Article Text

Abstract
Objective Although perturbations in mitochondrial function and structure have been described in the intestinal epithelium of Crohn’s disease and ulcerative colitis patients, the role of epithelial mitochondrial stress in the pathophysiology of inflammatory bowel diseases (IBD) is not well elucidated. Prohibitin 1 (PHB1), a major component protein of the inner mitochondrial membrane crucial for optimal respiratory chain assembly and function, is decreased during IBD.
Design Male and female mice with inducible intestinal epithelial cell deletion of Phb1 (Phb1iΔIEC ) or Paneth cell-specific deletion of Phb1 (Phb1ΔPC ) and Phb1fl/fl control mice were housed up to 20 weeks to characterise the impact of PHB1 deletion on intestinal homeostasis. To suppress mitochondrial reactive oxygen species, a mitochondrial-targeted antioxidant, Mito-Tempo, was administered. To examine epithelial cell-intrinsic responses, intestinal enteroids were generated from crypts of Phb1iΔIEC or Phb1ΔPC mice.
Results Phb1iΔIEC mice exhibited spontaneous ileal inflammation that was preceded by mitochondrial dysfunction in all IECs and early abnormalities in Paneth cells. Mito-Tempo ameliorated mitochondrial dysfunction, Paneth cell abnormalities and ileitis in Phb1iΔIEC ileum. Deletion of Phb1 specifically in Paneth cells (Phb1ΔPC ) was sufficient to cause ileitis. Intestinal enteroids generated from crypts of Phb1iΔIEC or Phb1ΔPC mice exhibited decreased viability and Paneth cell defects that were improved by Mito-Tempo.
Conclusion Our results identify Paneth cells as highly susceptible to mitochondrial dysfunction and central to the pathogenesis of ileitis, with translational implications for the subset of Crohn’s disease patients exhibiting Paneth cell defects.
- inflammatory bowel disease
- crohn's disease
- epithelial cells
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Previous studies suggest the involvement of epithelial mitochondrial dysfunction in the pathophysiology of inflammatory bowel disease, including Crohn’s disease and ulcerative colitis.
What are the new findings?
We identify Paneth cells as highly susceptible to mitochondrial dysfunction driven by Prohibitin 1 (Phb1) deletion and central to the development of ileitis.
Treatment of Paneth cell defects with Mito-Tempo during Phb1 deletion implicates a potential therapeutic application for abnormal Paneth cells via elimination of mitochondrial-derived reactive oxygen species.
Mito-Tempo also prevented the upregulation of interleukin-1β (IL-1β) and IL-18 in the ileum that were induced early after Phb1 deletion.
Phb1 deficiency induced loss of viability of the intestinal stem cell niche and Paneth cell defects in cultured enteroids.
How might it impact on clinical practice in the foreseeable future?
These are the first results that present a causative role of mitochondrial dysfunction in ileitis that initiates in Paneth cells.
Mitochondrial-targeted therapeutics may have translational utility in a subset of Crohn’s disease patients exhibiting Paneth cell defects.
Introduction
Crohn’s disease, an inflammatory bowel disease (IBD) characterised by recurring, incurable, chronic inflammation, is considered a global health problem with accelerating incidence in newly industrialized countries and stabilising, yet high prevalence in Western countries.1 Crohn’s disease is a multifactorial disease exhibiting loss of intestinal epithelial cell (IEC) barrier integrity and dysregulated immune cell responses due to unknown environmental triggers in genetically predisposed individuals.2 Genome-wide association studies have identified ~200 IBD risk loci,3 with 5% of these genes functionally linked to the maintenance of mitochondrial health.4 Mitochondria are dynamic organelles that readily respond to environmental stimuli and cellular demands for energy. Mitochondria are coordinators of cellular homoeostasis via their role in energy production and oxidative metabolism, induction of apoptosis, regulation of calcium, production of reactive oxygen species (ROS), and regulation of signal transduction and epigenomic intermediates. In the intestine, mitochondrial metabolism and function play key roles in immune cell activation, IEC barrier integrity and IEC differentiation programmes and stemness.5 6 Previous studies suggest the involvement of epithelial mitochondrial dysfunction in the pathophysiology of IBD, including Crohn’s disease and ulcerative colitis,7 8 but whether this is a cause or consequence of the pathogenesis of IBD is not known.
Prohibitin 1 (PHB1) belongs to a family of proteins that share an evolutionarily conserved stomatin/prohibitin/flotillin/HflK/C domain and serves diverse roles in cell function including regulation of cell cycle progression, apoptosis and transcription depending on its subcellular localisation. In IECs, PHB1 predominantly localises to the mitochondria.9 PHB1 is the major component protein of the inner mitochondrial membrane (IMM) where it forms a heterodimeric complex with PHB2 to exert chaperon function to stabilise mitochondrial DNA (mtDNA)-encoded proteins and regulate optic atrophy 1 (OPA1)-dependent IMM fusion.10 Additionally, PHB1 interacts with and is required for optimal activity of complexes I and IV of the electron transport chain (ETC).10 Expression of PHB1 is decreased in mucosal biopsies from IBD-afflicted patients.9 11 We previously showed that overexpression of epithelial PHB1 using genetic manipulation (Villin-Phb1 transgenic mice) or therapeutic delivery to the colon decreases oxidative stress and protects mice from experimental colitis.12 13 Given the known functions of PHB1 in mitochondrial structure and dynamics, we generated three novel mouse models of mitochondrial dysfunction via Phb1 deletion in the intestinal epithelium or specifically in Paneth cells. Here, we investigated the role of IEC mitochondrial dysfunction in intestinal inflammation.
Results
Phb1iΔIEC mice develop spontaneous ileitis
Genetic deletion of Phb1 results in embryonic lethality in mice and flies.14 To gain tissue and temporal control of PHB1 deletion, we ablated PHB1 in IECs of adult Phb1 floxed mice (Phb1fl/fl:Villin-CreERT2 referred to as Phb1iΔIEC mice) by tamoxifen administration. The absence of PHB1 protein in the epithelium was confirmed by western immunoblotting and immunohistochemistry (IHC) staining after tamoxifen injection (online supplementary figure S1). Beginning at 7 weeks after induction of Phb1 deletion, Phb1iΔIEC mice gained less body weight compared with Phb1fl/fl littermates (online supplementary figure S2A). Within 12 weeks after induction of Phb1 deletion, Phb1iΔIEC mice manifested spontaneous, discontinuous ileal inflammation (figure 1A), while sparing more proximal small intestine and colon (online supplementary figure S2B). Histological alterations in the ileum included infiltration of immune cells, thickening of the muscularis layers, crypt abscesses, crypt architectural changes including crypt branching, crypt elongation, and villus blunting (figure 1A,B, online supplementary figure S2C,D). Phb1iΔIEC mice exhibited splenomegaly and ileal infiltration of CD4+ T cells, CD11b+/CD11c+ dendritic cells and F4/80+ macrophages (figure 1C,D, online supplementary figure S2E). Expression of proinflammatory cytokines Tnfα, Interleukin-1β (IL-1β) and Interferon-γ (Ifnγ) was increased in Phb1iΔIEC ileum with a concomitant decrease in anti-inflammatory Il-10 (figure 1E). Gut microbiota was altered in Phb1iΔIEC mice compared with Phb1flfl littermates, with decreased bacterial diversity and significant decreases in abundance of Blautia, Roseburia, Coprococcus, Oscillibacter, as has been reported as decreased in IBD patients,15 16 as well as decreased abundance of Marvinbryantia, Acetatifactor and Pseudoflavonifractor (online supplementary figure S3). These bacterial alterations were observed 1 week after Phb1 deletion and decreases in abundance of Roseburia, Oscillibacter, Marvinbryantia and Acetatifactor and Shannon Diversity were sustained through 12 weeks after Phb1 deletion.
