Article Text

Abstract
Objective Reduced Paneth cell (PC) numbers are observed in inflammatory bowel diseases and impaired PC function contributes to the ileal pathogenesis of Crohn’s disease (CD). PCs reside in proximity to Lgr5+ intestinal stem cells (ISC) and mitochondria are critical for ISC-renewal and differentiation. Here, we characterise ISC and PC appearance under inflammatory conditions and describe the role of mitochondrial function for ISC niche-maintenance.
Design Ileal tissue samples from patients with CD, mouse models for mitochondrial dysfunction (Hsp60Δ/ΔISC) and CD-like ileitis (TNFΔARE), and intestinal organoids were used to characterise PCs and ISCs in relation to mitochondrial function.
Results In patients with CD and TNFΔARE mice, inflammation correlated with reduced numbers of Lysozyme-positive granules in PCs and decreased Lgr5 expression in crypt regions. Disease-associated changes in PC and ISC appearance persisted in non-inflamed tissue regions of patients with CD and predicted the risk of disease recurrence after surgical resection. ISC-specific deletion of Hsp60 and inhibition of mitochondrial respiration linked mitochondrial function to the aberrant PC phenotype. Consistent with reduced stemness in vivo, crypts from inflamed TNFΔARE mice fail to grow into organoids ex vivo. Dichloroacetate-mediated inhibition of glycolysis, forcing cells to shift to mitochondrial respiration, improved ISC niche function and rescued the ability of TNFΔARE mice-derived crypts to form organoids.
Conclusion We provide evidence that inflammation-associated mitochondrial dysfunction in the intestinal epithelium triggers a metabolic imbalance, causing reduced stemness and acquisition of a dysfunctional PC phenotype. Blocking glycolysis might be a novel drug target to antagonise PC dysfunction in the pathogenesis of CD.
- inflammatory bowel disease
- Crohn's disease
- intestinal stem cell
- energy metabolism
- intestinal epithelium
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Reduced Paneth cell (PC) numbers are frequently observed in inflammatory bowel diseases (IBD) and impaired PC function is a feature of ileal Crohn’s disease (CD).
PCs are involved in mucosal defence and support the intestinal stem cell (ISC) niche.
Mitochondrial dysfunction and alterations in energy metabolism in general are implicated during the onset and the course of IBD.
Mitochondrial function, metabolism and mitochondrial unfolded protein response are involved in intestinal epithelial cell differentiation and determine the cellular phenotype.
What are the new findings?
Reduced expression of the ISC marker Lgr5, in addition to reduced PC granularity, correlates with inflammation in patients with CD and CD-like TNFΔARE mice.
Morphological appearance of PCs and ISC in non-affected tissue margins predicts early postoperative endoscopic recurrence in patients with CD.
Induction of mitochondrial dysfunction in ISC by Hsp60 loss results in overall reduced Lgr5 expression and causes differentiation of Lgr5+ into aberrant PCs.
Reinforcing mitochondrial respiration by inhibition of glycolysis restores inflammation-imprinted dysfunction of the ISC niche.
How might it impact on clinical practice in the foreseeable future?
We provide evidence that impaired mitochondrial function is linked to the CD-associated loss of stemness and the generation of dysfunctional PC phenotypes. Demonstrating a proof-of-concept for targeting ISC alterations by implementing a drug-related metabolic shift, we rationalise a novel treatment approach for CD.
Introduction
Crohn’s disease (CD), belonging to the group of inflammatory bowel diseases (IBD), is characterised by transmural acute and chronic inflammation of intestinal tissue regions, typically involving the terminal ileum.1 In the pathogenesis of CD, multiple genetic risk factors together with environmental triggers result in a disturbed immune response towards a dysbiotic commensal microbiota.2 The intestinal epithelium as interface between microbiota and host critically contributes to intestinal homeostasis, and alterations in intestinal epithelial cell (IEC) subtypes including reduced numbers of goblet cells and Paneth cells (PCs) are frequently observed under inflammatory conditions.3 PCs are located in the crypt base of the small intestine, residing between Leucine-rich repeat-containing G-protein coupled receptor (Lgr) 5 crypt base columnar (CBC) intestinal stem cells (ISCs) and via secretion of antimicrobial peptides (AMPs) such as lysozyme, defensins (cryptdins), angiogenin-4 (Ang4) and secretory phospholipase A2, PCs contribute to pathogen clearance and shape the commensal microbiota.4 5
In addition to mucosal defence, PCs provide essential signals for maintenance of the ISC niche. Among those are Notch ligand (Dll4), and secreted factors like EGF and Wnt3,6 and also more recently identified metabolic signalling molecules including cyclic ADP ribose (cADPR)7 and lactate,8 facilitating optimal stem cell function. Moreover, on acute injury, PCs themselves serve as a reserve stem cell population, restoring Lgr5+ ISC via dedifferentiation, thereby contributing to tissue regeneration.9–11
Genetic risk variants of prominent CD-relevant genes involved in autophagy (ATG16L1), bacterial-sensing (NOD2), endoplasmic reticulum (ER) stress response (XBP1) and Wnt signalling (TCF4) were shown to affect PC function in terms of AMP production and secretion, and loss of PC defensins is particularly associated with ileal phenotypes of CD.3 12–16 Yet, it is controversial whether ileal CD is a specific disorder of PCs, or if loss of PC function is merely associated, but not causal for the development of CD. IEC-specific knockout models developing CD-like ileal inflammation including Caspase 8-/-,17 Atg16l1-/- 15 and Xbp1-/- 12 mice share the loss of PC function as a mechanism of pathogenesis; however, genetic ablation of PCs is not sufficient to generate a CD-associated phenotype.18 19 In line with a role of PCs in disease progression, TNFΔARE mice, a bacterial-driven model of chronic CD-like ileitis, display a loss of lysozyme-positive PCs subsequent to, but not preceding, the onset of TNF-driven tissue pathology. No increase in apoptosis could be observed in crypts from TNFΔARE mice, suggesting epithelial remodelling rather than PC-specific cell death.20 The cause of PC alterations as well as PC fate under chronic inflammatory conditions is currently unknown, though impairment of the exocytosis pathway in PC of mice with hypomorphic expression of the CD susceptibility gene Atg16l1 is paralleled by degenerating mitochondria.14 Interestingly, several genetic risk factors affecting mitochondrial function were identified for IBD,21 and intestinal inflammation has been suggested as energy-deficiency disease of IECs featuring alterations of the mitochondrial metabolism.22 23 Concomitantly, mitochondrial function and signalling pathways like the mitochondrial unfolded protein response (MT-UPR) have emerged as cellular checkpoint of metabolism, stemness and IEC differentiation programme.24 We previously demonstrated MT-UPR activation in IEC from patients with IBD and mouse models of intestinal inflammation25 and showed that activation of MT-UPR, induced by IEC-specific loss of the mitochondrial chaperone Hsp60, resulted in impaired mitochondrial respiration, and loss of ISC.26 However, the cellular origin and specific mechanisms that integrate mitochondrial function into CD pathology remain unknown. The cellular metabolism is increasingly recognised as a determiner of cellular phenotype and stem cell fate,24 and taking the essential role of PCs in the regulation of the ISC niche into account, we aimed at characterising PCs and Lgr5+ ISC in the context of mitochondrial function and inflammation.
In this study, we show that reduced PC granularity and reduced Lgr5 expression correlate with inflammation in patients with CD and TNFΔARE mice. Importantly, the morphological appearance of the ISC niche in non-affected tissue margins predicts early postoperative endoscopic recurrence in patients with CD, identifying an objective biomarker to select patients for preventive treatment. Induction of mitochondrial dysfunction in ISC by Hsp60 loss results in overall reduced Lgr5 expression and causes differentiation of Lgr5+ into aberrant PCs. Remarkably, inhibition of glycolysis is sufficient to override the inflammation-imprinted changes of the ISC niche ex vivo and rescued the ability of TNFΔARE mice-derived crypts to give rise to organoids.
Methods and material
All relevant methods and materials can be found in the online supplementary material.
