Article Text

Abstract
Objective The rs641738C>T variant located near the membrane-bound O-acyltransferase domain containing 7 (MBOAT7) locus is associated with fibrosis in liver diseases, including non-alcoholic fatty liver disease (NAFLD), alcohol-related liver disease, hepatitis B and C. We aim to understand the mechanism by which the rs641738C>T variant contributes to pathogenesis of NAFLD.
Design Mice with hepatocyte-specific deletion of MBOAT7 (Mboat7Δhep) were generated and livers were characterised by histology, flow cytometry, qPCR, RNA sequencing and lipidomics. We analysed the association of rs641738C>T genotype with liver inflammation and fibrosis in 846 NAFLD patients and obtained genotype-specific liver lipidomes from 280 human biopsies.
Results Allelic imbalance analysis of heterozygous human liver samples pointed to lower expression of the MBOAT7 transcript on the rs641738C>T haplotype. Mboat7Δhep mice showed spontaneous steatosis characterised by increased hepatic cholesterol ester content after 10 weeks. After 6 weeks on a high fat, methionine-low, choline-deficient diet, mice developed increased hepatic fibrosis as measured by picrosirius staining (p<0.05), hydroxyproline content (p<0.05) and transcriptomics, while the inflammatory cell populations and inflammatory mediators were minimally affected. In a human biopsied NAFLD cohort, MBOAT7 rs641738C>T was associated with fibrosis (p=0.004) independent of the presence of histological inflammation. Liver lipidomes of Mboat7Δhep mice and human rs641738TT carriers with fibrosis showed increased total lysophosphatidylinositol levels. The altered lysophosphatidylinositol and phosphatidylinositol subspecies in MBOAT7Δhep livers and human rs641738TT carriers were similar.
Conclusion Mboat7 deficiency in mice and human points to an inflammation-independent pathway of liver fibrosis that may be mediated by lipid signalling and a potentially targetable treatment option in NAFLD.
- NAFLD
- NASH
- liver fibrosis, lipidomics
Data availability statement
Data are available on reasonable request. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic fatty liver disease (NAFLD) results from a multifactorial pathogenesis including genetic risk factors, obesity, intestinal dysbiosis and insulin resistance.
Membrane-bound O-acyltransferase domain containing 7 (MBOAT7) has been previously reported as a risk gene in patients with liver diseases such as alcoholic and NAFLD, hepatitis B and C. Furthermore, reduced MBOAT7 protein in liver has been associated with altered phosphatidylinositol (PI) remodelling.
MBOAT7 deficiency in mice reshapes the hepatic levels of PI and lysophosphatidylinositol (LPI) and promotes hyperinsulinemia and hepatic insulin resistance.
Significance of this study
What are the new findings?
We report a hepatocyte specific deletion of MBOAT7 in mice using cre-loxP method and understanding its role in NAFLD.
Disturbance of the PI side chain remodelling pathway in hepatocytes alone is sufficient to elicit spontaneous steatosis under steady state and fibrosis upon an induction diet—that is, hepatocytes themselves may serve as pathogenetic driver of fibrosis.
Mboat7 deficiency in mice and humans defines a pathway to liver fibrosis that was independent of inflammation.
Commonly implicated lipid mediators such as free fatty acids and oxylipins remain unchanged.
Mboat7Δhep mouse model closely resembles the lipidomic changes in humans with the MBOAT7 risk variant.
The fibrotic phenotype might be driven by extracellular lipid mediators—namely LPI.
How might it impact on clinical practice in the foreseeable future?
Targeting PI signalling may be a novel approach to the treatment of NAFLD and liver fibrosis.
Patients carrying the MBOAT7 mutation may be prone to fibrosis development without significant hepatic inflammation.
