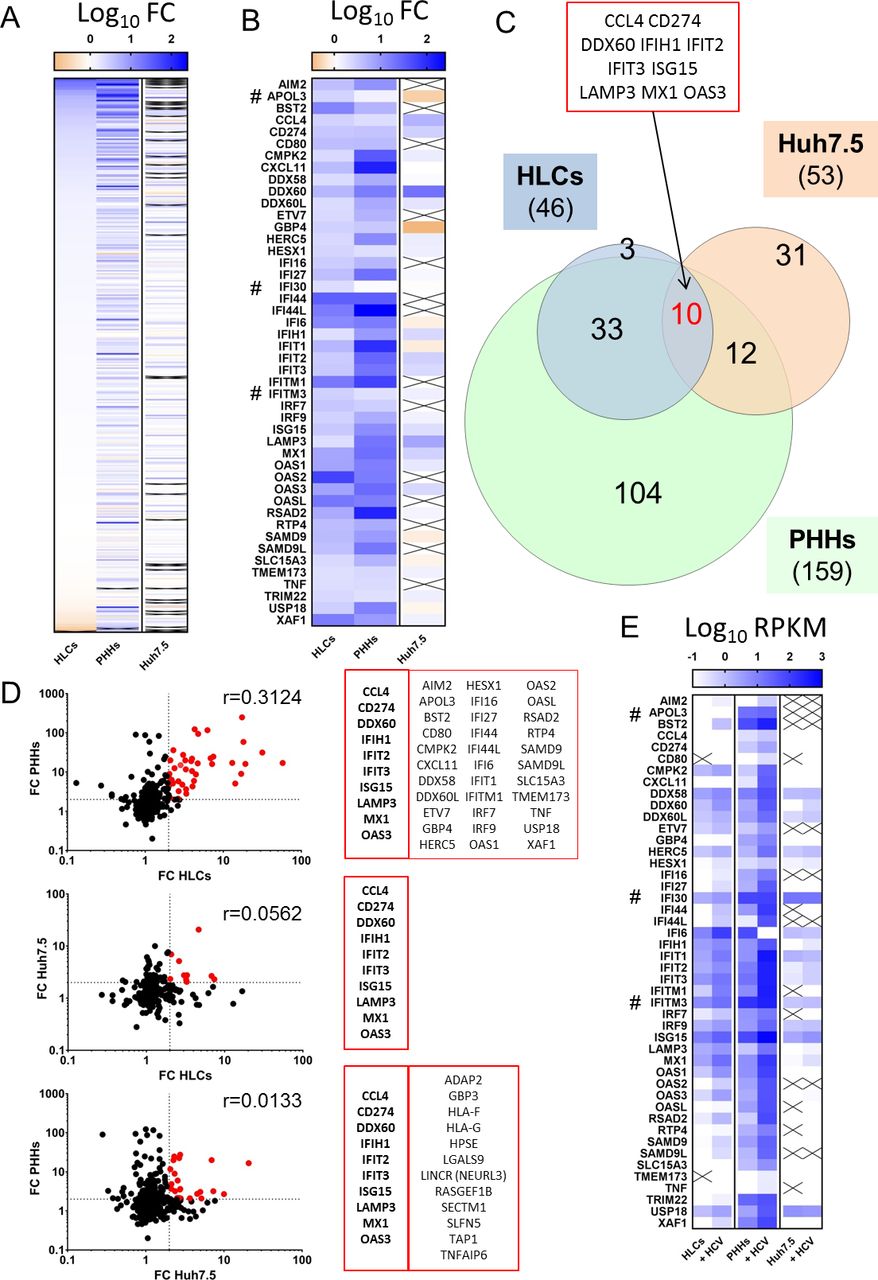
Interferon-regulated genes (IRGs) expression in stem cell-derived hepatocyte-like cells (HLCs) vs primary human hepatocytes (PHHs) vs Huh-7.5 and their induction by HCV infection (online supplementary table S4). PHHs and Huh-7.5 cells were inoculated with HCV Jc1 at an MOI 1, respectively. Cells were collected at 72 hours after inoculation and subjected to transcriptomic profiling. (A, B) Fold change on HCV infection of the 417 studied IRGs in HLCs, PHHs and Huh-7.5, ranked based on their overexpression in HCV-infected HLCs. Panel B illustrates the 46 IRGs upregulated more than twofold in HLCs (online supplementary table S5). # indicates the three IRGs upregulated in HLCs but not in PHHs. (C) Venn diagram of IRGs overexpressed in infected HLCs, Huh-7.5 and PHHs. (D) Correlation of expression fold change on HCV infection between HLCs, PHHs or Huh-7.5. Dotted line indicates fold change of 2. Red dots are IRGs upregulated in both cell models and are listed in the adjacent table. Pearson’s correlation coefficient r is displayed. (E) Average Reads Per Kilobase of transcript, per Million mapped reads (RPKM) values for IRGs upregulated more than twofold in infected HLCs and their respective expression in Huh-7.5 and PHHs, control or infected.