Supplemental material
Results
Reduced Paneth cell function and Lgr5-expression correlate with CD-like inflammation
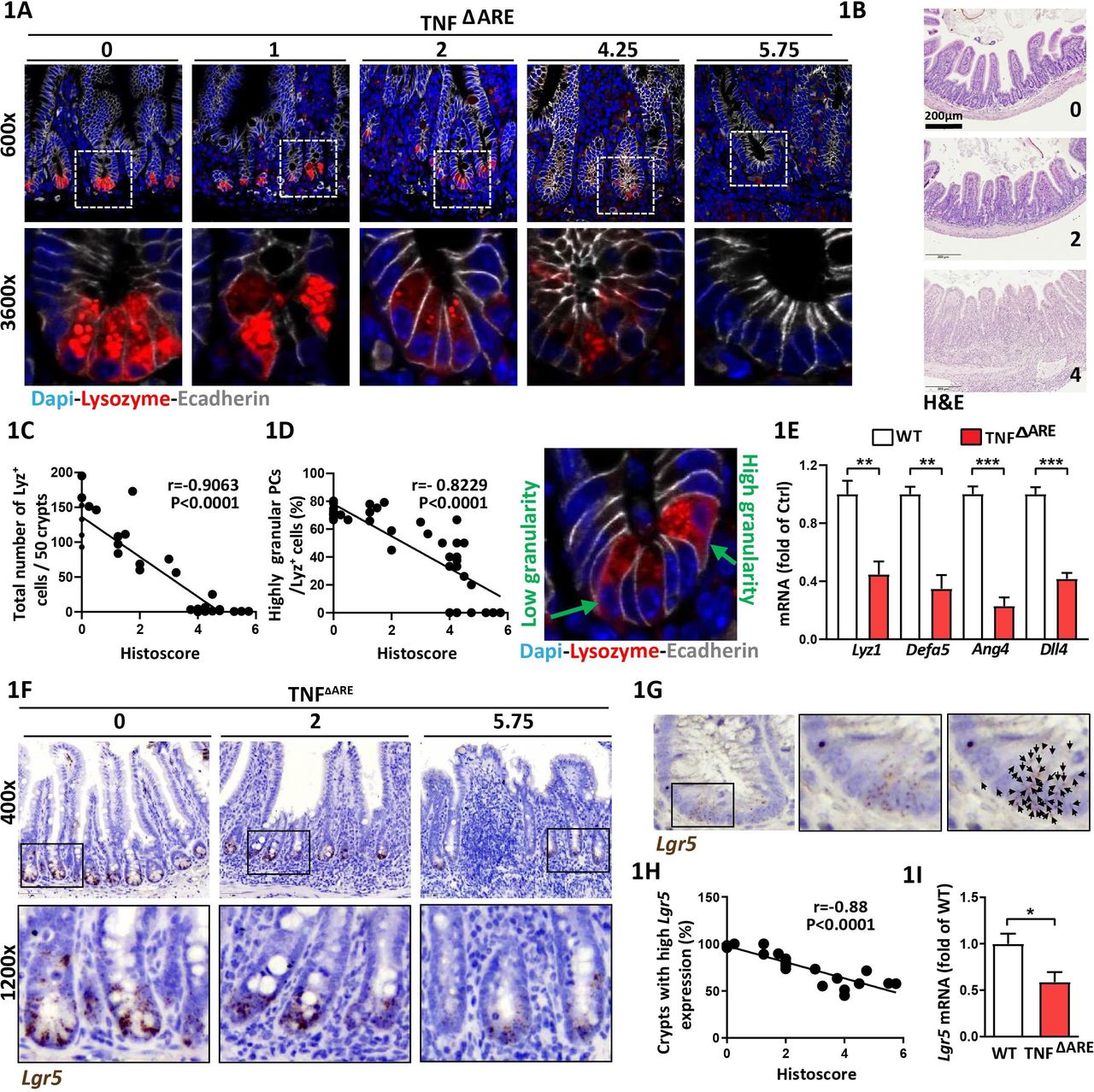
In TNFΔARE mice, a deletion in the tumour necrosis factor (TNF) AU-rich (adenosin-uracil) elements (ARE) leads to loss of translational control of TNF, resulting in a microbiota-driven transmural inflammation of the distal ileum.20 CD-like tissue inflammation gradually develops in TNFΔARE mice and has been linked to PC dysfunction.20 PCs contain distinct cytoplasmic granules for exocytosis of AMPs,27 and lysozyme-positive (Lyz+) secretory granule morphology is used as a functional marker for PC phenotype classification.13 14 We took advantage of the heterogeneity in the grade of CD-like inflammation in TNFΔARE mice to characterise PCs across a broad range of histopathological scores (HS). Ileal tissue sections derived from TNFΔARE mice with no (HS 0), moderate (HS <4) and severe (HS>4) inflammation were stained for Lyz and PCs were subdivided into a highly granular (≥2 Lyz+ cytoplasmic granules) and a lowly granular (<2 Lyz+ cytoplasmic granules and/or diffuse cytoplasmic Lyz staining) phenotype (figure 1A). Representative H&E stainings of tissue sections at different HS are given in figure 1B. Total numbers of Lyz+ PCs per crypt (figure 1C) and the percentage of highly granular PCs (figure 1D) inversely correlated with the grade of inflammation and signs of PC abnormalities were already visible in moderately inflamed mice (figure 1C,D). Comparing tissues from non-inflamed TNFΔARE mice and wild type (WT) littermates, we observed no differences in the total numbers of PCs. However, non-inflamed TNFΔARE mice showed a mild reduction in PC granularity (online supplementary figure 1A,B). Confirming reduced PC function in TNFΔARE mice, transcriptional levels of Lyz1 and PC-derived AMPs alpha-defensin 5 (Defa5) and Ang4 and ISC-niche supporting Dll4 were reduced (figure 1E). To depict the impact of ileitis-associated changes in PC function on CBC ISC marked by Lgr5 we performed in situ hybridisation for Lgr5 (figure 1F). The number of dots, representing Lgr5 transcripts, were counted (figure 1G) and crypts were classified as crypts with high Lgr5 expression (≥10 dots) and low Lgr5 expression (<10 dots), respectively. Consistent with reduced PC function, Lgr5 + ISCs were diminished under inflammatory conditions (online supplementary figure 1C) and numbers of crypts with high Lgr5 expression were inversely correlated with the degree of histopathology (figure 1H). Reduced stemness in inflamed TNFΔARE mice was confirmed by qPCR analysis of Lgr5 (figure 1I). No differences were observed in crypt morphology and Lgr5 expression between WT and non-inflamed TNFΔARE mice (online supplementary figure 1C,D). In situ hybridisation for Olfactomedin (Olfm) 4, a broader marker of active ISCs, confirmed reduced stemness under inflammatory conditions (online supplementary figure 1E-G). The parallel decrease of PC function and stemness indicates the tight functional interrelation of the ISC niche under inflammatory conditions.
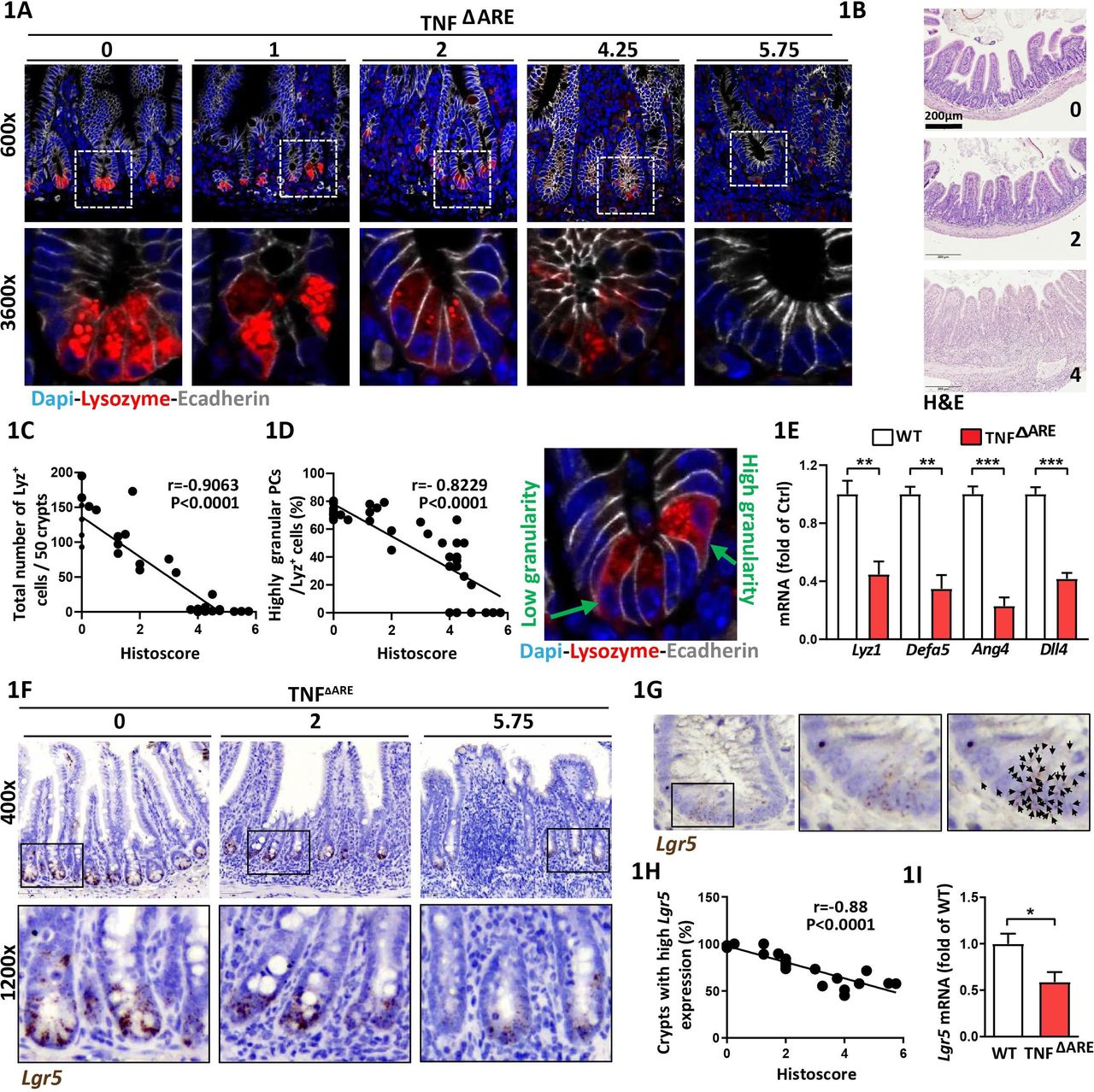
Paneth cell dysfunction and reduced Lgr5-expression correlate with CD-like inflammation in TNFΔARE mice Ileal tissue sections from TNFΔARE mice with different levels of inflammation and IEC isolates derived from TNFΔARE mice and WT littermates were analysed. (A) Immunofluorescence (IF) costaining of Lysozyme (red) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue), lower panel: higher magnification of the indicated sections. Numbers above the pictures indicate the HS of the respective tissue section. (B) Representative H&E images for HS 0 (non-inflamed), 2 (moderate inflammation), 4 (severe inflammation). (C) Correlation analysis (Pearson) of the number of Lysozyme positive (Lyz+) cells and HS and (D) proportions of highly granular Lyz+ cells (≥2 granules) and HS. Right: representative Lyz staining depicting Lyz+ cells with low and high granularity, respectively. (E) qRT-PCR analysis of IECs for genes involved in PC function (n=5) (F) Representative pictures of Lgr5 in situ hybridisation, including magnifications; numbers indicate the respective HS. (G) Illustration of Lgr5 transcript quantification; each dot, indicated by a black arrow represents one Lgr5 transcript. (H) Correlation analysis (Pearson) of the proportion of crypts with high Lgr5 expression (≥10 transcripts) and HS. (I) qRT-PCR analysis of IECs for Lgr5 (n=5). Statistics were performed by unpaired t-test. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. ARE,AU-rich (adenosin-uracil) elements; CD, Crohn’s disease; HS, histopathological score; IEC, intestinal epithelial cell; PC, Paneth cell; TNF, tumour necrosis factor; WT, wild type.