Introduction
Non-alcoholic fatty liver disease (NAFLD) affects up to 30% of adults and 70%–80% of obese and diabetic individuals worldwide.1 NAFLD encompasses a spectrum of liver pathologies from simple steatosis (non-alcoholic fatty liver, NAFL) to non-alcoholic steatohepatitis (NASH) and eventually liver cirrhosis.2 3 Histopathological characteristics of NASH include steatosis, ballooning, inflammation and fibrosis.3 4 A recent study indicated that severe fibrosis may be present in the absence of inflammation.5 NASH can further progress to cirrhosis, end-stage liver disease and hepatocellular carcinoma.1
Human genetic association studies have identified variation in several genes to be associated with NAFLD pathology, such as patatin-like phospholipase domain-containing 3, transmembrane 6 superfamily member 2 and also more recently membrane-bound O-acyltransferase domain containing 7 (MBOAT7).4 6 7 MBOAT7, a multispanning transmembrane protein, participates in acyl chain remodelling of phosphatidylinositol (PI) in the Lands cycle, by adding arachidonoyl-CoA (20:4) to 1-stearoyl (18:0) lysophosphatidylinositol (LPI) thereby generating sn-1-stearoyl-sn-2-arachidonoyl PI species.8–11 The identified human risk variant, MBOAT7 rs641738T, has been associated with alcohol-related cirrhosis, NAFLD and with inflammation and fibrosis in chronic hepatitis B and C.12–18
In carriers of the rs641738C>T variant, reduced levels of MBOAT7 protein in the liver14 and hepatic-PI remodelling18 have been observed. Additionally, Mboat7 deficiency in mice reshapes the hepatic levels of PI and LPI and promotes hyperinsulinemia and hepatic insulin resistance.19 20 However, the mechanisms by which the rs641738C>T variant leads to NAFLD development and especially hepatic fibrosis are not sufficiently understood. Human genetic data on MBOAT7 point to a strong association of fibrosis across a range of different liver diseases.13 14 16–18 Since fibrosis is the key prognostic marker for NAFLD21 and the endpoint of many current clinical studies on NASH,22 23 investigation of Mboat7-related disease pathways holds promise to provide additional insight into fibrosis progression of chronic liver disease.
Here, we present a combined analysis of a mouse model with hepatocyte-specific Mboat7 deletion with genetic and lipidomic analysis of human liver biopsies across the spectrum of NAFLD. We demonstrate that hepatocyte-specific deletion of Mboat7 in mice is sufficient to cause hepatic-steatosis at steady-state. On nutritional triggering with an NASH-inducing diet, MBOAT7 deficiency resulted in a lipid-driven and inflammation-independent pathway towards liver fibrosis.
Materials and methods
Human NAFLD cohort
A total of 846 adult Caucasian NAFLD patients from tertiary referral centres in Austria (n=258), Germany (n=537) and Switzerland (n=51) who underwent percutaneous or surgical liver biopsy were included into this study (online supplementary tables 1–2). NASH was defined by the NAFLD Activity Score. Presence of fibrosis was assessed histologically according to Kleiner classification24 (stage-F0:no fibrosis; stage-F1:perisinusoidal fibrosis to portal/periportal fibrosis; stage-F2:perisinusoidal and portal/periportal fibrosis; stage-F3:bridging fibrosis). The liver biopsies were read by experienced pathologists in a blinded fashion. Portions of this NAFLD cohort have been described previously.25–27 All patients gave their written consent.
Supplemental material
Human lipidomic cohort
A total of 280 human liver samples were obtained from patients in Germany, in whom an intraoperative liver biopsy was indicated on clinical grounds such as during scheduled liver resection, exclusion of liver malignancy during major oncologic surgery or assessment of liver histology during bariatric surgery. Samples were snap frozen immediately in liquid nitrogen ensuring an ex vivo time of less than 40 seconds. Patients with evidence of viral hepatitis, haemochromatosis or alcohol consumption >20 g/day (women) and >30 g/day (men) were excluded. For all samples, MBOAT7 rs641738C>T genotype as well as full phenotypic and histological information24 generated by a single pathologist (CR) blinded to the lipidomic analysis was available (table 1). Phenotypic groups (table 1) were defined on the basis of clinical and histological parameters.28
Overview of the patients in the lipidomic cohort
Mouse model
C57BL/6 mice with hepatocyte-specific deletion of Mboat7 (Mboat7Δhep) were derived from crossing mice with a floxed Mboat7 allele with mice expressing Cre recombinase under the control of Albumin promoter (Albumin-Cre). Mboat7 floxed mice, MBOAT7tm1c(KOMP)Wtsi, were purchased from MRC Harwell, Oxford, UK. Cre negative littermate mice with Mboat7 floxed alleles (Mboat7WT) were used as controls. Mice were fed normal diet (Chow diet V1534-300, Ssniff Spezialdiäten) or a high-fat, methionine-low, choline-deficient diet (HFCDD, 60% kcal fat, 0.1% methionine and choline-deficient diet; A06071302, research diets composition in online supplementary table 3) ad libitum. All experiments were performed according to the German animal welfare law and approved by the Landesdirektion Sachsen, Germany.