Supplemental material
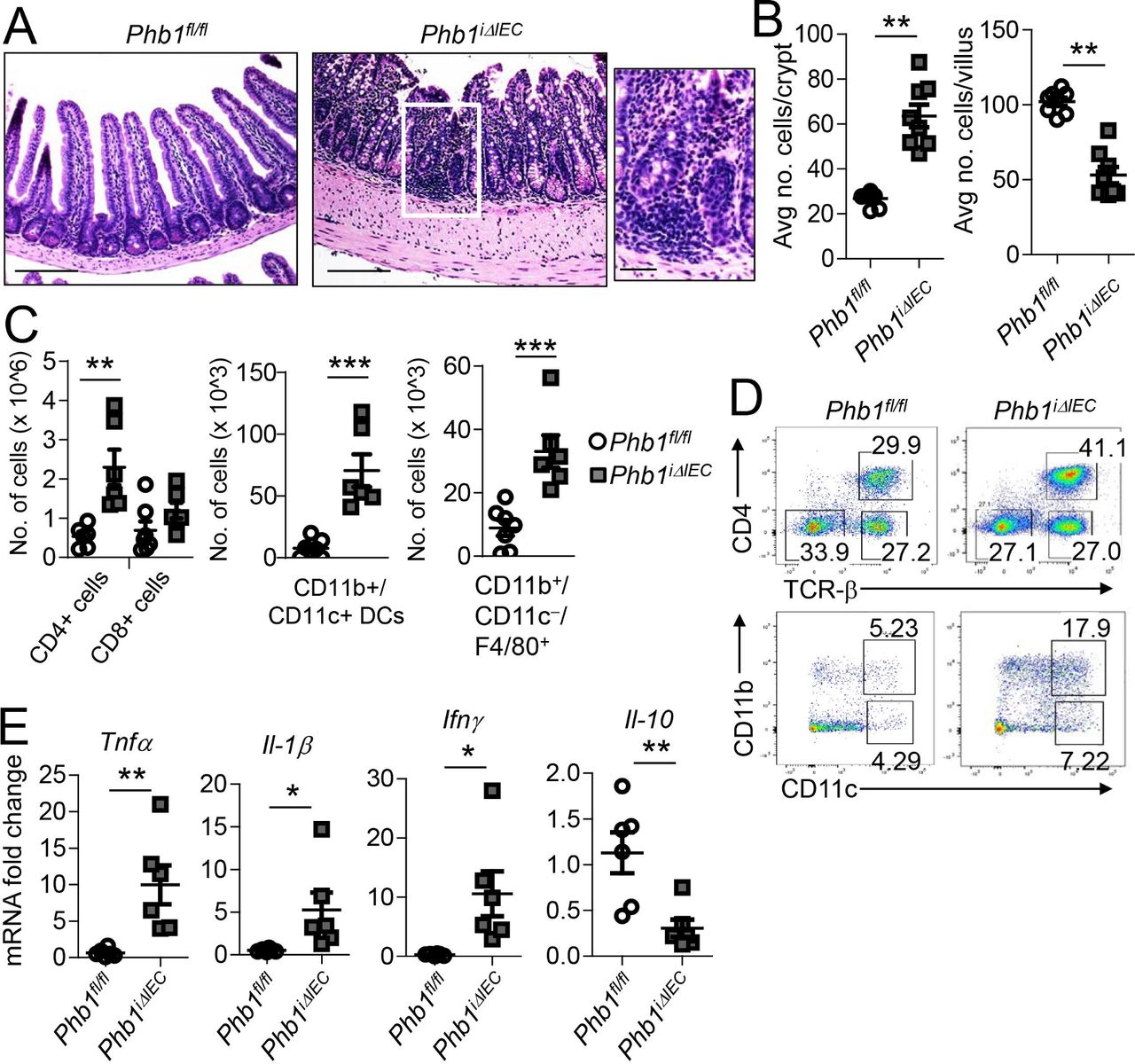
Phb1i ΔIEC mice develop spontaneous ileitis 12 weeks after Phb1 deletion. (A) H&E-stained sections showing ileum histology. Scale bars: 250 µm, boxed pullout: 75 µm. (B) The number of epithelial cells across 50 crypts or 50 villi per mouse. (C) Absolute number of ileal lamina propria (LP) immune cells calculated of total LP cells isolated from ileum. Data are representative of three independent experiments. (D) Representative flow cytometry plots gated on CD45+ cells (top) or gated on CD45+MHCII+Gr-1-B220-Cells (bottom) (E) mRNA expression in ileum measured by qPCR. Results are presented as individual data points±SEM of 10 mice (B), 6-7 mice (1 Phb1i ΔIEC outlier removed after Grubbs’ test) (C), or 6 mice (E) per group. *P<0.05, **P<0.01, ***P<0.001 by unpaired, two-tailed Student’s t-test. IFN-γ, interferon-γ; IL-10, interleukin-10; Phb1i ΔIEC, intestinal epithelial cell deletion of Phb1; Phb1 ΔPC, Paneth cell-specific deletion of Phb1; TNFα, tumour necrosis factor α.
Ileal IECs exhibit mitochondrial dysfunction during deletion of Phb1
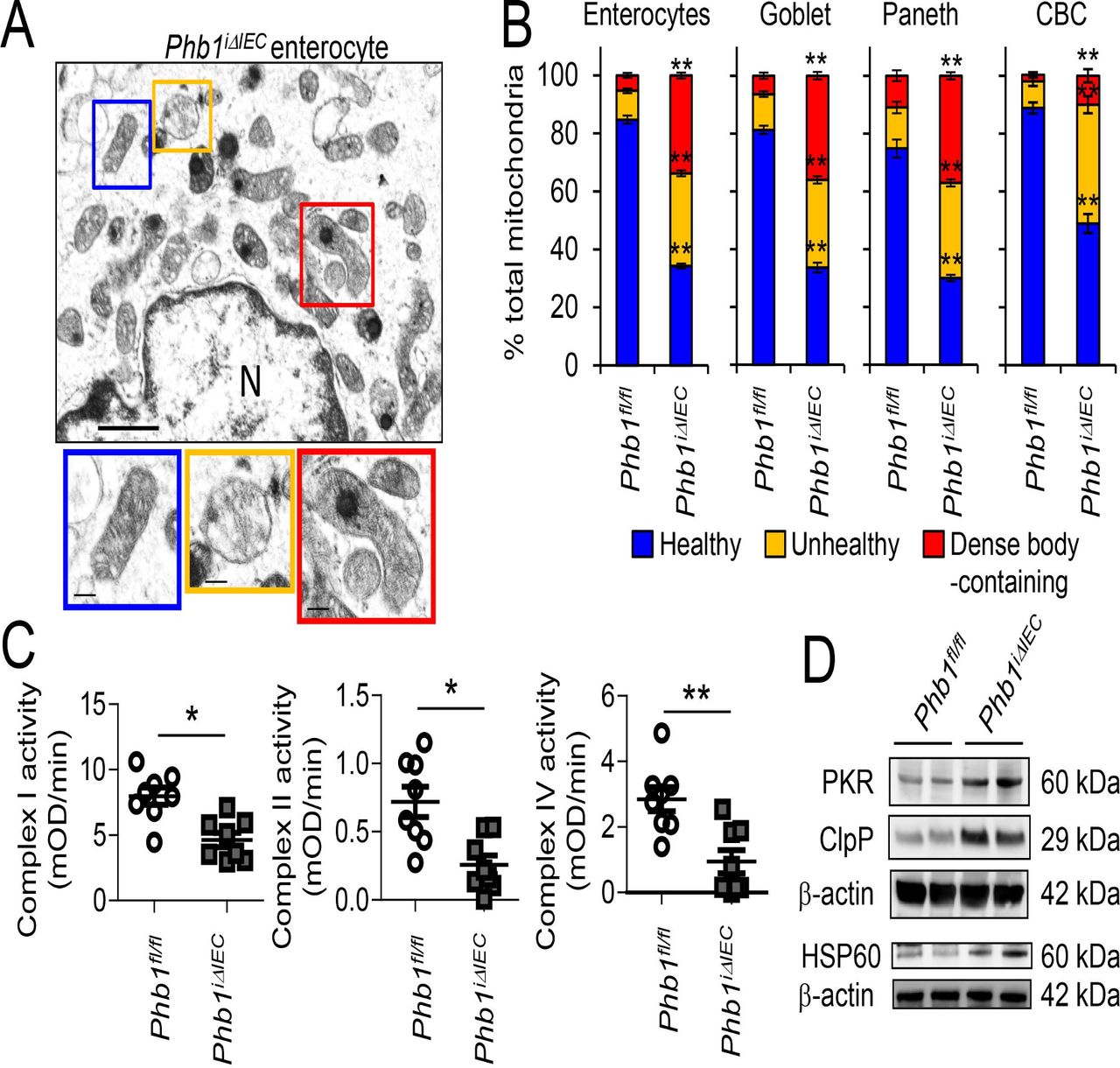
Since mitochondrial morphology is closely linked to function, mitochondria in epithelial cells of the ileum were visualised by transmission electron microscopy (TEM). Twelve weeks after induction of Phb1 deletion, ileal epithelial cells demonstrated loss of microvilli and increased vacuolation (online supplementary figure S4). TUNEL+ staining in ileal crypts and villi of Phb1iΔIEC mice suggest apoptosis and/or necrosis of the epithelium, confirmed by increased Cleaved caspase 3 expression (online supplementary figure S5A–C). A statistically significant proportion of the mitochondria of enterocytes, goblet cells, Paneth cells and crypt base columnar (CBC) stem cells of Phb1iΔIEC mice were unhealthy (swollen, dissolution/delocalisation of cristae) or contained electron-dense inclusion bodies as a reaction to injury17 (figure 2A,B). Mitochondrial dysfunction in Phb1iΔIEC ileal IECs was also evident by decreased activity of ETC complexes I, II and IV (figure 2C) and the induction of the mitochondrial unfolded protein response (mtUPR; figure 2D), which is triggered on accumulation of unfolded proteins within the mitochondrial matrix and is evident in epithelial cells from Crohn’s disease and ulcerative colitis patients.8 These mitochondrial changes did not significantly alter ATP concentration in isolated ileal IECs (online supplementary figure S6A), as has been shown previously in other cells with Phb1 deficiency.18 19

Phb1iΔIEC ileal IECs exhibit mitochondrial dysfunction. Time point shown is 12 weeks after Phb1 deletion. (A) TEM of ileal enterocyte. Representative healthy (blue box), unhealthy (orange box), and dense inclusion body-containing (red box) mitochondria. Scale bars: 500 nm. N, nucleus. (B) % healthy, unhealthy, and dense inclusion body-containing mitochondria visualised by TEM. n=200 enterocytes, 200 goblet cells, 50 Paneth cells, and 50 CBC cells with an average of 42 mitochondrial/cell. Enteroendocrine