Experimentally controlled clearance of chronic HLC infection by HCV p100
We then examined if it is possible to establish a chronic, long-term HCV infection of HLCs. We previously described that HLCs could be maintained for several weeks31 and could support chronic infection with HBV.9 In the present study, we maintained HLCs for 3 weeks, with only limited loss of hepatic maturation (online supplementary figure S11). The HLCs maintained an active lipoprotein secretion pathway (online supplementary figure S11D), an important host factor for production of infectious progeny viruses. Ruxolitinib had no effect on the level of maturation of the cells even after 3 weeks of treatment (online supplementary figure S11A,C).
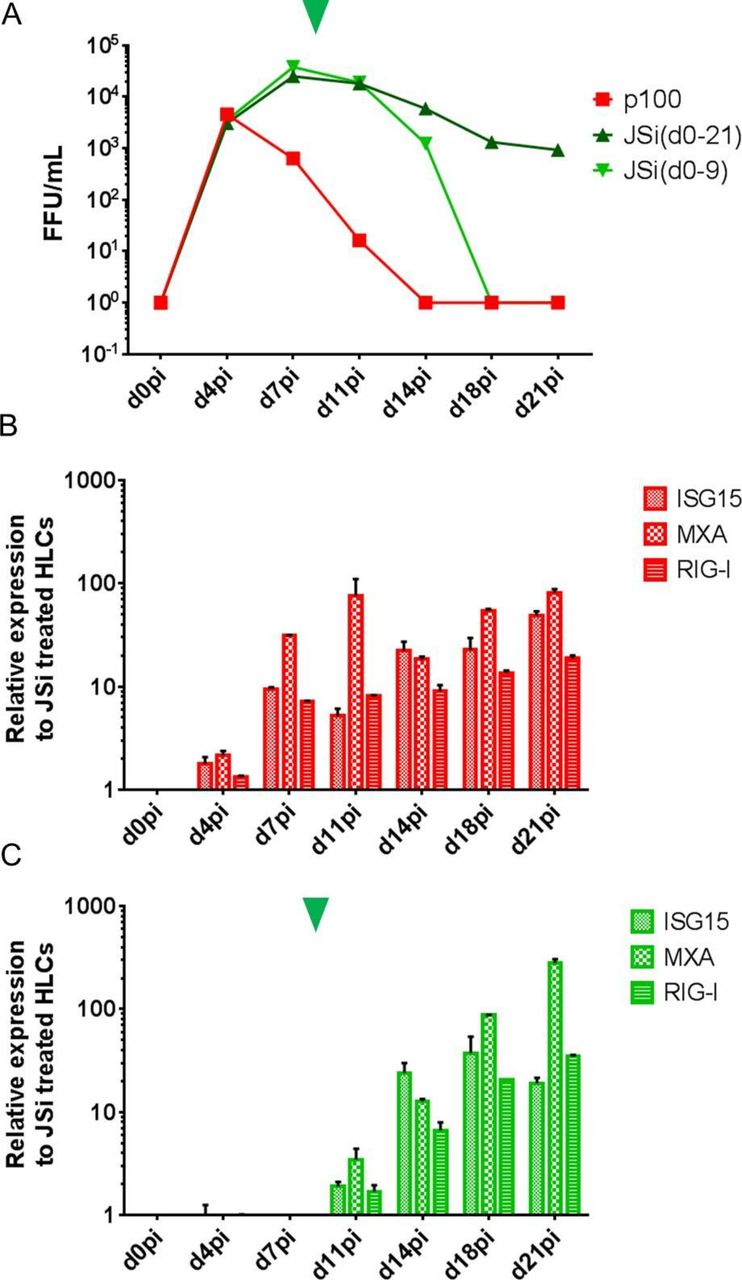
When HLCs were infected with p100 HCV and cultured without ruxolitinib, the induction of innate immune responses cleared the virus within 2 weeks (figure 5A,B, red). On the other hand, when kept under ruxolitinib treatment, p100 infection was maintained with constant infectious virus production for at least 21 days (figure 5A, dark green), without IRG induction (figure 5B). If ruxolitinib was removed from the culture medium (figure 5A,C, arrowhead), infectious titres subsequently dropped (figure 5A, light green line), concomitant with an induction of IRGs (figure 5C) mimicking the innate immune response observed at earlier time points in infected non-treated HLCs (figure 5B).