Aberrant PC phenotype and LGR5-expression correlate with active ileal CD
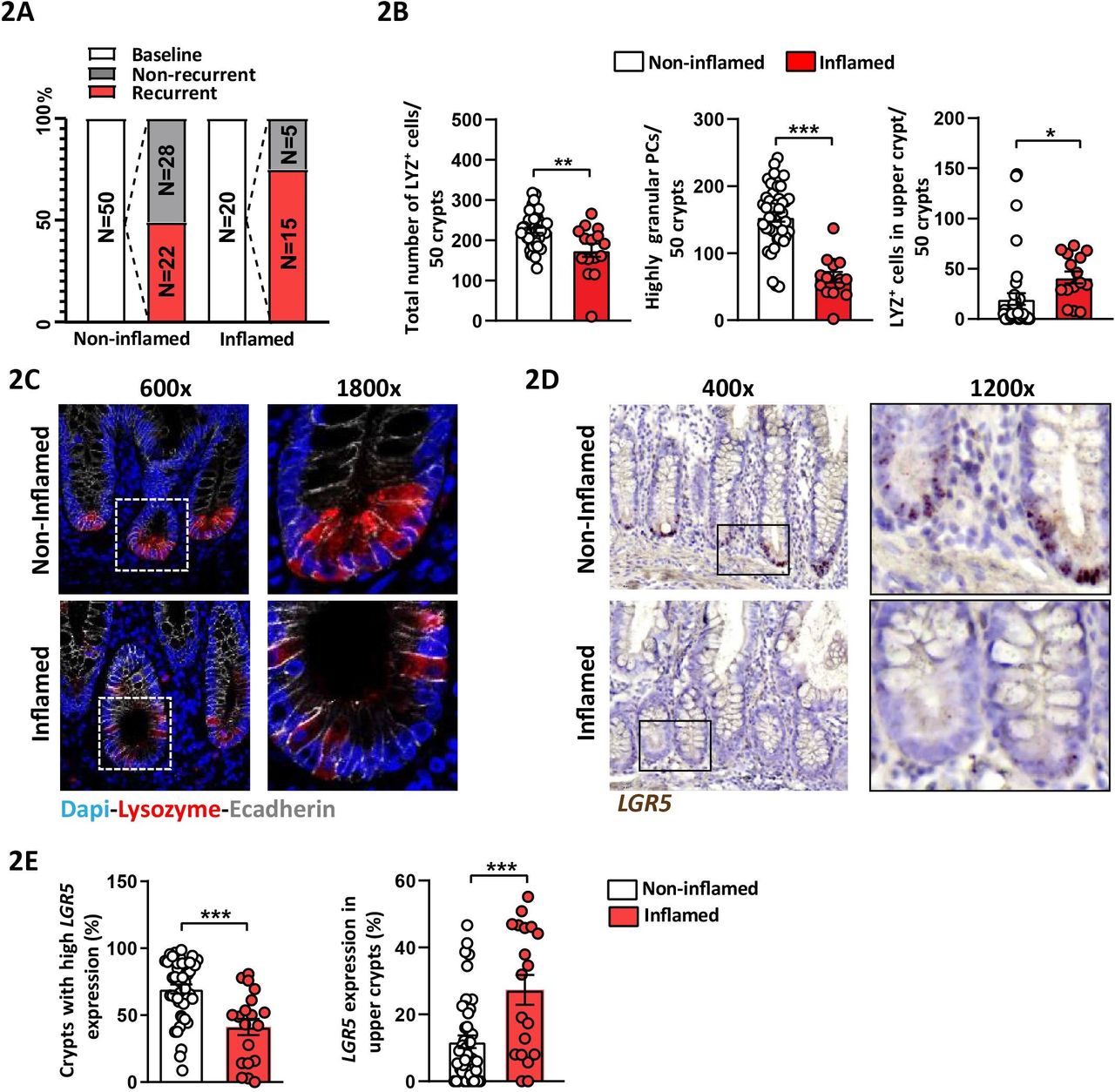
To verify the relevance of TNFΔARE mice, we obtained samples from 70 patients with CD undergoing surgical resection from a previously published prospective study.28 Early postoperative lesions observed by ileocolonoscopy best predict the course of disease and ileocolonoscopy is currently the gold standard to assess the risk of clinical recurrence.29 Ileal tissue margins were collected during resection surgery and a postoperative endoscopy was performed 6–12 months later to assess the endoscopic recurrence according to the Rutgeerts score. Endoscopic recurrence was defined as a Rutgeerts score ≥i2. Classification of patients with CD according to the presence or absence of inflammation in the resected tissue samples at baseline, and classification of postoperative endoscopic recurrence, is schematically shown in figure 2A, and patients’ characteristics at the time of surgery are compiled in online supplementary table 1. Consistent with TNFΔARE mice, numbers of LYZ+ PCs and highly granular PCs (≥2 LYZ+ cytoplasmic granules) were significantly reduced in tissue margins classified as inflamed at time of surgery compared with those classified as non-inflamed (figure 2B,C). Interestingly, the number of LYZ+ cells in ‘upper crypt’ (above +6 position) significantly increased in inflamed compared with non-inflamed tissue margins, suggesting an aberrant ISC niche architecture under inflammatory conditions (figure 2B, right). Reflecting the CD-like mouse model, inflamed tissue margins displayed a reduction in crypts with high LGR5 expression (≥15 dots), and concomitant to LYZ+ cells above the +6 position, LGR5 expression in upper crypts was induced (figure 2D,E).