cells were too sparse to quantitate. (C) ETC complex activity in isolated ileal IECs. (D) Representative Western blots of mtUPR markers in isolated ileal IECs. Results are presented as pooled data means±SEM (B) or as individual data points±SEM (C) of four mice per group (B) of eight mice per group (C, D). *P<0.05, **P<0.01 by 1-way ANOVA followed by Bonferroni’s test (B) or by unpaired, 2-tailed Student’s t-test (C). ANOVA, analysis of variance; CBC, crypt base columnar; ETC, electron transport chain; HSP, heat shock protein; IECs, intestinal epithelial cells; mtUPR, mitochondrial unfolded protein response; Phb1iΔIEC , intestinal epithelial cell deletion of Phb1; PKR, protein kinase R; SEM, standard error mean; TEM, transmission electron microscopy.
Since mitochondrial dysfunction demonstrated in Phb1-deficient ileal IECs could be secondary to inflammation, we next determined whether earlier time points after the induction of Phb1 deletion exhibited mitochondrial dysfunction. Mitochondrial ultrastructural abnormalities and mtUPR were induced in IECs as early as 1 week following Phb1 deletion and was sustained through 12 weeks (online supplementary figure S6B–E). Upregulation of Opa1, which is a master regulator of mitochondrial fusion and bioenergetics, but not other genes controlling mitochondrial function (Polg1, Polg2, Tfam or Pgc1), was evident early after Phb1 deletion in ileal IECs (online supplementary figure S6F). Additionally, endoplasmic reticulum (ER) stress and mitochondrial dysfunction can be interrelated during chronic inflammation with communication between the two organelles.20 Indeed, upregulation of ER UPR markers coincides with mitochondrial dysfunction in Phb1iΔIEC ileal IECs (online supplementary figure S7A,B). Very few mice displayed histological inflammation of the ileum at early time points (1 or 3 weeks) following Phb1 deletion (table 1), suggesting that mitochondrial dysfunction precedes ileitis in this model.
Proportion of Phb1iΔIEC mice that manifest mitochondrial dysfunction, abnormal Paneth and goblet cells, and histological inflammation
Mitochondria are the primary source of ROS with 0.2%–2.0% of oxygen consumed converted to superoxide by normal ETC activity. Mitochondrial dysfunction causes accumulation of mitochondrial-derived ROS (mtROS) due to blockade of forward electron flow through the ETC, subsequently causing more electrons to leak to oxygen. Oxidative damage to lipids and DNA is evident in the ileum of Phb1iΔIEC mice as early as 1 week after Phb1 deletion (online supplementary figure S7C,D). Additionally, ROS-generating mitochondria have been shown to activate the NLRP3 inflammasome, which in turn leads to caspase-1 dependent secretion of proinflammatory cytokines IL-1β and IL-18.21 Phb1iΔIEC mice exhibit increased expression of active Caspase-1, IL-1β and IL-18 in ileum 1 week after Phb1 deletion without altered expression of other proinflammatory cytokines or chemokines commonly upregulated in intestinal inflammation at this early time point (online supplementary figure S8).