Lipidomic analysis
Liver tissue (~25 mg) was homogenised and protein concentration was determined by BCA/Pierce 660 assay. Lipids were extracted from aliquots containing 50 µg of total protein and quantified by shotgun lipidomics, as previously described.29 30 Lipidomix, Berlin, performed oxylipin and free fatty acid (FFA) measurement in liver (~30 mg) using liquid chromatography electrospray ionisation tandem mass spectrometry (LC/ESI-MS/MS).31
Statistical analysis
Statistical analyses were performed using GraphPad software. For comparison of quantitative measurements, the Mann-Whitney U test or unpaired t test was used. Data are expressed as mean±SE of the mean.
See online supplementary section for detailed explanation on methods. Mouse data are additionally represented in the form of dot plots (online supplementary figures 13–19)
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Results
Analysis of allelic imbalance
Association of MBOAT7 rs641738C>T variant with lower mRNA expression and lower protein levels of MBOAT7 in human liver has been reported.14 As the downregulation of MBOAT7 might be either caused by direct effects of the genomic variation or secondary effects, we analysed the relative allelic expression of rs8736 (LD to rs641738T: r2=0.98) in rs641738T heterozygous individuals. Transcripts bearing rs8736 in the 3’UTR of MBOAT7 show a significantly (p<0.0001) lower expression than transcripts bearing the ancestral allele (figure 1A), thereby indicating a cis down-regulatory effect on the haplotype carrying the rs641738T allele.

Allelic imbalance and spontaneous steatosis phenotype of Mboat7Δhep in 10 weeks old mice fed with normal diet. (A) Relative allelic hepatic mRNA expression of MBOAT7 transcripts in rs641738C>T heterozygous individuals using the cSNP rs8736 that is in LD to rs641738T. Shown is the ratio of mutation carrying transcripts compared with transcripts bearing the ancient allele (left column). DNA (right column) was used as a control. n=37. (B–J) Data derived from Mboat7WT and Mboat7Δhep mice which were fed a normal diet. Data are presented as mean±SEM. **P<0.01 (Mann-Whitney U test). (B) mRNA expression of Mboat7 was determined by qRT-PCR in Mboat7WT (n=6) and Mboat7Δhep (n=7) mice. (C) H&E staining of livers from 10-week-old Mboat7WT and Mboat7Δhep mice. (D) Oil red O staining of livers from 10-week-old Mboat7WT and Mboat7Δhep mice. Scale bars, 100 µm. (E) Hepatic-concentrations of triglyceride (TAG) and cholesterol ester (CE) (n=5 mice). (F–I) Flow cytometry analysis was performed in non-parenchymal cells (NPCs). (F) Total number of NPCs (n=6–7 mice). (G–I) Hepatic number of monocytes (CD45+/Ly6G-/CD11b+/F4/80-), macrophages (CD45+/Ly6G-/CD11b+/F4/80+) and neutrophils (CD45+/Ly6G+/CD11b+) in livers of Mboat7WT and Mboat7Δhep mice (n=6–7 mice). (J) liver RNA sequencing in Mboat7WT and Mboat7Δhep mice. GSEA shows a positive correlation between ECM related genes and Mboat7 deficiency. qRT-PCR, quantitative real time-polymerase chain reaction; CE, cholesteryl ester; ECM, extracellular matrix; GSEA, gene set enrichment analysis; LD,linkage disequilibrium; MBOAT7, membrane-bound O-acyltransferase domain containing 7; NS, not significant; SEM, SE of the mean.