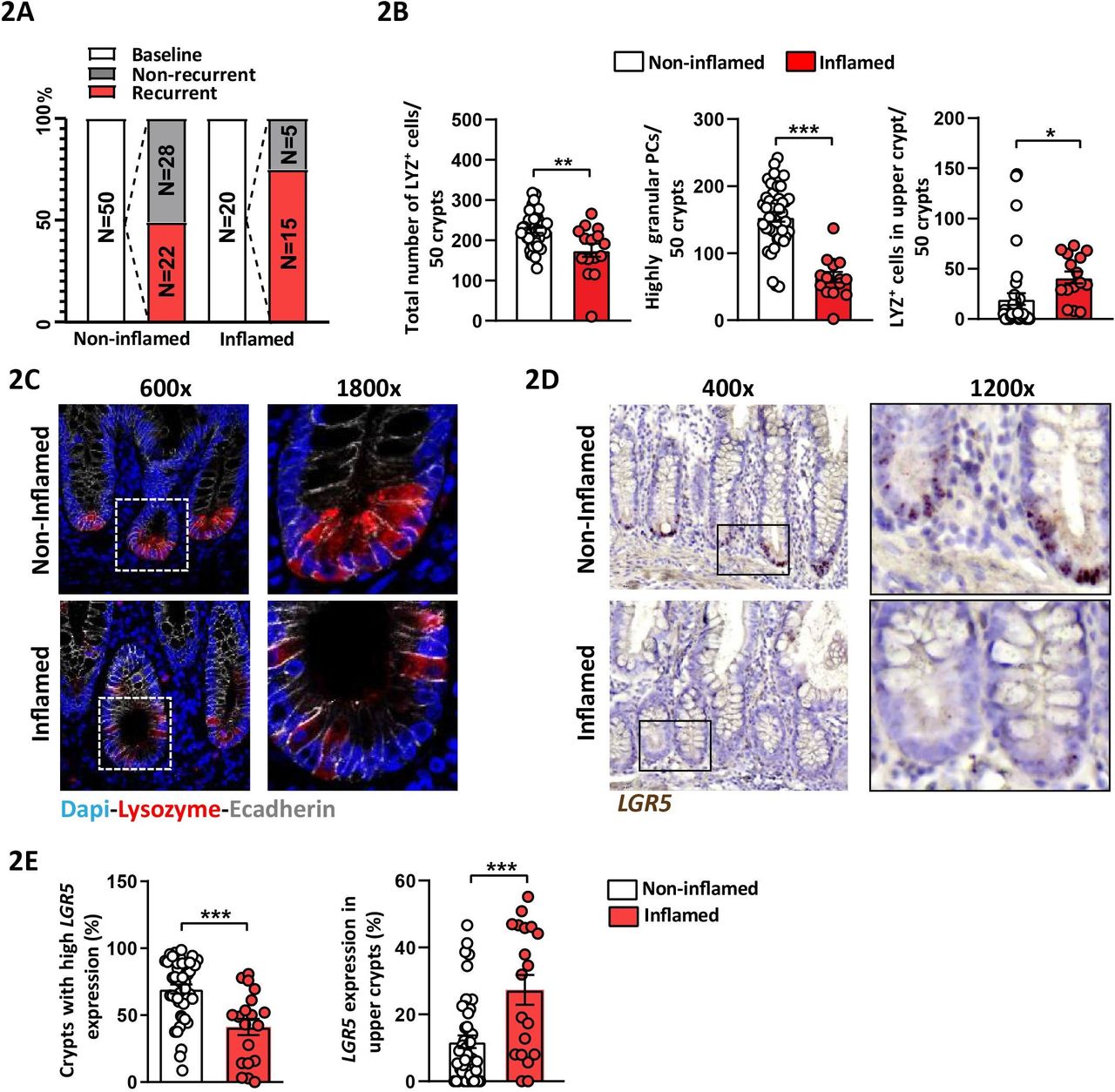
Aberrant Paneth cell phenotype and LGR5-expression correlate with disease activity in ileal tissue margins of patients with CD Ileal tissue sections of patients with CD undergoing resection surgery were analysed and tissue margins classified as non-inflamed and inflamed, respectively, at time of surgery were compared. (A) Overview of CD patient numbers for baseline and endoscopic follow-up disease classification. (B) Quantification of the total number of LYZ+ cells, highly granular LYZ+ cells (≥2 granules) and number of LYZ+ cells in upper crypt based on LYZ IF staining. (C) Representative pictures of IF costaining of Lysozyme (red) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue), right panel: higher magnification of the indicated sections. (D) Representative pictures of LGR5 in situ hybridisation, including magnifications. (E) Quantification of LGR5 in situ hybridisation giving the proportion of crypts with high LGR5 expression (≥15 LGR5 transcripts) and of crypts with LGR5 expression in upper crypt. Statistics were performed by unpaired t-test. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. CD, Crohn’s disease; IEC, intestinal epithelial cell; LYZ+, Lysozyme positive.
Paneth cell phenotype and LGR5-expression pattern predict recurrence in patients with CD
As previously shown,28 inflammation in resected ileal tissue at baseline was predictive for endoscopic recurrence, yet 44% of the analysed patients with CD with non-inflamed tissue margins still developed recurrent disease (figure 2A). Analysing regions of the intestine free of severe active or chronic inflammation has been proposed to more accurately reflect disease development due to the potential presence of early molecular and pathologic changes.13 Therefore, we tested in ileal margins classified as non-inflamed at the time of surgery if abnormal PC phenotypes and aberrant LGR5+ expression predict endoscopic recurrence after 6–12 months. Indeed, a low proportion of highly granular PCs (figure 3A,B) and high numbers of LYZ+ cells in upper crypts (figure 3C,D) correlated with the risk of recurrence, while the total number of LYZ+ PCs in crypts were not different between recurrent and non-recurrent groups (online supplementary figure 2A,B). In line, a low proportion of crypts with high LGR5 expression (figure 3E,F) and enhanced LGR5 expression in upper crypts (figure 3G,H) were further predictive for endoscopic recurrence. Assigning numbers of highly granular PCs, PCs in upper crypt, highly Lgr5 expressing crypts and Lgr5 expression in upper crypts as risk factors (0–4), we demonstrated a strong cumulative effect of the number of risk factors on the probability of disease recurrence (online supplementary figure 2C). Smoking, that had been identified previously as a risk factor for recurrence in this cohort,28 was associated with reduced PC granularity, a low proportion of crypts with high LGR5 expression and enhanced LGR5 expression in upper crypts (online supplementary figure 3A–C). Yet, within patients with CD experiencing recurrence, smoking had no additional impact on the risk factors analysed (online supplementary figure 3D,E). CD-associated risk alleles of NOD2 and ATG16L1 have been reported to impact PC phenotype;13 however, we observed no impact of the single nucleotide polymorphisms ATG16L1 rs6752107 and NOD2 rs2066845 or rs2066844 on the ISC niche appearance, probably due to a high proportion of risk allele carriers (online supplementary figure 3F-H). Contrarily, in tissue margins classified as inflamed at the time of surgery, aberrant PC phenotypes and stemness failed to better stratify disease recurrence (online supplementary figure 4). In summary, these data show a strong predictive value for alterations of the ISC niche for early endoscopic recurrence, indicating these phenotypic changes as first molecular signs of inflammatory changes, preceding macroscopic lesions.

PC phenotype and LGR5-expression predict disease recurrence in patients with non-inflamed CD Ileal tissue sections classified as non-inflamed at time of surgery of patients with CD undergoing resection surgery were analysed. Tissue sections were stained for LYZ by IF and for LGR5 by in situ hybridisation, respectively, and expression patterns were quantified. Numbers of highly granular LYZ+ cells (≥2 granules), numbers of LYZ+ cells in upper crypt, proportion of crypts with high LGR5 expression (≥15 LGR5 transcripts) and proportion of crypts with LGR5 expression in upper crypt were determined. Patients with CD with endoscopic recurrence (Rutgeerts score ≥i2) 6–12 months after surgery were compared with patients with CD not experiencing recurrence. (A,C,E,G) Overall comparison of recurrent versus non-recurrent patients for the respective marker; (B,D,F,H) From left to right: distribution of the respective marker among patients with recurrent (red circles) and non-recurrent (black circles) CD with median indicated; probability of patients with CD to experience recurrence if above or below the median for the respective marker; representative pictures showing sections from patients with CD above or below median. (A,B) Number of highly granular LYZ+ cells (≥2 granules), (C,D) Number of LYZ+ cells in upper crypt, (B,D) IF costainings of Lysozyme (red) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue). (E,F) Proportion of crypts with high LGR5 expression (≥15 LGR5 transcripts), (G,H) proportion of crypts with LGR5 expression in upper crypt, (F,H) LGR5 in situ hybridisation. (A,C,E,G) Statistics were performed by unpaired t-test. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. (B,D,F,H) Statistical analysis was performed via χ² test. CD, Crohn’s disease; IEC, intestinal epithelial cell; LYZ+, Lysozyme positive; PC, Paneth cell.
Inflammation in TNFΔARE mice is associated with impaired mitochondrial function
Phenotypic changes of PCs have been described concomitantly to structurally impaired mitochondria,14 and mitochondrial stress signalling (MT-UPR) is apparent in IEC from patients with IBD and mouse models of intestinal inflammation.25 Accordingly, adenosine triphosphate (ATP) levels were reduced in isolated ileal crypts from TNFΔARE mice, along with increased levels of the MT-UPR marker proteins Hsp60 and (dsRNA-activated protein kinase) Pkr25 under inflammatory conditions (figure 4A–D). Concomitantly, transcriptional levels of genes involved in signalling pathways related to disturbed mitochondrial function, including MT-UPR (Trb3, Atf5, Chop), antioxidative response (Hif1a) and low ATP levels (Prkaa2, AMP-kinase) were upregulated in ileal crypts (figure 4E), while expression of Grp78, a surrogate marker of ER stress remained unaltered (figure 4F). In line, transmission electron microscopy showed markedly reduced numbers of PCs with typical morphological features at the ileal crypt bases of inflamed TNFΔARE mice. The few remaining Paneth-like cells often exhibited secretory granules with broadened halos, intracytoplasmic vacuolations, dilation of the rough ER, as well as degenerative mitochondrial alterations, including mitochondrial swelling with dissolution and disruption of cristae, loss of matrix density and occasionally formation of intramitochondrial electron-dense inclusions (figure 4G). Autophagy contributes to the mobilisation of cellular energy stores and alterations in autophagy-related proteins have been linked to the disruption of the PC granule exocytosis pathway and degenerating mitochondria.14 Thus staining for the autophagy marker LC3, an increase in LC3 expression in crypt bases of inflamed TNFΔARE mice was observed accompanying metabolic and morphologic alterations (online supplementary figure 5A,B).