Phb1iΔIEC mice exhibit ileal Paneth cell abnormalities early after Phb1 deletion
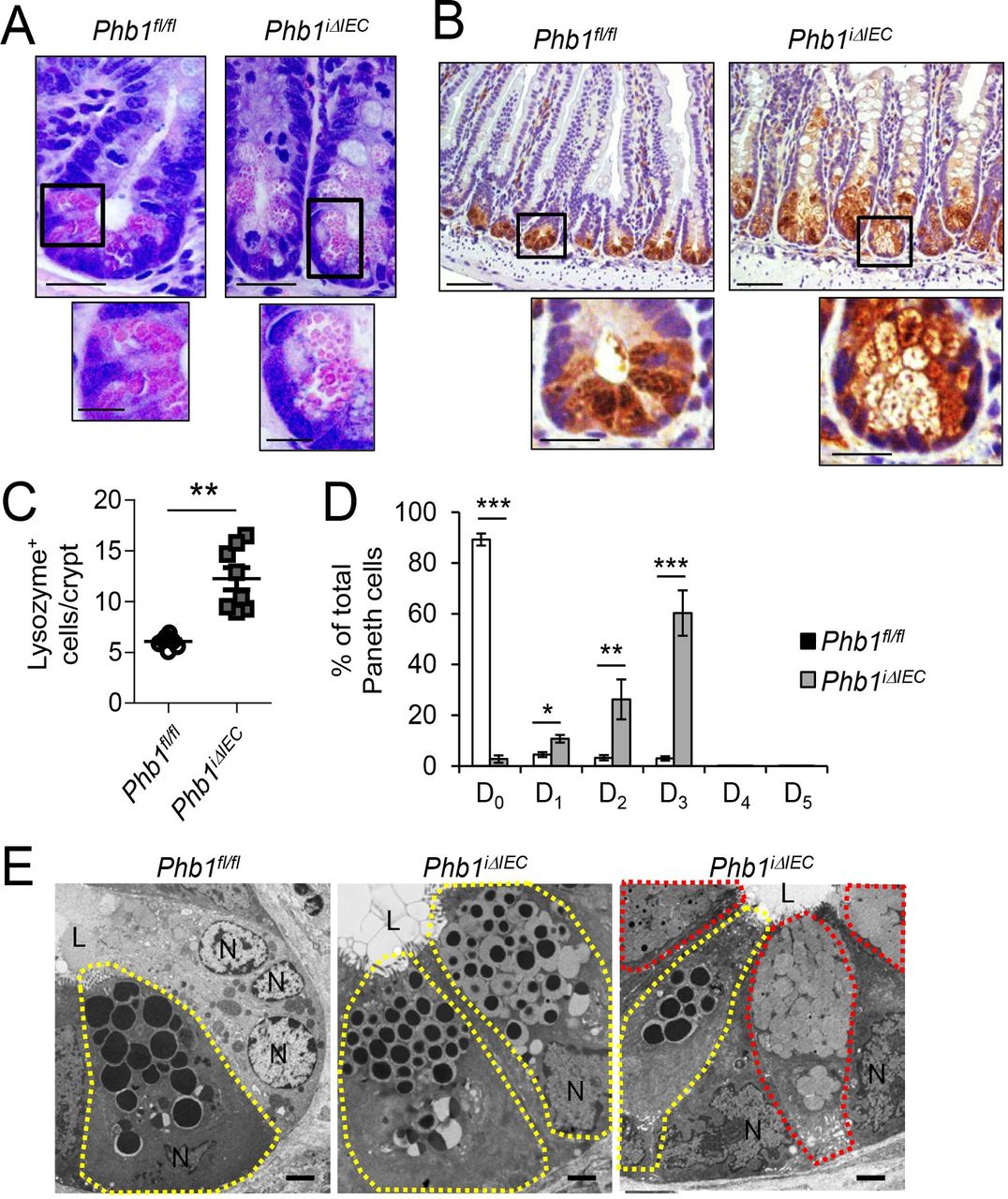
During histological examination of H&E-stained ileum sections, alterations in Paneth cells, such as smaller secretory granules, were obvious in Phb1iΔIEC mice 12 weeks after Phb1 deletion (figure 3A). Paneth cells contribute to gut homoeostasis by synthesising antimicrobial peptides and proteins such as lysozyme and defensins (also called cryptdins) and intestinal stem cell (ISC) niche factors.22 IHC staining demonstrated that lysozyme+ cells were expanded in number in Phb1iΔIEC mice (figure 3B,C). Lysozyme is normally efficiently packaged into secretory granules in Paneth cells as demonstrated in Phb1fl/fl mice (figure 3B). Ileal Paneth cells in Phb1iΔIEC mice exhibited altered patterns of lysozyme allocation (as previously described by23 24), with the majority showing diminished or diffuse lysozyme staining (figure 3B,D). Using these same criteria, 20%–50% of Crohn’s disease patients have ≥20% abnormal Paneth cells (called Type I Paneth cell phenotype), which was associated with gut microbiota alterations and poor clinical outcomes such as early postoperative recurrence after resection.25–27 Paneth cell granules in Phb1iΔIEC mice visualised by TEM were smaller and composed of an electron dense core and an enlarged electron-lucent peripheral halo compared with Phb1fl/fl mice (figure 3E). Previous studies have identified this electron-lucent halo in murine Paneth cells as predominantly containing Mucin2.28 Phb1iΔIEC mice exhibited decreased mRNA expression of antimicrobials produced by Paneth cells such as RegIIIγ, Cryptdin3, Cryptdin5 and Ang4 (online supplementary figure S9A), suggesting loss of Paneth cell function vs simply degranulation of Paneth cell enzymes on a sustained basis.

Phb1ΔIEC mice exhibit ileal Paneth cell abnormalities. Time point shown is 12 weeks afetr Phb1 deletion. (A) H&E-stained sections showing Paneth cells. Scale bars: 50 µm, boxed pullout: 20 µm. (B) Lysozyme IHC staining. Scale bars: 250 µm, boxed pullouts: 50 µm (C) The number of lysozyme+ cells across 50 crypts per mouse. (D) % of Paneth cells displaying each pattern of lysozyme expression. D0, normal; D1, disordered; D2, diminished; D3, diffuse; D4, excluded; D5, enlarged. 500 cells quantitated each mouse. (E) TEM of crypt base. Paneth cells, yellow outline; goblet-like cells, red outline. N, nucleus; L, lumen. Scale bars: 2 µm. Results are presented as individual data points±SEM (C) or as pooled data means± SEM (D) of 8 mice per group. **P<0.01, ***p<0.001 by unpaired, 2-tailed Student’s t-test. (B) Or by 1-way ANOVA followed by Bonferroni’s test (D). ANOVA, analysis of variance; Phb1 ΔIEC, intestinal epithelial cell deletion of Phb1.
Goblet-like cells containing dense core mucin granules, or a combination of mucin granules and abnormal Paneth cell granules, were observed in the crypt base of Phb1iΔIEC mice by TEM (figure 3E). Alcian blue (AB)–periodic acid schiff staining confirmed an increase in the number of AB+ cells in the crypt base during Phb1 deletion (figure 4A,B). Colocalisation of MUC2 (goblet cell marker) and lysozyme (Paneth cell marker) staining in the Phb1iΔIEC crypt base, evident in areas with and without inflammation, suggest these cells are ‘intermediate’ goblet/Paneth cells (figure 4C). These intermediate cells have been proposed to be Paneth cells undergoing transformation to goblet cells, goblet cells in the process of being converted to Paneth cells, or a precursor of both lineages. This altered differentiation programme in Phb1iΔIEC crypts was accompanied by increased TUNEL+ staining and cell proliferation (online supplementary figure S5), suggesting increased turnover of the intestinal epithelium.

Ileal Paneth cells resemble goblet/Paneth intermediate cells and goblet cell number and size are increased in Phb1 iΔIEC mice. Time point shown is 12 weeks after Phb1 deletion (A) AB-PAS-stained sections. Dashed line denotes crypt base. Scale bars: 250 μm, boxed pullouts: 50 μm. (B) The number of AB+ cells in the crypt base (below dashed line in A) or above the crypt base across 50 crypts per mouse. (C) MUC2 (goblet cell marker) and Lysozyme (Paneth cell marker) immunofluorescene staining. Scale bars: 250 μm, boxed pullout showing co-localisation: 75 μm. (D) Quantitation of average mucin area/goblet cell above the crypt base. 250 cells quantitated each mouse. (E) TEM of villus goblet cells. Mucin granules, yellow outline. Scale bars: 1 μm. Results are presented as individual data points ± SEM of 8 mice per group. **P<0.01, ***p<0.001 by unpaired, 2-tailed student’s t test. AB-PAS, alcian blue–periodic acid schiff; L, lumen; MUC2, mucin2; N, nucleus; Phb1 iΔIEC, intestinal epithelial cell deletion of Phb1; SEM, standard error mean; TEM, transmission electron microscopy.