Hepatocyte deletion of MBOAT7 in mice results in spontaneous steatosis characterised by increased cholesterol ester content
Previous data13 14 and our allelic imbalance analysis in human biopsies suggest, that reduced levels of MBOAT7 protein are the main locus-specific mechanism of contribution to NAFLD pathogenesis in rs641738C>T carriers. We, therefore, generated mice with hepatocyte-specific deletion of Mboat7 using loxP-flanked (‘floxed’) alleles of Mboat7 exon 5 (online supplementary figure 1A) and Cre-mediated deletion (Alb-Cre; Mboat7 fl/fl mice; hereafter referred to as Mboat7Δhep mice). Littermate Cre negative Mboat7 fl/fl mice were used as controls (Mboat7WT). Efficient deletion of Mboat7 in liver was verified at mRNA level (figure 1B and online supplementary figure 1B) and Cre-loxP-mediated recombination was confirmed in primary hepatocytes by genomic-PCR (online supplementary figure 1C). We analysed liver histology in 10-week-old mice which were fed a normal diet. While there was no difference in liver weight (online supplementary figure 2A), livers from Mboat7Δhep animals contained more lipids than Mboat7WT mice as shown by H&E and Oil red O staining (figure 1C,D). Mboat7Δhep primary hepatocytes showed increased lipid accumulation as compared with Mboat7WT cells, as shown by Oil red O staining (online supplementary figure 2B). Quantitative lipidomic analysis revealed the presence of steatosis in the Mboat7Δhep livers and primary hepatocytes was associated with the accumulation of cholesteryl esters (CEs) but not triglycerides (TAGs) (figure 1E, online supplementary figure 2C). Specifically, CE 16:1, CE 16:0, CE 18:2 and CE 18:1 were significantly increased in Mboat7Δhep livers (online supplementary figure 2D). Flow cytometry analysis revealed no differences in the number of non-parenchymal cells (NPCs), monocytes, macrophages and neutrophils between Mboat7Δhep and Mboat7WT livers (figure 1F–I, online supplementary figure 2E–G). Quantitative real time PCR (qRT-PCR) analysis showed that mRNA expression of interferon-γ (IFN-gamma), tumour necrosis factor α (TNF-alpha), interleukin-1 (IL1)-beta and IL6 were unaltered between the two groups (online supplementary figure 2H). Protein levels of inflammatory mediators in the liver were unaltered between two groups (online supplementary figure 2I).
Supplemental material
Supplemental material

Mboat7-deficiency promotes hepatic fibrosis in mice on a high fat, methionine-low, choline-deficient diet in an inflammation independent manner. (A, C, D, F, J–L) data derived from Mboat7WT and Mboat7Δhep which were fed a HFCDD for 6 weeks. Data are presented as mean±SEM. *P<0.05, **P<0.01. (Mann-Whitney U test). (A) liver weight of Mboat7WT and Mboat7Δhep mice (n=7–8 mice). (B) H&E staining. Scale bars, 200 µm. (C) Hepatic-concentrations of TAG and CE (n=7–8 mice). (D, E) quantification (D) and representative images (E) of picrosirius red positive area in livers of Mboat7WT and Mboat7Δhep mice. Scale bars, 100 µm. (n=7–8 mice). (F) quantification of hydroxyproline (n=4–5 mice). (G–I) RNA sequencing results in livers of Mboat7WT and Mboat7Δhep mice. (G) heat map of significantly upregulated genes (FDR <0.05) in Mboat7Δhep livers from the extracellular matrix organisation pathway (from online supplementary figure 5A). (H) GSEA shows a positive correlation between the extracellular matrix related genes and Mboat7 deficiency. (I) heat map of collagen subtypes that were significantly (FDR <0.05) upregulated in the Mboat7Δhep livers. (J) qRT-PCR results (n=4–5 mice). (K, L) flow cytometry analysis of NPCs, (total NPCs, monocytes, macrophages, neutrophils, CD4, CD8 T cells) isolated from livers of Mboat7WT and Mboat7Δhep mice. (n=4–5 mice). CE, cholesteryl ester; GSEA, gene set enrichment analysis; HFCDD, high-fat, methionine-low, choline-deficient diet; MBOAT7, membrane-bound O-acyltransferase domain containing 7; FDR, False discovery rate; NPCs, non-parenchymal cells; SEM, SE of the mean; TAG, triglyceride.
Supplemental material
RNA sequencing of livers from Mboat7WT and Mboat7Δhep mice fed a normal diet was performed. Eighty-eight genes were significantly downregulated and 98 genes were significantly upregulated following Mboat7 deletion (online supplementary table 4). Gene set enrichment analysis (GSEA) revealed a negative correlation between genes involved in fatty acid metabolism and hepatocyte specific Mboat7 deficiency (online supplementary figure 3A). Fatty acid oxidation-related genes such as Acaa1a, Acox1, Cpt1a, and Ehhadh were diminished and a lipogenic gene, Scd1 was upregulated in Mboat7Δhep livers (online supplementary figure 3B). Significantly altered genes from RNA sequencing, involved in fatty acid metabolism and cholesterol (Chol) metabolism were confirmed by qRT-PCR (online supplementary figure 3C). Furthermore, Angptl3, which is involved in lipoprotein metabolism was upregulated in Mboat7Δhep livers (online supplementary figure 3B and D); however, the circulating levels of Angptl3 protein was unaltered between the two groups (online supplementary figure 3E). GSEA revealed a positive correlation between genes involved in production of extracellular matrix (ECM) and Mboat7 deficiency (figure 1J).