Inflammation in TNFΔARE mice is associated with mitochondrial dysfunction in ileal crypts Isolated ileal crypts and tissue sections from TNFΔARE mice and WT littermates were analysed. (A) ATP content of primary isolated crypts relative to life cell protease activity measured by a fluorescence assay (n=10/7). (B) Overview of crypt structure and illustration of the area used for protein quantification. (C) IF images of Hsp60 (green) counterstained with Dapi (nuclei, blue), including magnifications. Numbers above the pictures indicate the HS of the respective tissue section. Right: corresponding quantification. (D) Costaining of Pkr (green) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue), including magnifications. Numbers above the pictures indicate the HS of the respective tissue section. Right: corresponding quantification. (E) qRT-PCR analysis of primary ileal crypts for genes involved in mitochondrial MT-UPR, mitochondrial signalling and (F) ER stress (n=6). Statistics were performed by unpaired t-test. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. (G) Transmission electron microscopy of ileal crypt bases. Panel A: WT; A1-2: PCs display abundant, apical, electron-dense SGs with narrow halos (arrow). Asterisks mark secretory granules with small electron dense cores and wide rims of flocculent material of low electron-density. A-3. Unaltered appearance of rER and mitochondria (M) in a WT-PLC. B: non-inflamed TNFΔARE mice; B-1–6: PC ultrastructure essentially resembles WT mice. N: nucleus. C: inflamed TNFΔARE mice. C-1–2: Few remaining cells with PLC-typical location and morphology often show vacuolation (V) and broadened halos of secretory granules. IC: infiltrating inflammatory cell C-4–6: Mitochondrial lesions in PLC include intramitochondrial inclusions (C-4, C-5), mitochondrial swelling with dissolution and disruption of cristae, and loss of matrix density and also distension of the rER (C-6). ARE,AU-rich (adenosin-uracil) elements; CD, Crohn’s disease; HS, histopathological score; IEC, intestinal epithelial cell; PC, Paneth cell; rER, rough endoplasmic reticulum; SG, secretory granule; TNF, tumour necrosis factor; WT, wild type.
Mitochondrial impairment in ISC causes transition towards dysfunctional Paneth cells
PC and Lgr5 + ISC alterations concurrent to mitochondrial impairment were present under inflammatory conditions in TNFΔARE mice. Hence, we characterised the role of mitochondrial function on ISC niche appearance using a mouse model in which Hsp60 can be specifically deleted in Lgr5 + ISC (Hsp60 flox/flox × Lgr5-eGFP-IRES-CreERT2-Tg) via administration of tamoxifen (Hsp60Δ/ΔISC).26 Hsp60 represents a target gene of MT-UPR signalling and constitutes the main chaperone of the mitochondrial matrix.30 Thus, loss of Hsp60 leads to disturbed proteostasis in mitochondria and subsequent activation of MT-UPR signalling.26 Consequently, Hsp60 deficiency results in mitochondrial dysfunction, including reduced mitochondrial respiration and a drop in cellular ATP content.26 Confirming MT-UPR activation on Hsp60 loss, transcription of the cochaperone and surrogate marker of MT-UPR, Hsp10 increased early after induction of Hsp60 deletion in parallel with induction of Trb3, Chop, Hif1a and Prkaa2 (figure 5A). Following Lgr5 and Lyz expression in the ileum of Hsp60Δ/ΔISC mice for up to 6 days after the end of tamoxifen treatment, transient changes of the ISC niche were characterised. At day 2, the proportion of highly Lgr5 expressing crypts dropped (figure 5B,C). Parallel efforts to characterise PC phenotypes demonstrated that Lgr5 - Lyz+ cell numbers decreased, while numbers of Lgr5 + Lyz+ double positive cells increased, starting at day 0 after end of tamoxifen treatment (figure 5B–D). Along with diminished expression of markers for ISCs and PCs, PC granularity and expression of PC-derived AMPs were reduced in response to Hsp60 loss-induced mitochondrial dysfunction in ISCs (figure 5E–G). Reflecting inflamed TNFΔARE mice, LC3 staining in crypt bases of Hsp60Δ/ΔISC mice depict induction of autophagy (online supplementary figure 5C) along with metabolic and morphological alterations of the ISC niche. To exclude the possibility of mitochondrial dysfunction-mediated cell death of Lgr5 + ISC and Lyz+ cells, tissue sections from day 2, when loss of Lgr5 expression was most pronounced, were costained for TUNEL and eGFP or Lyz, respectively. Furthermore, tissue sections were stained for the apoptosis marker cleaved caspase (CC) 3, confirming absence of enhanced apoptosis at day 2 after the end of tamoxifen treatment, but indicating increased apoptosis in crypt basis at days 4 and 6 (online supplementary figure 6). Hsp60 deficiency in Lgr5 + ISCs was associated with a complete loss of proliferation as indicated by Ki67 staining (online supplementary figure 7A). However, expression of Olfm4 and Hopx, a marker of +4 reserve stem cells potentially contributing to Lgr5+ ISC replenishment,31 was retained and transiently enhanced in crypts (online supplementary figure 7B,C). In line, cells above the crypt basis remained Hsp60 and Ki67 positive throughout the observed time points (online supplementary figure 7A). Crypt-based cells regained Hsp60 and Ki67 expression from day 4 on, indicating apoptotic cell death and extrusion of Hsp60 deficient cells. Lgr5 + ISC can differentiate into PCs, and vice versa, dedifferentiation of PCs and acquisition of stem-like features has been described following loss of CBC ISCs.9 10 To determine if dysfunctional PCs arise from mitochondrial function-impaired ISCs, we stained for Lyz and Hsp60. Hsp60 negative cells directly originated from Lgr5 + ISCs that had suffered from mitochondrial dysfunction. The ratio of Lyz+ cells in which Hsp60 could not be detected was increased at day 0 and day 2 (figure 5H,I), indicating Lgr5 +, Hsp60- ISCs to acquire Lyz expression via mechanisms initiated by mitochondrial dysfunction. Furthermore, reduced PC granularity and diffuse Lyz staining was associated with lack of Hsp60 in PCs (figure 5J). In summary, these data suggest that mitochondrial impairment, including reduced mitochondrial respiration, initiates a transition of Lgr5 + ISCs towards a PC-like phenotype. However, the inability of these Lgr5 + Lyz+ double positive cells to adjust their mitochondrial function to the new cell-phenotypic demand (due to lack of Hsp60), seems to hamper differentiation into functionally mature PCs (figure 5K). Interestingly, costaining of Hsp60 with Mucin 2 (Muc2), Chromogranin A (ChgA) or doublecortin-like kinase 1 (dclk1) as markers for the secretory IEC types (goblet cells, enteroendocrine cells and tuft cells, respectively) indicated that ISC suffering from mitochondrial dysfunction only acquired Lyz expression, since Muc2, ChgA and dclk1 expressing cells remained Hsp60 positive (online supplementary figure 8A–C). At day 6 after tamoxifen treatment, ISCs and PCs regained normal phenotypes comparable to Hsp60 flox/flox mice (figure 5B,C,E,F,H). At this time point, Hsp60 mRNA expression in IEC returned to control levels (figure 5A). The complete regain of Hsp60+, Lgr5 + ISCs and a normal ISC niche phenotype indicates replenishment of Lgr5 + ISCs from reserve ISC populations as described before.9 32