In addition to the appearance of intermediate cells in the ileal crypts of Phb1iΔIEC mice, AB+ cells above the crypt base were increased in number and size with increased cytoplasmic mucin/cell (figure 4B–E). Examination of ileum at earlier time points after the induction of Phb1 deletion demonstrated that crypt intermediate cells are evident before abnormal villus AB+ cells (table 1 and online supplementary figure S9B,C). As early as 1 week after Phb1 deletion, mRNA expression of goblet cell genes Muc2 and Muc4 are increased with a concomitant increase in Klf4 and Elf3 (online supplementary figure S9A), which are transcription factors involved in goblet cell differentiation. Phb1iΔIEC mice exhibited decreased mRNA expression of Hes1 and increased Math1 (online supplementary figure S9A), a situation that has been shown to promote secretory cell differentiation.29 Sox9, which controls, at least in part, Paneth cell differentiation, and Lgr5, the intestinal CBC stem cell marker, were not altered in Phb1iΔIEC mice (online supplementary figure S9A). FABP6 immunostaining suggests that enterocyte numbers are decreased by 12 weeks after Phb1 deletion but not as early as 1 week, while enteroendocrine cells (marked by chromogranin A+ expression) were unaffected in Phb1iΔIEC mice (online supplementary figure S10).
To determine the sequence of pathological events driving ileal inflammation in Phb1iΔIEC mice, the proportion of mice that manifest ileitis, Paneth cell or villus AB+ cell abnormalities, or mitochondrial dysfunction at 1, 3, 6 or 12 weeks after the induction of Phb1 deletion were quantitated. Mitochondrial dysfunction was evident in all mice at 1 week, Paneth cell abnormalities in the majority of mice beginning at 3 weeks, followed by villus AB+ cell abnormalities and spontaneous ileitis, which developed in the majority of mice after 6 weeks (table 1). Collectively, these results demonstrate that the loss of Phb1 causes mitochondrial dysfunction and subsequent Paneth cell defects prior to the onset of ileitis.
Mito-Tempo ameliorates mitochondrial dysfunction, Paneth cell defects and ileitis during loss of Phb1
To determine whether a mitochondrial-targeted antioxidant has therapeutic potential in Paneth cells, the dysfunction of which plays a role in the pathogenesis of Crohn’s disease,22 a subset of mice were treated daily for 3 weeks with Mito-Tempo concurrent with tamoxifen injections to induce Phb1 deletion. Mito-Tempo is a mitochondrial-targeted superoxide dismutase 2 mimetic shown to have antioxidant properties.30 Mito-Tempo ameliorated mitochondrial ultrastructural abnormalities and mtUPR and ER UPR responses in Phb1iΔIEC ileal IECs (figure 5A–C). Additionally, Mito-Tempo restored lysozyme staining and Paneth cell antimicrobial expression such as RegIIIγ, Cryptdin3 and Cryptdin5 in Phb1iΔIEC mice (figure 5D,E). AB+ crypt cells and expression of Muc2 and Muc4 in Phb1iΔIEC mice treated with Mito-Tempo was similar to Phb1fl/fl mice (figure 5D,E), suggesting that Mito-Tempo prevented differentiation changes favouring intermediate cells during Phb1 deletion. Mito-Tempo also prevented the upregulation of Il-1β and Il-18 in the ileum that was induced during Phb1 deletion (figure 5F). We continued administration of Mito-Tempo for an additional 9 weeks to assess severity of ileitis (figure 5G). Only 20% (2/10 mice) of Phb1iΔIEC mice treated with Mito-Tempo through 12 weeks exhibited histological evidence of inflammation in the ileum, compared with 80% (8/10 mice) of Phb1iΔIEC mice treated with vehicle.
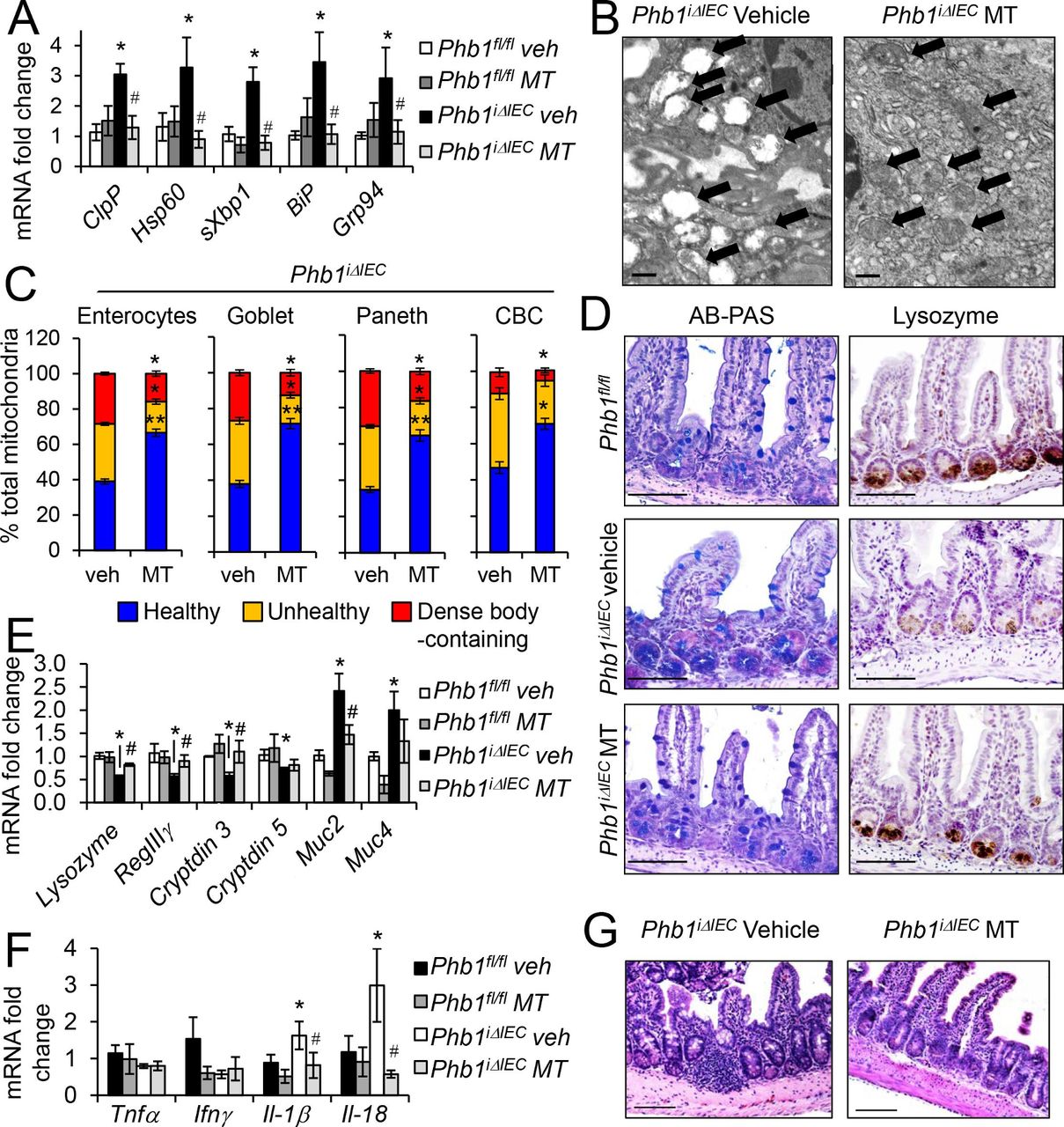
Mito-Tempo (MT) ameliorates mitochondrial dysfunction, Paneth cells defects, and ileitis in Phb1 iΔIEC ileum. (A) mRNA expression of mtUPR (ClpP, Hsp60) and ER UPR (sXbp1, BiP, Grp94) markers in isolated ileal IECs measured by qPCR. (B) TEM of ileal enterocytes. Arrows indicate mitochondria. Scale bars: 500 nm. (C) % healthy, unhealthy (swollen, dissolution of cristae), and dense inclusion body-containing mitochondria visualized by TEM. n=200 enterocytes, 200 goblet cells, 50 Paneth cells, and 50 CBC cells with an average of 53 mitochondrial/cell. (D) AB-PAS and Lysozyme immunohistochemistry staining. Scale bars: 250 μm. mRNA expression in isolated ileal IECs (E) or whole ileum (F) measured by qPCR. (G) H&E-stained sections showing ileum histology at 12 weeks. Scale bars: 250 μm. Results are presented as pooled data means±SEM of 6 mice per group (A, E, F) or 4 mice per group (C). *P<0.05, **p<0.01 vs Phb1 fl/fl veh, #p<0.05 vs Phb1 iΔIEC veh by 1-way ANOVA followed by Bonferroni’s test. AB-PAS, alcian blue–periodic acid schiff; ANOVA, analysis of variance; CBC, crypt base columnar; ER, endoplasmic reticulum; IECs, intestinal epithelial cells; IFN-γ, interferon-γ; IL-18, interleukin-18; mtUPR, mitochondrial unfolded protein response; Phb1 iΔIEC, intestinal epithelial cell deletion of Phb1; TEM, transmission electron microscopy; TNFα, tumour necrosis factor α.