Supplemental material
Mboat7 deficiency promotes hepatic fibrosis on a high fat, methionine-low, choline-deficient diet in an inflammation independent manner
Mice were fed a HFCDD for 6 weeks to induce NAFLD. Both Mboat7Δhep and Mboat7WT mice accumulated lipids and showed no difference in liver weight (figure 2A,B). Although there was no visible difference in hepatic-lipid accumulation (figure 2B), Mboat7Δhep livers accumulated more CE with no changes in TAG levels (figure 2C). CE 16:1, CE 16:0, CE 18:3, CE 18:2, CE 18:1, CE 18:0, CE 20:5 and CE 20:3 species were elevated in Mboat7Δhep livers (online supplementary figure 4A).
Supplemental material
In the Mboat7Δhep mice we observed increased aspartate transaminase (AST) activity (online supplementary figure 4B) and alanine aminotransferase (ALT) activity showed a non-significant trend towards increase in Mboat7Δhep mice (online supplementary figure 4C). Increased fibrosis was assessed by picrosirius red staining and western blot for TIMP1 in Mboat7Δhep mice (figure 2D,E and online supplementary figure 4D). The hepatic-hydroxyproline concentrations were increased by ~45% in the Mboat7Δhep as compared with Mboat7WT livers (p=0.031, figure 2F). On liver mRNA sequencing, 466 genes were upregulated and 112 genes were downregulated in Mboat7Δhep livers following HFCDD feeding (online supplementary table 5). In accordance with the picrosirius red and hydroxyproline measurements, pathways related to ECM, extracellular structure organisation, wound healing and tissue remodelling were significantly upregulated in Mboat7Δhep livers (figure 2G and online supplementary figure 5A). GSEA analysis showed a positive correlation between genes involved in production of ECM and Mboat7 deletion (figure 2H). Transcripts of collagen subtypes such as Col6a3, Col4a5, Col4a4, Col6a1, Col6a2, Col1a2, Col1a1, Col3a1, Col5a2, Col15a1, Col8a1 and Acta2 were elevated in the Mboat7Δhep livers (figure 2I and online supplementary figure 5B). Several ECM-related genes were further confirmed by qRT-PCR with consistent results (figure 2J). Various lipid metabolic pathways and ‘inflammatory response’ pathways were downregulated in livers of Mboat7 deficient mice following HFCDD feeding (online supplementary figure 5C). Intriguingly, there were no differences in the number of NPCs, macrophages, neutrophils, CD4 T cells and CD8 T cells between the two groups on flow cytometry analysis (figure 2K,L). Nominal numbers of monocyte population were reduced in Mboat7Δhep livers (figure 2L). However, the percentage of inflammatory cells were unchanged (online supplementary figure 5D). Inflammatory mediators such as TNF-alpha, IL6, IL1-beta and IFN-gamma were all unchanged at the mRNA level (online supplementary figure 5E) and on mRNA sequencing (data not shown). Protein levels of TNF-alpha and KC/GRO were increased and IL2 was decreased in Mboat7Δhep livers, whereas IFNγ, IL1β, IL4, IL5, IL6, IL10 and IL12p70 were unaltered between the two groups (online supplementary figure 5F). Markers of ER stress were not altered in the Mboat7Δhep liver (online supplementary figure 6A–K).