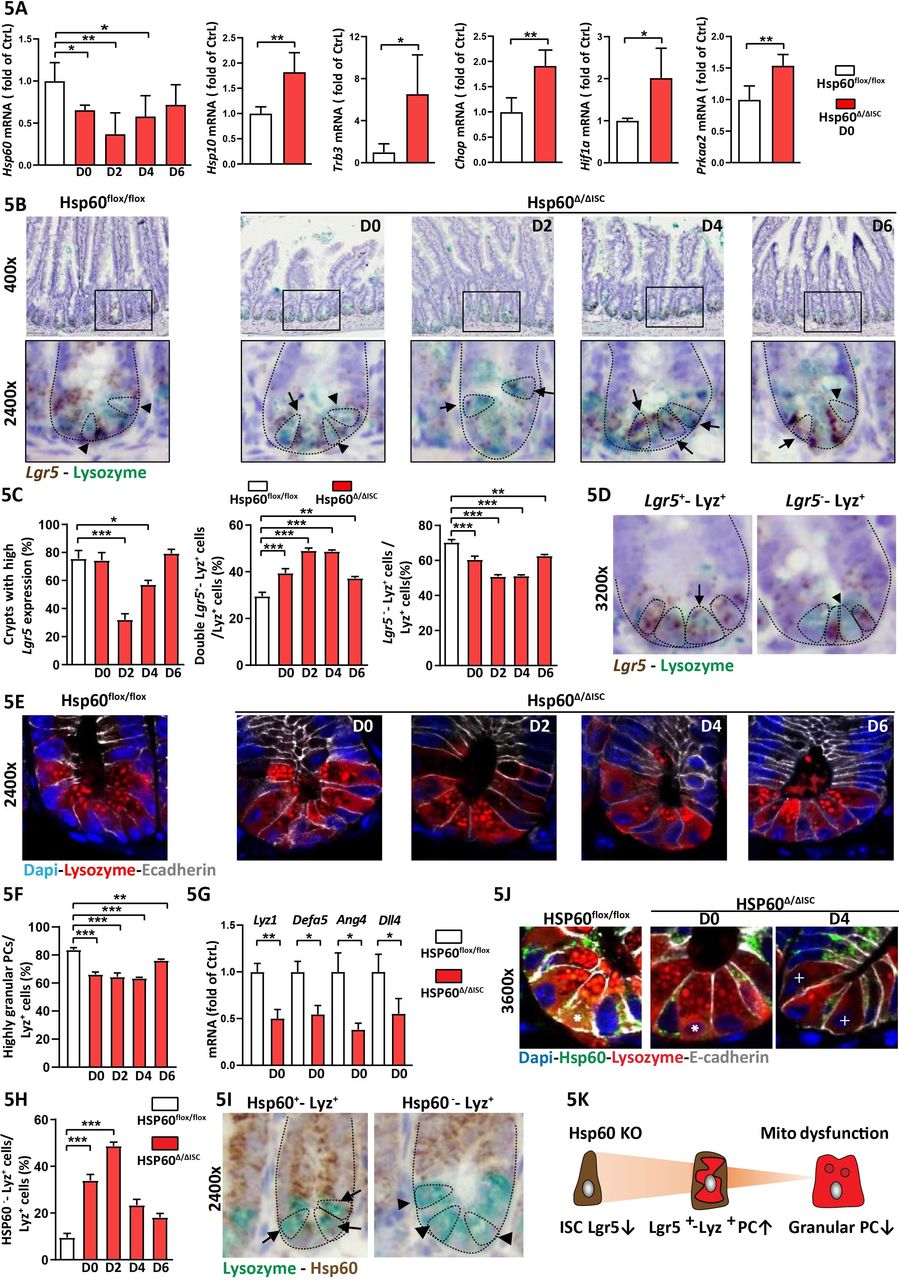
Mitochondrial impairment in ISC causes transition towards dysfunctional Paneth cells Ileal tissue sections and IEC isolates of Hsp60flox/flox mice and Hsp60Δ/ΔISC mice were analysed at different time points after end of tamoxifen treatment. (A) qRT-PCR analysis of IECs for Hsp60 and genes involved in mitochondrial MT-UPR and mitochondrial signalling at day 0. (B) Lgr5 in situ hybridisation (brown) and lysozyme (Lyz, turquoise) immunehistochemistry (IHC) costaining, lower panel: magnifications, dotted lines indicate crypt and cell borders, arrow-heads indicate Lyz+ cells, arrows indicate Lgr5—Lyz double-positive cells. (C) Quantification of the proportion of crypts with high Lgr5 expression (≥10 Lgr5 transcripts), the proportion of Lgr5—Lyz double-positive cells, and the proportion of Lgr5 negative, Lyz single-positive cells over time. (D) Representative pictures of Lgr5—Lyz double-positive cells, and Lgr5 negative, Lyz single-positive cells; dotted lines indicate crypt and cell borders, arrows indicate Lgr5—Lyz double-positive cells, arrow-heads indicate Lgr5 negative, Lyz single-positive cells. (E) IF costaining of Lyz (red) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue) showing granular and non-granular staining pattern. (F) Quantification of the proportion of highly granular Lyz+ cells (≥2 granules). (G) qRT-PCR analysis of IECs from day 0 for PC-derived AMP and Dll4. (H) Quantification of the proportion of Hsp60 negative, Lyz single positive cells. (I) Representative pictures of IHC costaining for Hsp60 (brown) and Lyz (turquoise) used for quantification; dotted lines indicate crypt and cell borders, arrow indicates Hsp60—Lyz double positive cells, arrow-heads indicate Hsp60 negative, Lyz single-positive cells. (J) IF costaining of Hsp60 (green), Lyz (red), and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue), including magnifications. Asterisks indicate Hsp60—Lyz double positive, highly granular Paneth cells, crosses indicate Hsp60 negative, Lyz single-positive cells, depicting a diffuse Lyz staining. (K) Schematic representation of the main findings in this figure. Statistical analyses were performed by one-way ANOVA followed by Tukey test or unpaired t-test. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. AMP, antimicrobial peptides; ANOVA, analysis of variance; IEC, intestinal epithelial cell; MT-UPR,mitochondrial unfolded protein response;
Crypts from inflamed TNFΔARE mice fail to grow into organoids
Further investigating the role of mitochondrial function on ISC niche-regulation under inflammatory conditions, primary crypts of non-inflamed and inflamed TNFΔARE mice were isolated and seeded in medium without Wnt factors.33 Addition of Wnt factors to the medium is not essential for small intestinal (SI) organoid culture and has been linked to the presence of PCs providing these signals naturally.34 Ileal crypts from inflamed mice displayed reduced Lgr5 and Lyz expression, confirming figure 1 (figure 6A,B). Consistently, ileal crypts from inflamed, but not non-inflamed TNFΔARE mice almost completely failed to grow into organoids. Remaining organoids showed a severe defect in de-novo crypt formation (budding) (figure 6C). In non-inflamed TNFΔARE mice and WT littermates, seeding efficiency and budding were comparable (online supplementary figure 9A), confirming inflammation and not genotype as responsible for impaired growth. Of note, crypts derived from the jejunum, directly adjacent to the inflamed ileum but showing no tissue pathology in TNFΔARE mice, also displayed a reduced ability to form organoids (figure 6D). Applying an intestinal organoid culture medium supplemented with Wnt factors directly after crypt isolation did not rescue growth (online supplementary figure 9B). Hence, it is likely that disturbances of both Lgr5 + ISCs and PCs contribute to the inflammation-induced growth defect.
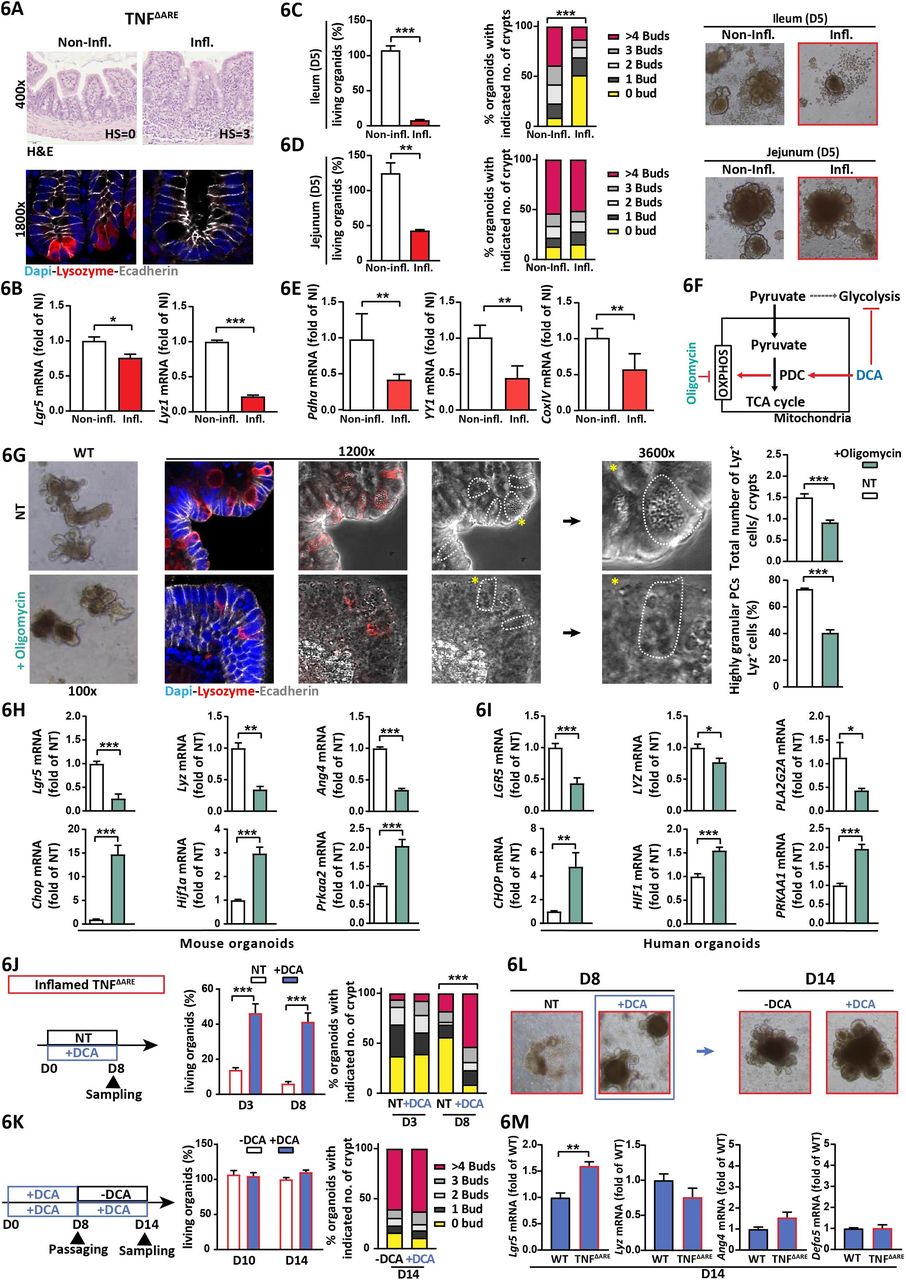
Mitochondrial respiration determines stemness and PC functionality under inflammatory conditions (A–E) Ileal tissues and crypts derived from non-inflamed and inflamed TNFΔARE mice were analysed. (A) Representative pictures of ileal sections; top: H&E staining with histopathological score indicated; bottom: IF costaining of Lyz (red) and E-cadherin (grey) counterstained with Dapi (nuclei, blue) showing absence of PCs in inflamed tissue. (B) qRT-PCR analysis of primary ileal crypts for Lgr5 and Lyz (n=6). (C,D) Characterisation of intestinal organoids derived from ileal (C) and jejunal (D) crypts at day 5 of ex vivo culture. Left: Proportion of living organoids; middle: quantification of de novo crypt formation (budding); right: representative bright field pictures. (E) qRT-PCR analysis of primary ileal crypts for metabolism-determining genes (n=6). (F) Schematic representation of oligomycin and DCA targets. (G,H) Ileal organoids derived from WT mice were treated with oligomycin for 24 hours. (G) Left: bright field and IF costaining of Lyz (red) and E-cadherin (IEC borders, grey) counterstained with Dapi (nuclei, blue). Dotted lines indicate cell borders of Lyz+ cells; asterisks indicate Lyz+ cells magnified in the pictures on the right side. Right: quantification of Lyz+ cell numbers per crypt (upper graph) and the proportion of highly granular (≥2 granules) Lyz+ cells (lower graph). (H) qRT-PCR analysis of intestinal organoids for Lgr5 and Paneth cell function-associated genes (upper panel) and for genes associated with mitochondrial signalling (lower panel, n=6). (I) Same analysis as in (H) for human organoids derived from the small intestine and treated with oligomycin for 24 hours (n=6). (J–L) Ileal organoids derived from inflamed TNFΔARE mice were analysed (n=6). (J) From left to right: experimental scheme (D0=isolation of primary crypts and start of culture), non-treated and DCA-treated organoids were compared; proportion of living organoids at day 3 and day 8 of culture; quantification of de novo crypt formation at day 3 and day 8. (K) From left to right: experimental scheme, DCA-treated organoid cultures were passaged at day 8, and subsequently cultured in control (-DCA) and DCA-containing medium, respectively, until day 14; proportion of living organoids at day 10 and day 14 of culture; quantification of de novo crypt formation at day 14. (L) Representative bright field pictures of organoids at day 8 (before passaging) and at day 14 of culture. (M) WT and inflamed TNFΔARE mice-derived ileal organoids were treated with DCA for 14 days. qRT-PCR analysis for Lgr5 and Paneth cell function-associated genes. Statistics were performed by unpaired t-test when comparing two groups and by one-way ANOVA followed by Tukey test for three-group comparisons, respectively. Statistics for budding analyses were performed by Kruskal–Wallis test on ranks. Bars represent mean+SEM. Asterisks indicate significant differences *p<0.05, **p<0.01, ***p<0.001. ANOVA, analysis of variance; ARE,AU-rich (adenosin-uracil) elements; DCA, dichloroacetate; Lyz+, Lyz positive; OXPHOS, oxidative phosphorylation; WT, wild type.