Since Tempo not targeted to the mitochondria has been shown to affect microbiota,31 we administered Mito-Tempo in vitro using ileal enteroids to eliminate microbiota as a confounding factor and to examine epithelial cell-intrinsic responses. Enteroids derived from Phb1iΔIEC mice formed fewer crypt buds and exhibited significant death during the 7-day culture period compared with enteroids derived from Phb1fl/fl mice (online supplementary figure S11A–C). Phb1iΔIEC enteroids also displayed activation of the mtUPR and ER UPR, increased production of mitochondrial-derived superoxide and loss of expression of Paneth cell antimicrobials RegIIIγ, Cryptdin3, Cryptdin5 and Ang4 (online supplementary figure S11F,G). Mito-Tempo decreased Phb1iΔIEC enteroid death, mitochondrial-derived superoxide production, mtUPR and ER UPR activation and restored expression of Paneth cell antimicrobials to that of Phb1fl/fl enteroids (online supplementary figure S11E–G). Mito-Tempo had no effect on crypt budding (online supplementary figure S11D). These results suggest that mtROS contributes to Paneth cell defects and loss of viability of the ISC niche during IEC Phb1 deficiency.
Mice with Paneth cell-specific deletion of Phb1 develop spontaneous ileitis
To delineate the role of mitochondrial dysfunction specifically in Paneth cells and its effect on ileitis, we generated two mouse lines with Paneth cell-specific deletion by crossing Phb1fl/fl mice to Defα6-Cre mice (Phb1Defα6ΔPC ) and to Mist1-CreERT2 mice (Phb1iMist1ΔPC ). MIST1 (BHLHA15) is a critical transcription factor expressed in mature exocrine secretory cells like Paneth cells.32 Robust expression of PHB1 was demonstrated in Phb1fl/fl mice and confirmed to be deficient in Phb1Defα6ΔPC and Phb1iMist1ΔPC mice (online supplementary figures S12A and S14A). Phb1Defα6ΔPC and Phb1iMist1ΔPC mice developed spontaneous, discontinuous ileitis with similar characteristics to that seen in Phb1iΔIEC mice such as infiltration of CD4+ T cells, CD11b+/CD11c+ dendritic cells and F4/80+ macrophages, crypt elongation, villus blunting and thickening of the muscularis layers (figure 6A,B, online supplementary figures S12B, S14B–D). Inflammation was restricted to the ileum (online supplementary figures S12C and S14C). Despite these similarities, the penetrance of ileitis across Phb1Defα6ΔPC and Phb1iMist1ΔPC was lower than in Phb1iΔIEC mice (table 1 and online supplementary table S1). Much like Phb1iΔIEC mice, inflammation in Phb1Defα6ΔPC and Phb1iMist1ΔPC mice was associated with less body weight gain, enlarged spleens, and an early increase in Il-1β and Il-18 expression followed by later increased Tnfα and Ifnγ and decreased Il-10 (figure 6C, online supplementary figures S12D, S12E, S14E-F). Paneth cell abnormalities were evident as early as 1 week after the induction of Phb1 deletion in Phb1iMist1ΔPC mice and by 8 weeks of age in Phb1Defα6ΔPC , with altered lysozyme staining allocation pattern and increased AB+ staining (figure 6E–F, online supplementary figures S13A–C and S15A–F). Unlike Phb1iΔIEC mice, the number of lysozyme+ cells in Phb1ΔPC crypts was decreased compared with Phb1fl/fl mice (figure 6D and online supplementary figure S15C). Goblet cells above the crypt base in Phb1Defα6ΔPC and Phb1iMist1ΔPC mice appeared similar to Phb1fl/fl mice throughout all time points studied (figure 6F, online supplementary figures S13D, S15F). Altered expression of Paneth cell antimicrobials, Muc2 and Muc4, and Hes1 and Math1 in Phb1Defα6ΔPC mice matched the expression pattern displayed by Phb1iΔIEC mice (online supplementary figure S13E).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
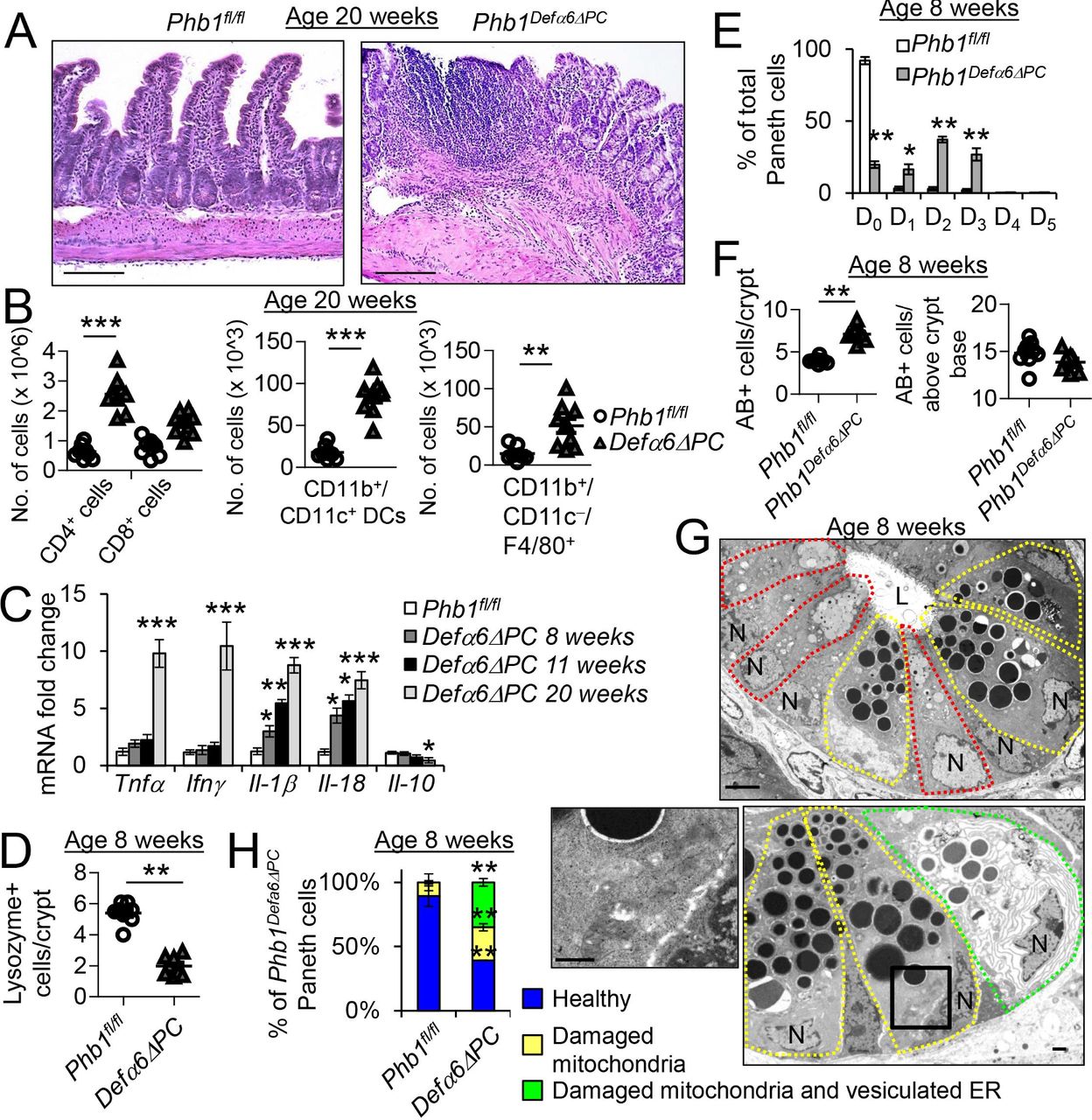
Phb1 Defα6ΔC mice develop mitochondrial dysfunction, Paneth cell defects, and spontaneous ileitis. (A) H&E-stained ileum sections at 20 weeks of age. Scale bars: 250 μm. (B) Absolute number of ileal LP immune cells calculated of total ileal LP cells. (C) mRNA expression in ileum measured by qPCR. (D) The number of Lysozyme+ cells across 50 crypts per mouse. (E) % of Paneth cells displaying each pattern of Lysozyme expression. 50 cells quantitated each mouse. (F) The number of AB+ cells in the crypt base or above the crypt base across 50 crypts per mouse. (G) TEM of Phb1 Defα6ΔPC ileum crypt base. Paneth cells, yellow outline; goblet-like cells, red outline; Paneth cell with vesiculated ER, green outline. Scale bars: 1 μm. Box denotes area of higher magnification showing unhealthy mitochondria. (H) % of Paneth cells displaying vesiculated ER. Results are presented as individual data points ± SEM (B, D, F) or as pooled data means ± SEM (C, E) of 9 mice per group. *P<0.05, **p<0.01, ***p<0.001 vs PHB1fl/fl by unpaired, 2-tailed Student’s t test (B, D, F) or by 1-way ANOVA followed by Bonferroni’s test (C, E). ANOVA, analysis of variance; ER, endoplasmic reticulum; IL-10, interleukin-10; IFN-γ, interferon-γ; L, lumen; N, nucleus; TEM, transmission electron microscopy; TNFα, tumour necrosis factor α.