Taken together, these data show that the innate immune response to HCV infection can be controlled by using ruxolitinib, thus permitting a chronic HCV infection. Withdrawal of ruxolitinib experimentally restores innate immunity and control of the infection, making this a valuable tool to monitor and manipulate the hepatocyte innate immune response to HCV.
Discussion
Here, we report that stem cell-derived HLCs represent a tractable and immunocompetent model for the study of the cellular response to HCV. Tailored use of ruxolitinib, an inhibitor of the JAK/STAT signalling pathway, allows experimentally controlled induction of innate immunity and in turn viral clearance or persistence.
HCV infection studies in HLCs are challenging due to low infection rates and poor replication levels. In this regard, utilisation of p100, a Jc1-derived virus population with elevated fitness in Huh-7.5 cells is a key improvement, as infection with this virus population permits robust recapitulation of the entire viral life cycle in HLCs. It is not clear which features of p100 are important for mediating enhanced infection rate in the HLC culture. Sequencing analysis of p100 revealed nine coding mutations across the viral polyprotein.29 Four of them map to the NS3-4A protease complex, the NS5A phosphoprotein and the NS5B RNA-dependent RNA polymerase, and may directly affect RNA replication, and/or presentation of viral dsRNA to cellular PRRs. The point mutation in NS5A, a protein that has been implicated in inhibiting PKR function,36 could modify PKR-dependent repression of innate immunity.32–34 In line with this notion, p100 infection of Huh-7.5 cells is associated with enhanced phosphorylation of PKR and cellular protein translation shut off.29 Treatment of HLCs with the C16 PKR inhibitor decreased replication of p100 during the first days pi, suggesting that PKR activation may participate in the initial establishment of the p100 infection of HLCs. However, at later time points, C16 no longer exerted a significant effect. Collectively, these findings imply that viral determinants govern infection efficiency in this model and that early activation of PKR may facilitate infection. A broader comparison of HCV strains including primary virus populations from patients could help to characterise viral features critical for infection of these cells.
Modulation of the JAK/STAT pathway has been discussed previously as a way to improve viral replication in HLCs.16–18 Here, we monitored in detail the induction of innate immunity and its influence, on the complete replication cycle over extended periods. To modulate the JAK/STAT pathway, we used ruxolitinib, a kinase inhibitor that may have additional effects on the treated cells. However, analyses of gene expression data excluded a major impact of ruxolitinib on the level of HLC maturation, or expression of HCV replication co-factors (online supplementary figure S7). We also showed that ruxolitinib specifically blocked induction of IFNs and IRGs upon triggering of RLR (online supplementary figure S4). Finally, blocking IFNAR-dependent signalling, or silencing of STAT1 expression enhanced infection (figure 2). These data emphasise the role of IFNs in control of HCV infection of HLCs and support the conclusion that ruxolitinib enhances HCV infection by blocking JAK/STAT signalling.