Mitochondrial respiration is required to maintain stemness and PC functionality
The metabolism of cells constituting the ISC niche is coordinated to support each other’s demand and particularly mitochondrial oxidative phosphorylation (OXPHOS) is fine tuned to meet the requirements of different IEC subtypes.8 24 Since crypts derived from an inflammatory environment and Hsp60Δ/ΔISC organoids both display mitochondrial dysfunction, ISC niche alterations and a growth defect (figures 4 and 6A–D, online supplementary figure 9C,D), we determined mRNA expression levels of key factors involved in OXPHOS and glycolysis in primary crypt derived from non-inflamed and inflamed TNFΔARE mice.
Pdha, belonging to the pyruvate dehydrogenase complex (PDC), Yy1, a transcriptions factor regulating mitochondrial complex I genes and cytochrome c oxidase (Cox) IV were consistently reduced (figure 6E). PDC acts as gatekeeper of metabolism by linking cytoplasmic glycolysis to the TCA cycle and OXPHOS, Hence, oligomycin, blocking OXPHOS and dichloroacetate (DCA), targeting PDC to shift ATP generation from glycolysis to OXPHOS35 (figure 6F), were used to characterise the role of metabolism on ISC niche function. Murine ileal WT organoids and human SI organoids were treated with sublethal doses of oligomycin (online supplementary figure 9E) or DCA. Organoids treated with oligomycin depict lower numbers of Lyz+ cells per crypt and reduced PC granularity (figure 6G). Concomitantly, transcriptional levels of Lgr5, Lyz and Ang4 were reduced, while Chop, Hif1a and Prkaa were upregulated (figure 6G). Noteworthy, these results were mirrored in human intestinal organoids (figure 6H). In contrast, DCA had only minimal effects on these readouts (online supplementary figure 10).
Reinforcing mitochondrial respiration restores inflammation-imprinted dysfunction of the ISC niche
Finally, we tested if the inflammation-associated growth defect in TNFΔARE mice-derived crypts could be reversed by DCA. Indeed, addition of DCA to the organoid culture medium directly after seeding rescued the ability of inflamed TNFΔARE mice-derived ileal crypts to grow into organoids and form de-novo crypts (figure 6J). Strikingly, DCA withdrawal subsequent to passaging at day 8 of culture conferred no negative effects to organoid growth compared with continued DCA treatment (figure 6K,L). Comparing DCA-exposed organoids derived from inflamed TNFΔARE mice and WT mice, transcriptional level of ISC and PC-associated genes largely converged (figure 6M). These data demonstrate that activation of OXPOHOS via inhibition of glycolysis is sufficient to restore inflammation-imprinted metabolic dysfunction of the ISC niche.
The persisting regain of stemness after withdrawal of DCA underlines the reversible nature of ISC alterations under inflammatory conditions, making ISC niche metabolism an attractive target for therapeutic interventions (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
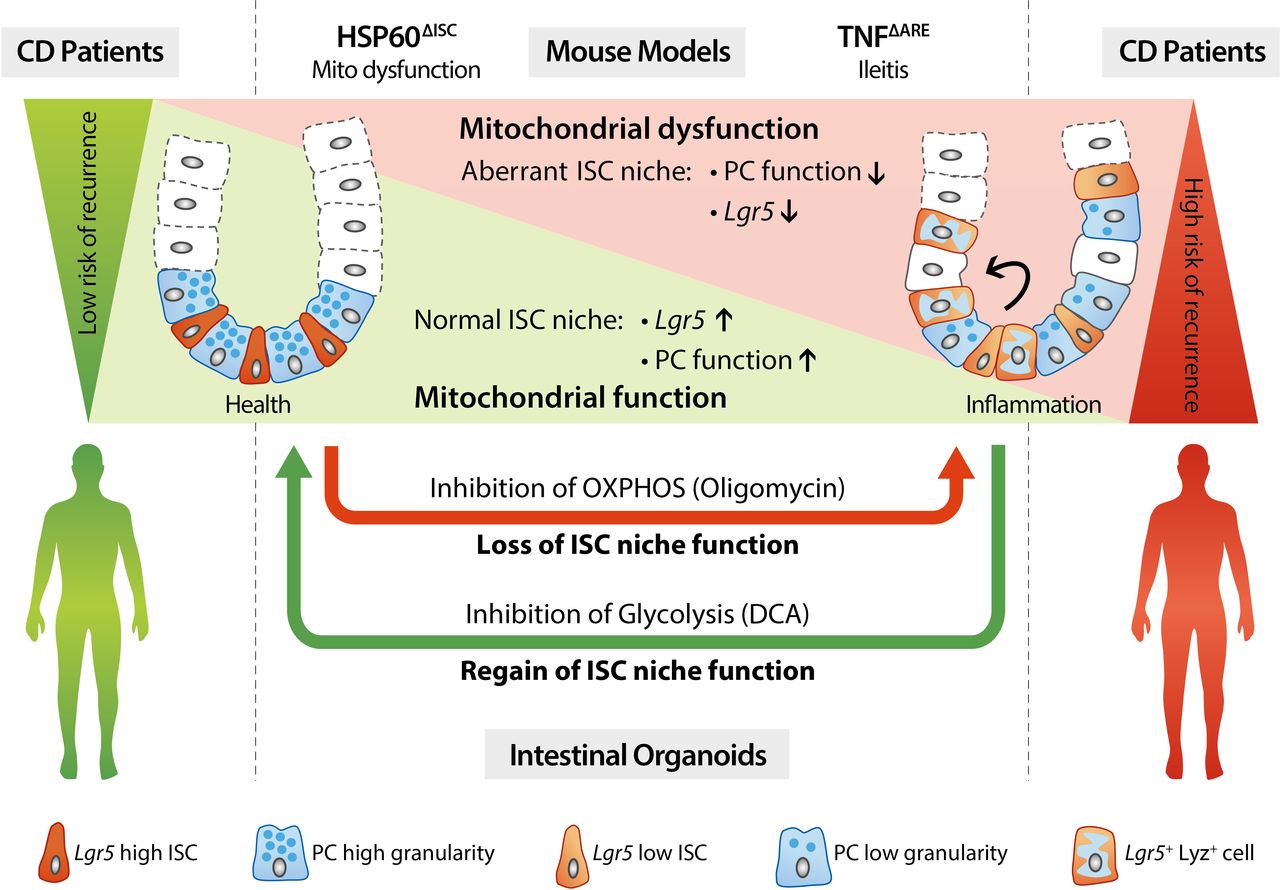
Mitochondrial impairment drives ISC transition towards dysfunctional PCs predicting Crohn’s disease recurrence Schematic representation of the main findings of this work. Targeted disruption of mitochondrial function in ISCs leads to transition of ISCs into dysfunctional PCs. Under inflammatory conditions, mitochondrial impairment in the ISC niche results in ISC exhaustion and generation of dysfunctional PCs characterised by loss of Lyz positive granules, concomitant to aberrant Lgr5 and Lyz expression in upper crypts. These alterations precede tissue pathology and serve as predictive markers for early endoscopic recurrence in CD. Ex vivo, the inhibition of mitochondrial respiration (OXPHOS) in intestinal organoids reflects the impact of an inflammatory environment on the ISC niche, whereas reinforcement of OXPHOS by inhibition of glycolysis is able to override inflammation-imprinted changes of the ISC niche. ARE,AU-rich (adenosin-uracil) elements; CD, Crohn’s disease; ISC, intestinal stem cell; Lyz, Lysozyme; OXPHOS, oxidative phosphorylation; PC, Paneth cell; TNF, tumour necrosis factor.