TEM analysis 1 week after Phb1 deletion in Phb1Mist1ΔPC mice and at 8 weeks of age in Phb1Defα6ΔPC mice demonstrated similar mitochondrial ultrastructural changes in Paneth cells, but not other IECs, as those demonstrated in Phb1iΔIEC mice (figure 6G, online supplementary figures S13F, S15G, and S15H). Enteroids derived from Phb1Defα6ΔPC or Phb1iMist1ΔPC mice exhibited decreased crypt budding similar to Phb1iΔIEC enteroids, but less severe enteroid death and mitochondrial superoxide production compared with Phb1iΔIEC enteroids that was further decreased by Mito-Tempo treatment (online supplementary figure S11B–F). Additionally, Paneth cells of Phb1Defα6ΔPC and Phb1iMist1ΔPC mice exhibited secretory granule alterations and the appearance of goblet/Paneth intermediate cells similar to Phb1iΔIEC mice (figure 6G and online supplementary figure S15H). Interestingly, a significant proportion of Phb1Defα6ΔPC and Phb1iMist1ΔPC Paneth cells manifested vesiculated ER in addition to damaged mitochondria, suggesting severe ER stress which was corroborated by elevated ER UPR markers (figure 6G,H, online supplementary figures S13G, S15H, and S15I). A similar progression to ileitis was evident in Phb1Defα6ΔPC and Phb1iMist1ΔPC mice as was noted in Phb1iΔIEC mice with initial mitochondrial dysfunction, followed by Paneth cell defects and subsequent ileal inflammation (table 1 and online supplementary table S1). Collectively, these results suggest that mitochondrial dysfunction due to Phb1 deletion in Paneth cells is sufficient to drive ileitis.
Discussion
Mitochondrial dysfunction is central to many chronic diseases, including arthritis, neurodegeneration, cardiovascular disease and cancer. Previous studies suggest a link between mitochondrial dysfunction and Crohn’s disease. The paediatric RISK stratification study using RNA-sequencing analysis on Crohn’s disease mucosal biopsies demonstrated that of patients who were at risk of stricturing, those who exhibited an enriched mitochondrial function gene signature remained complication free through 36-month follow-up.33 In a proteome study of paediatric Crohn’s disease patients, impaired mitochondrial function was implicated and correlated with increased disease severity.34 It was recently demonstrated that mtDNA is released into the serum in IBD patients and acts as a proinflammatory signalling molecule.7 Deletion of Irgm, Slc22A5, Pgc1α or Mdr1, genes that serve roles in regulating mitochondrial health, cause spontaneous colitis or increase susceptibility to experimental colitis.4 5 Well-characterised mouse models of spontaneous ileitis such as SAMP1/Yit or TNFΔARE mice have not been examined for IEC mitochondrial dysfunction, but ileal histological alterations of Phb1iΔIEC or PhbΔPC mice were reminiscent to that of SAMP1/Yit mice.35 Expression of PHB1 is decreased in mucosal biopsies from IBD-afflicted patients and in mouse models of colitis,9 11 but the mechanism leading to decreased PHB1 expression in human IBD is unknown. IBD GWAS studies have not identified genetic mutation of PHB1. Genetic polymorphisms of PHB1 have been associated with gastric, breast, ovarian and skin cancers with some studies implicating regulation at the 3′ UTR,36 however, this has not been identified in IBD. Further studies are necessary to elucidate the mechanism of PHB1 deficiency during IBD.
Our results identify Paneth cells as highly susceptible to mitochondrial dysfunction driven by loss of Phb1 and central to the pathogenesis of ileitis. A total of 70%–80% of Crohn’s disease patients have inflammation involving the ileum, with or without colonic involvement. Mitochondrial dysfunction in IECs during Phb1 deletion was evidenced by mitochondrial ultrastructural abnormalities, decreased activity of ETC complexes, increased mtROS and oxidative damage to lipids and DNA. These mitochondrial changes did not significantly alter ATP concentration in isolated ileal IECs, suggesting that IECs as a population have adequate energy production. Future studies will access individual IEC types like Paneth cells for ATP production. Mitochondrial health may be especially important in intestinal secretory cells (Paneth, goblet and enteroendocrine cells) which are mitochondria-rich to sustain energy-expending secretory functions. Additionally, mitochondrial dysfunction is likely to be especially deleterious in terminally differentiated long-lived cells, such as Paneth cells, in which damaged organelles are not diluted by cell replication.22 Type I Paneth cell phenotype, characterised by ≥20% abnormal Paneth cells, defined by lysozyme granule staining characteristics (disordered, diminished, diffuse, enlarged or excluded) using mucosal biopsy samples,27 occurs in approximately 20% of adult ileal Crohn’s disease patients and is independent of active inflammation.25 26 Crohn’s disease-associated ATG16L1 T300A and NOD2 risk alleles were shown to correlate with type I Paneth cell phenotype.23 27 ATG16L1 and NOD2 play important roles in autophagy, an evolutionarily conserved catabolic pathway that removes cytoplasmic components, including damaged organelles, through lysosomal degradation. The autophagy pathway is crucial to suppress ileitis during Xbp1 deletion in Paneth cells.37 It is likely that mitophagy (autophagy of mitochondria) plays an important role in intestinal homoeostasis, but this has yet to be demonstrated in patients with IBD. Interestingly, patients with Crohn’s disease with type I Paneth cell defects exhibited decreased expression of oxidative phosphorylation genes, suggesting this subset of patients may have altered mucosal mitochondrial function, but this was not further elucidated.26 Our current results provide a mechanistic link between loss of PHB1, mitochondrial dysfunction and Paneth cell defects, which are three characteristics demonstrated in patients with Crohn’s disease. Future studies will elucidate whether type I Paneth cell patients exhibit mitochondrial dysfunction, altered mitophagy or altered PHB1 expression.