Time-dependent administration of ruxolitinib provided several interesting insights. When added concomitant with virus inoculation, kinetics of infectious virus accumulation were very similar in the presence or absence of the drug for the first days of infection, with some minor fluctuation between experiments observed (figures 1A, 5A, online supplementary figures S5 and S6). These data suggest that, during this time period, HCV efficiently escapes or does not trigger JAK/STAT-signalling-dependent innate immune defenses. In line with this notion, highly sensitive RT-qPCR analysis of HCV-infected HLCs revealed only minimal IFN and IRG mRNAs induction until d4pi, despite of readily detectable de novo infectious virus production. We cannot completely rule out that HCV infection is sensed immediately after infection but in too few cells (as illustrated by fluorescence microscopy, figure 1E and online supplementary figure S5D) to be detected by our assays.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chronic stem cell-derived hepatocyte-like cell (HLC) infection by HCV p100 and its JAK/STAT-dependent clearance. A representative experiment of three is given. (A) Production of infectious progeny virions by HLCs infected with HCV p100 (MOI: 0.5), cultured for 21 days in absence (red) or presence of ruxolitinib (JAK/STATpathway inhibitor (JSi)) from d0pi to d21pi (dark green) or from only d0pi to d9pi (light green, arrowhead indicates time of withdrawal of JSi). Expression of selected interferon-regulated genes (IRGs) (B) in infected, non-treated HLCs, and (C) in infected HLCs treated with ruxolitinib from d0pi to d9pi, normalised to infected, ruxolitinib-treated HLCs, assessed by RT-qPCR.
Clearly, during virus transmission in vivo, HCV frequently overcomes these liver intrinsic innate restrictions and establishes a chronic infection. In the HLC-p100 model, infection is typically cleared between day 11 and 14 after inoculation, unless pharmacological ablation of IRG induction. This difference could relate to distinct HCV replication levels, and different robustness of innate immune responses between HLCs ex vivo and hepatocytes in vivo. Nevertheless, this novel HCV-HLC infection model provides unique opportunities to dissect the importance of IFN-dependent and IFN-independent mechanisms of HCV control on natural HCV infection and with endogenously expressed viral sensors and effectors. The innate immune response mounted during the second week pi closely mirrors the one observed on HCV Jc1 infection of PHHs. While the induction of the anti-HCV innate immunity in infected HLCs seemed quantitatively lower than the one in infected PHHs, detailed comparative pathway analysis showed that the responses in HLCs and PHHs are qualitatively similar (online supplementary table S3). It confirms the authenticity of HLCs as an in vitro model for studying the hepatocyte reaction to HCV infection.
While working with PHHs presents strong technical limitations, working with hPSCs could hold the key to better experimental approaches. For example, hPSCs can be genetically engineered using CRISPR/Cas9 and subsequently differentiated in HLCs reproducing the edited phenotype.37 Moreover, hiPSCs can be derived from patients with interesting genetic background, for example, polymorphisms in the type III IFNs genes, associated with spontaneous clearance and response to treatments.38 While this strategy has been proposed,11 it has not been so far used to decipher patient-specific innate immune response on in vitro HCV infection. We sequenced our hPSCs for single nucleotide polymorphisms of interest (online supplementary figure S12). Interestingly, H9ESC are homologous for the rs368234815(TT/ΔG) non-favourable ΔG allele, which leads to the expression of IFNL4, and is in patients associated with higher IRGs induction.39 Future studies involving p100, which robustly infects HLCs, and tailored ESC or iPSC cells representing these and other loci may help to discover new principles that govern the highly variable courses and outcomes of HCV infection, thereby instructing personalised infection medicine.
Acknowledgments
The authors would like to thank Dr KG Chen and Dr TJ Liang (NIH, USA) for the generation and transfer of the H9ESC adapted to monolayer culture; Professor CM Rice (Rockefeller University, USA) for the Huh-7.5 cells and the 9E10 anti-NS5A antibody; Professor E Domingo (Center for Molecular Biology 'Severo Ochoa', Spain) for the p100 HCV population; Michael Engelmann, Dr Gisa Gerold and the HZI Genome Analytics research centre for technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AC and TP designed the study concept, analysed data and wrote the manuscript. AC also performed experiments. JS and RB performed experiments, analysed data and edited the manuscript. FWRV provided essential reagents and edited the manuscript.
Funding This study is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project ID 158989968—SFB900, subprojects A6 and under the Germany’s Excellence Strategy—EXC 2155 'RESIST'—Project ID 39087428.
Disclaimer The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request to: Dr Arnaud Carpentier, arnaud.carpentier@twincore.de, ORCID identifier 0000-0002-6994-4689; Professor Thomas Pietschmann, thomas.pietschmann@twincore.de, ORCID identifier 0000-0001-6789-4422.
RNA-seq data are publicly available through the NIH GEO platform (https://www.ncbi.nlm.nih.gov/geo/): GEO accession GSE132606 and GSE132548.