Discussion
IBDs, including CD and ulcerative colitis, constitute a global health problem.36 A major challenge in the treatment of CD is the heterogeneity of the disease, with only subsets of patients responding to therapies like administration of anti-TNF antibodies.37 The great number of susceptibility genes identified along with a range of different environmental triggers, might account for these difficulties, implicating diverse mechanisms contributing to the variable phenotypes of CD. Several CD-relevant pathways converge on the level of PCs, like ER stress, autophagy and bacterial recognition and consistently, the presence of NOD2 and ATG16L1 disease-associated alleles as well as bacterial infection have been demonstrated to affect PC phenotype.12 14 15 38 Hence, aberrant PC phenotypes have been proposed as a biomarker to stratify patients with CD according to similar disease mechanisms in order to yield better treatment outcomes.13 The data presented here implicate that determining ISC niche appearance improves risk stratification of patients with CD, and that targeting the underlying mitochondrial dysfunction evolves as a novel strategy for therapy.
Reduced PC functionality has been extensively described for CD in the context of AMP production and alterations of the intestinal microbiota.3 5 15 27 However, PCs serve as multifunctional guardians of the crypt, also providing essential signals for the ISC niche.27 PCs evolve directly above the crypt base, and Lgr5 + ISCs compete for available surface of their own direct progeny,34 implicating ISC changes concomitant to PC abnormalities. Consequently, we found diminished Lgr5 expression in patients with CD and in inflamed TNFΔARE mice, reflecting the inflammation-associated gradual loss of PC function. Furthermore, in patients with CD, LYZ and LGR5 showed a distorted expression pattern with PCs and ISCs abundantly present in upper crypts. So far, Lgr5 + ISC loss has been described in the primary response to DSS-induced inflammation and on high-dose γ-irradiation.9 39 The temporary depletion and rapid restoration of these cells following acute injury seems to underlie the regenerative response of crypts. Under these conditions, PCs contribute to the repair and regeneration of damaged intestinal tissue by acquiring stem like features.9–11 However, irradiation and DSS-induced inflammation cause acute mucosal injury and do not reflect the chronic inflammatory conditions of IBD pathology. Despite reduced Lgr5 expression, crypts derived from DSS-treated mice show an enhanced capacity to form organoids,9 in contrast to crypts from inflamed TNFΔARE mice that fail to give rise to organoids. Hence, the phenotypic changes of the ISC niche under chronic inflammation are likely not associated with healing processes but rather indicate stemness exhaustion as a feature of pathology. Moreover, ISC niche abnormalities were detected before onset of severe tissue pathology in TNFΔARE mice and in non-affected tissue margins of patients with CD, suggesting that these changes most likely represent early, molecular marker of inflammatory changes.
Additionally, we provide experimental evidence for a direct role of mitochondria in the development of dysfunctional PCs using an ISC-specific mouse model of mitochondrial dysfunction and ex vivo inhibition of OXPHOS in organoid cultures. Experimental disturbance of mitochondrial function reduced stemness and PC functionality, demonstrating dysfunctional PCs as direct descendants of ISCs suffering from mitochondrial impairment. Thus, while mitochondrial dysfunction in ISC seems to disturb the balance between self-renewal and differentiation, the inability to fine-tune mitochondrial function subsequently seems to interfere with differentiation processes, impeding normal PC maturation.
This is in line with the pivotal role of mitochondria in ISC fate through regulation of the metabolic switch during differentiation.24 In Lgr5 + ISCs isolated from the mouse small intestine, OXPHOS highly contributes to cellular bioenergetics, and mitochondrial activity further increases with differentiation and de-novo crypt formation in intestinal organoids,8 indicating why ISC and ISC differentiation processes might be particularly vulnerable to mitochondrial impairment. OXPHOS inhibition and the subsequent decrease in ISC and PC marker genes was associated with transcriptional activation of Chop, hypoxia-inducible factor 1-α and AMP kinase in murine and human intestinal organoids. All of these genes are involved in metabolic regulation and link mitochondrial homeostasis to CD-relevant pathomechanisms including MT-UPR, ROS signalling and energy sensing.24 40–42
Most remarkable, ex vivo culture experiments using intestinal crypts derived from inflamed TNFΔARE mice suggest (I) that the inflammatory environment imprints the ISC niche towards reduced stemness and PC function, (II) that these changes persist under normal culture conditions and cannot be overcome by addition of Wnt factors, (III) that a targeted metabolic intervention using DCA treatment rescues stemness and reverses the inflammatory imprinting, allowing organoids to propagate under normal culture conditions. DCA treatment results in diminished glycolysis and improved mitochondrial respiration, an ability used in the therapy of several different solid tumours to reverse the Warburg effect in cancer cells.35 Identification of target metabolites conferring to ISC niche homeostasis could be a promising target of future research.
The cause of mitochondrial disturbances present in patients with CD and mouse models of intestinal inflammation is currently unknown. In general, enterocytes of patients with IBD have been reported to display swollen mitochondria with irregular cristae indicative of impaired function,43 and inflammation is associated with hypoxia. Infiltrating immune cells, invading pathogens and the increased energy demand of resident cells limit the available oxygen,44 and together with reduced blood supply,45 these changes contribute to the hypoxic conditions under chronic inflammation. Furthermore, polymorphisms in genes impacting mitochondrial functions, mitochondrial carrier protein uncoupling protein (UCP) 2 and SLC22A5, encoding the carnitine transporter OCTN2, have been described as risk factors in IBD.46 47 UCP2 is proposed to control the speed of the TCA cycle, and production of mitochondrial ROS, and to promote the metabolic shift from glucose to fatty acid oxidation.48 OCTN2 transports long-chain fatty acids conjugated to carnitine into mitochondria for β-oxidation. β-oxidation is particularly implicated in CD pathogenesis, and consequently, pharmacological inhibition of intestinal fatty acid β-oxidation as well as genetic ablation of OCTN2 results in experimental colitis.49 50 These genetic risk factors might act in concert with other CD-relevant pathways described above and might render IECs particularly sensitive to environmental triggers of inflammation.
In patients with CD, the proportion of PCs with aberrant granule structure and diffuse LYZ distribution has been shown to correlate with the cumulative number of NOD2 and ATG16L1 risk alleles,13 linking multiple CD genetic susceptibility loci to a defined PC phenotype. High proportions of abnormal PCs were associated with shorter time to disease recurrence after surgery in patients with a more aggressive clinical disease course.13 Our findings on PC granularity corroborate these findings in an independent patient cohort refining the endpoint of prediction to early postoperative lesions observed by ileocolonoscopy 6–12 months after surgery, and expand the set of predictive cellular markers to Lgr5+ ISC and to the location of expression. Abnormal PC morphology has also been linked to an activated immune response gene signature in crypts from patients with CD13 and to a transcriptional profile of cytokine stimulation in mice with hypomorphic expression of Atg16l1.14 Together with the evidence presented here, this suggests an interrelated role of molecular inflammation and metabolism for ISC and PC phenotype. Furthermore, our data indicate some kind of signal propagation from adjacent, inflamed tissue regions, as jejunal organoids derived from inflamed TNFΔARE mice display impaired growth.
In conclusion, we identified ISC niche alterations as target of molecular inflammatory changes and predictive marker of early endoscopic recurrence in CD. In this context, we demonstrated that impaired mitochondrial function is linked to CD-associated loss of stemness and the generation of dysfunctional PC phenotypes. Mitochondria-derived signals might collaborate with IBD susceptibility genes to impact on ISC niche functionality, and in concert with environmental factors, such as intestinal microbiota or diet, contribute to the loss of ISC niche homeostasis observed in ileal CD. By implementing a drug-related shift towards mitochondrial respiration, we provide a proof-of-concept for the importance of mitochondrial metabolism in regulating ISC and PC functions and thereby rationalise a novel treatment approach for CD.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SK and ER contributed equally.
Contributors SK, ER and DH designed the experiments, performed data analysis and wrote the manuscript. SK, ER and EB performed mouse and organoid culture experiments. SK performed tissue analysis of patients. AB performed the transmission electron microscopy. EG, NW, AM and PG supported analyses. MA and NH provided patient samples.
Funding DH received funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB 1371 (Projektnummer 395357507; P01) and Priority Programme SPP 1656. DH and MA received funding from the Helmsley Cheritable Trust (IBDOT).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Additional data are available on request.
Linked Articles
- Inflammatory bowel disease