mtROS are well-established primary downstream signalling molecules and, if excessive, mtROS can lead to cellular damage. Treatment of Phb1iΔIEC mice with the mitochondrial-targeted antioxidant Mito-Tempo ameliorated Paneth cell defects and ileitis during loss of Phb1. Mito-Tempo also prevented the upregulation of Il-1β and Il-18 in the ileum of Phb1iΔIEC mice, suggesting that mtROS contributes to early increased secretion of these cytokines. Our results indicate that epithelial mitochondrial dysfunction evident 1 week after Phb1 deletion concomitantly occurs with enhanced secretion of IL-1β and IL-18. Future studies will determine whether the mtROS signalling via the NLRP3 inflammasome is involved in enhanced secretion of IL-1β and IL-18 in this model and the cellular source of these cytokines. Additionally, in Phb1-deficient enteroids, Mito-Tempo increased viability, decreased mtROS, decreased mtUPR and ER UPR activation, and restored expression of Paneth cell antimicrobials to that of Phb1fl/fl enteroids, suggesting epithelial cell-intrinsic responses to Mito-Tempo. These results demonstrate that mtROS contributes to Paneth cell defects and loss of viability of the ISC niche during IEC Phb1 deficiency and present a druggable pathway for therapeutic targeting.
Mitochondrial function is a crucial modulator of stem cell fate in relatively quiescent stem cell populations, including haematopoietic stem cells and neural stem cells, as well as more active ISCs requiring more bioenergetics activity due to frequent turnover of the intestinal epithelium.38 Phb1iΔIEC mice demonstrated Paneth/goblet intermediate cells early after Phb1 deletion prior to signs of histological inflammation, followed by later increased number and size of goblet cells and decreased enterocytes. This altered differentiation programme in Phb1iΔIEC crypts was accompanied by increased crypt cell proliferation and apoptosis in crypts and villi, likely as a consequence of tissue repair induction. Goblet and Paneth cells differentiate from a common secretory cell lineage progenitor that is distinct from the enterocyte cell lineage progenitor.29 Differentiation towards the secretory cell lineage is mediated by at least two transcription factors, Hes1 and Math1, which are altered in Phb1iΔIEC mice in a manner shown to favour secretory cell differentiation. Additionally, Klf4 is increased in Phb1 iΔIEC ileum and has been shown to be required for goblet cell-differentiation.39 Intermediate cells in Phb1iΔIEC mice could originate from changes in the Paneth cell itself to promote transformation to a goblet cell, goblet cells being converted to Paneth cells, or stem cell responses driving altered lineage differentiation. It was previously shown that ISC mitochondrial dysfunction and decreased ability to produce ATP results in altered ISC self-renewal and proliferation.38 We speculate that altered Paneth/goblet cell differentiation in ileum of Phb1iΔIEC mice could be due to (1) PHB1 directly regulating the Notch-1 (upstream of Hes1/Math1) and/or Klf4 pathways, (2) modulation of Notch-1 and/or Klf4 by redox-dependent signalling or mitochondrial dysfunction subsequent to PHB1 deletion and/or (3) a response to inflammation, however, this seems unlikely since altered secretory allocation was demonstrated preceding histological signs of inflammation. Additionally, IL-18 plays an important role in goblet cell differentiation40 41 and future studies will assess whether the Paneth/goblet intermediate cells observed in Phb1iΔIEC mice are driven by IL-18 to prevent goblet cell maturation.
Paneth cell-specific deletion of Phb1 induced a similar progression to ileitis as was demonstrated in Phb1iΔIEC mice with initial mitochondrial dysfunction and Paneth cell defects. At early time points, mitochondrial dysfunction was evident in only Paneth cells in Phb1ΔPC mice, and not other IECs, confirming the importance of a healthy mitochondrial pool in Paneth cells in maintaining intestinal homoeostasis. Penetrance of inflammation across Phb1ΔPC mice was less than Phb1iΔIEC mice, suggesting mitochondrial dysfunction throughout the intestinal epithelium, as in Phb1iΔIEC mice, accelerates the progression to inflammation. Indeed, enteroids derived from Phb1iΔIEC mice exhibited more severe death and mitochondrial superoxide production compared with Phb1Defα6ΔPC or Phb1iMist1ΔPC enteroids. Unlike in Phb1iΔIEC mice, AB+ cells above the crypt base in Phb1Defα6ΔPC and Phb1iMist1ΔPC mice appeared normal throughout all time points examined with no alteration of Klf4 or Elf3 expression. This suggests Phb1 deletion in ISCs or in goblet cells themselves drives goblet cell abnormalities in Phb1iΔIEC mice that is not recapitulated during Paneth cell-specific Phb1 deletion. Similar to Phb1iΔIEC mice, Phb1 iMist1ΔPC mice developed Paneth cell defects and the appearance of intermediate cells at a time point (1 week) prior to normal turnover of Paneth cells (every 30–60 days22). This suggests that mitochondrial dysfunction in Paneth cells could trigger Paneth cell-intrinsic dedifferentiation to intermediate cells. Alternatively, death of Paneth cells could be accelerated in the Phb1 deficient mice and as a repair response, an ISC-derived effect could give rise to intermediate cells. Given the close proximity of Paneth cells and ISCs and the known interrelationship of Paneth cell mitochondrial metabolism and ISC niche homoeostasis,42 loss of Phb1 in Paneth cells could in turn signal to alter ISC differentiation response. Future studies using reporter mice will elucidate whether Paneth cells or ISCs trigger altered IEC differentiation during loss of Phb1.
In summary, our findings further our understanding of intestinal homoeostasis originating at the cellular (Paneth cell) level and organellar (mitochondria) level. We identify Paneth cells as highly susceptible to mitochondrial dysfunction driven by Phb1 deletion and central to the development of ileitis. Treatment of Paneth cell defects with Mito-Tempo during Phb1 deletion implicates a potential therapeutic application for abnormal Paneth cells via elimination of mtROS. Mitochondrial-targeted therapeutics may have translational utility in a subset of patients with Crohn’s disease exhibiting Paneth cell defects. These are the first results presenting a causative role of mitochondrial dysfunction in ileitis that initiates in Paneth cells.
Materials and methods
Materials and methods are included in the online supplementary materials.
Supplemental material
Acknowledgments
We thank Jie Han and Arwa S Kathiria for technical assistance and Beth Cook for histology processing (Baylor Scott & White Research Institute). We are grateful to P Kay Lund and Amanda Mah (University of North Carolina at Chapel Hill, NC) for assistance with enteroid culturing. We thank Ricardo Olivarez and Stephanie Kara at the Anatomic Pathology Laboratory at Children's Health Medical Centre Dallas, Texas, USA, for assistance with electron microscopy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Study concept and design: TD, LAF, RFS, RSB, KV and ALT. Acquisition of data: DNJ, MP, WLN, KT, LT-S and ALT. Analysis and interpretation of data: MP, WLN, KT, BLC, LT-S, KV and ALT. Drafting the manuscript: DNJ, MP and ALT. Critical revision of the manuscript for important intellectual content: TD, LAF, RFS, JCM, RSB, KV and WET.
Funding This work was supported by National Institutes of Health grants R01-DK117001 (ALT), R01-DK105129 (JCM), R01-DK088199 (RSB), R03-DK098229 (ALT) and Litwin IBD Pioneers Crohn’s Colitis Foundation 301869 (ALT).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval Baylor Scott & White Research Institute Institutional Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplementary information. All mice described in this study must be obtained through an MTA.