Supplemental material
Supplemental material
Supplemental material
The MBOAT7 rs641738C>T variant is associated with hepatic fibrosis in humans with a body mass index ≤35 independent of the presence of inflammation
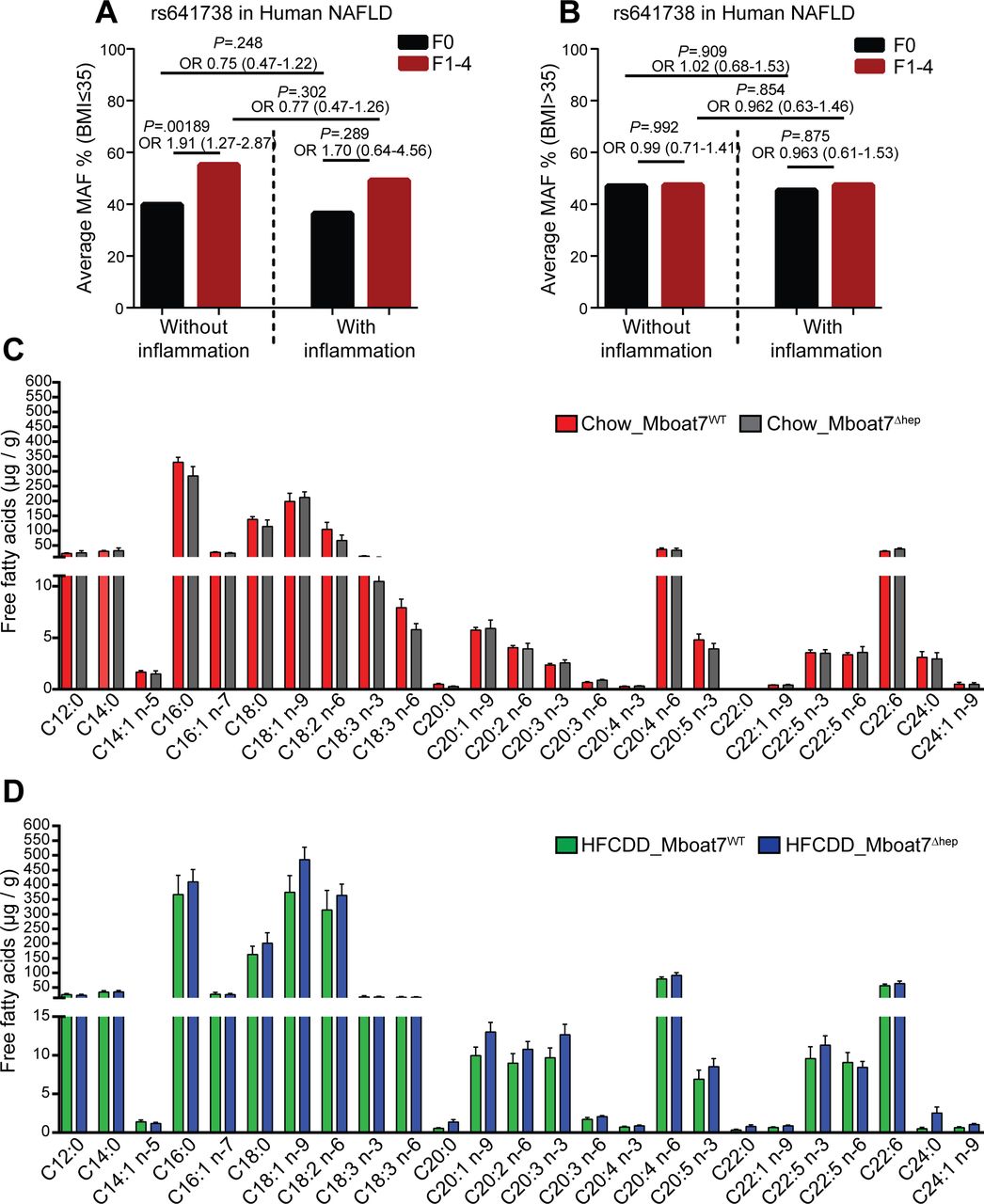
We analysed the frequency of the rs641738C>T allele in patients (separately in body mass index (BMI) ≤35 and BMI >35) from a cross-sectional NAFLD liver biopsy cohort stratified by the presence of liver fibrosis and lobular inflammation. In patients with BMI ≤35, the rs641738C>T allele was significantly associated with the presence of fibrosis (F1-F4) in the absence of lobular inflammation (p=0.001; OR=1.91 (1.27–2.87), figure 3A). In the presence of lobular inflammation (1–3), the association of the rs641738C>T allele with the presence of fibrosis yielded a similar odds ratio (p=0.289; OR=1.70 (0.64–4.56)), suggesting the development of fibrosis may be independent of inflammation in patients carrying the rs641738C>T variant (figure 3A). Interestingly, in morbidly obese patients with a BMI >35, no association of rs641738C>T genotype with the presence of histological fibrosis was observed (figure 3B).

The MBOAT7 rs641738T variant associates with fibrosis independent of inflammation. Hepatic Mboat7 deficiency is not associated with altered free fatty acids in mice. (A, B) the frequency of the MBOAT7 rs641738C>T allele in NAFLD patients (A) BMI ≤35 (without inflammation, F0, n=215; without inflammation, F1–4, n=74; with inflammation, F0, n=10; with inflammation, F1–4, n=62) and (B) BMI >35 (without inflammation, F0, n=231; without inflammation, F1–4, n=96; with inflammation, F0, n=64; with inflammation, F1–4, n=94) from a cross-sectional liver biopsy cohort, stratified by the presence of liver fibrosis and lobular inflammation. Genetic analyses were calculated using an additive model. Differences between the groups were compared by logistic regression analysis adjusted for sex, age, BMI and presence of T2DM (online supplementary table 1 & 2). (C, D) hepatic free-fatty acid levels on normal diet (C) and HFCDD diet (D). Data are presented as mean±SEM. (n=6–8 mice). BMI, body mass index; HFCDD, high-fat, methionine-low, choline-deficient diet; MBOAT7, membrane-bound O-acyltransferase domain containing 7; NAFLD, non-alcoholic fatty liver disease; T2DM, Type 2 Diabetes mellitus; MAF, Minor allele frequency; SEM, SE of the mean.
Mboat7Δhep mice do not exhibit altered hepatic levels of FFA and oxylipins
As Mboat7 functions by adding arachidonoyl-CoA (20:4) to 1-stearoyl (18:0) LPI (online supplementary figure 7),9 32 33 we hypothesised that Mboat7 deficiency could lead to altered levels of FFA and fatty acid metabolites (oxylipins) in the liver. Therefore, we measured the hepatic concentrations of FFA and oxylipins in Mboat7WT and Mboat7Δhep mice. We found no significant differences in the levels of FFA between the two groups both on a normal diet and on HFCDD feeding (figure 3C,D). On correction for multiple testing across both diets, there was no difference in the hepatic oxylipin concentrations between the Mboat7WT and Mboat7Δhep mice on normal diet (online supplementary table 6) and after HFCDD (online supplementary table 7).
Supplemental material
Lipidomic analysis of PI and LPI species in mouse liver
Given the biochemical function of Mboat79 32 33 (online supplementary figure 7), hepatocyte-specific knockout of the enzyme might have an impact on the molecular composition and molar abundance of LPI and PI. In normal diet fed mice, total LPI levels were increased from 0.59±0.11 pmol/µg of total protein in Mboat7WT livers to 2.72±0.26 pmol/µg in Mboat7Δhep livers (figure 4A). LPI 20:4 was diminished in Mboat7Δhep livers while LPI 16:0, LPI 18:1 and LPI 18:0 were increased (figure 4A,B). Of note, LPI 18:1 was not detected in Mboat7WT but was induced in Mboat7Δhep livers (figure 4B). Total PI levels were reduced in Mboat7Δhep livers (figure 4A). As reported previously, PI 38:4 was the most abundant PI species accounting for 88% of total PI in Mboat7WT livers (data not shown).19 PI38:4 was strikingly downregulated with ~60% reduction in Mboat7Δhep livers as compared with Mboat7WT livers (figure 4C). In addition to PI38:4, PI36:4 and PI38:5 were also significantly reduced in the Mboat7Δhep livers (figure 4C). In addition to PI and LPI, phosphatidylglycerol (PG), lysophosphatidylglycerol (LPG) and phosphatidic acid (PA) were elevated in the livers of Mboat7 deficient as compared with Mboat7 sufficient mice (figure 4A). The hepatic levels of other phospholipid classes such as PC, PE, PS, PC-O and lysophospholipids such as lysophosphatidylethanolamine (LPE) and lysophosphatidylcholine (LPC), ceramides, sphingomyelin (SM), diacylglycerol and Chol were unaltered (figure 4A). In order to identify alterations in the hepatocyte lipidome, we performed lipidomic analysis in isolated primary hepatocytes from Mboat7WT and Mboat7Δhep mice (online supplementary figure 8A). Similar to total liver, we found that LPI and PG levels were enhanced in Mboat7Δhep hepatocytes as compared with Mboat7WT hepatocytes (online supplementary figure 8A), demonstrating that the significant alterations observed in the liver lipidome (figure 4A) mainly originated from the hepatocytes.
Supplemental material
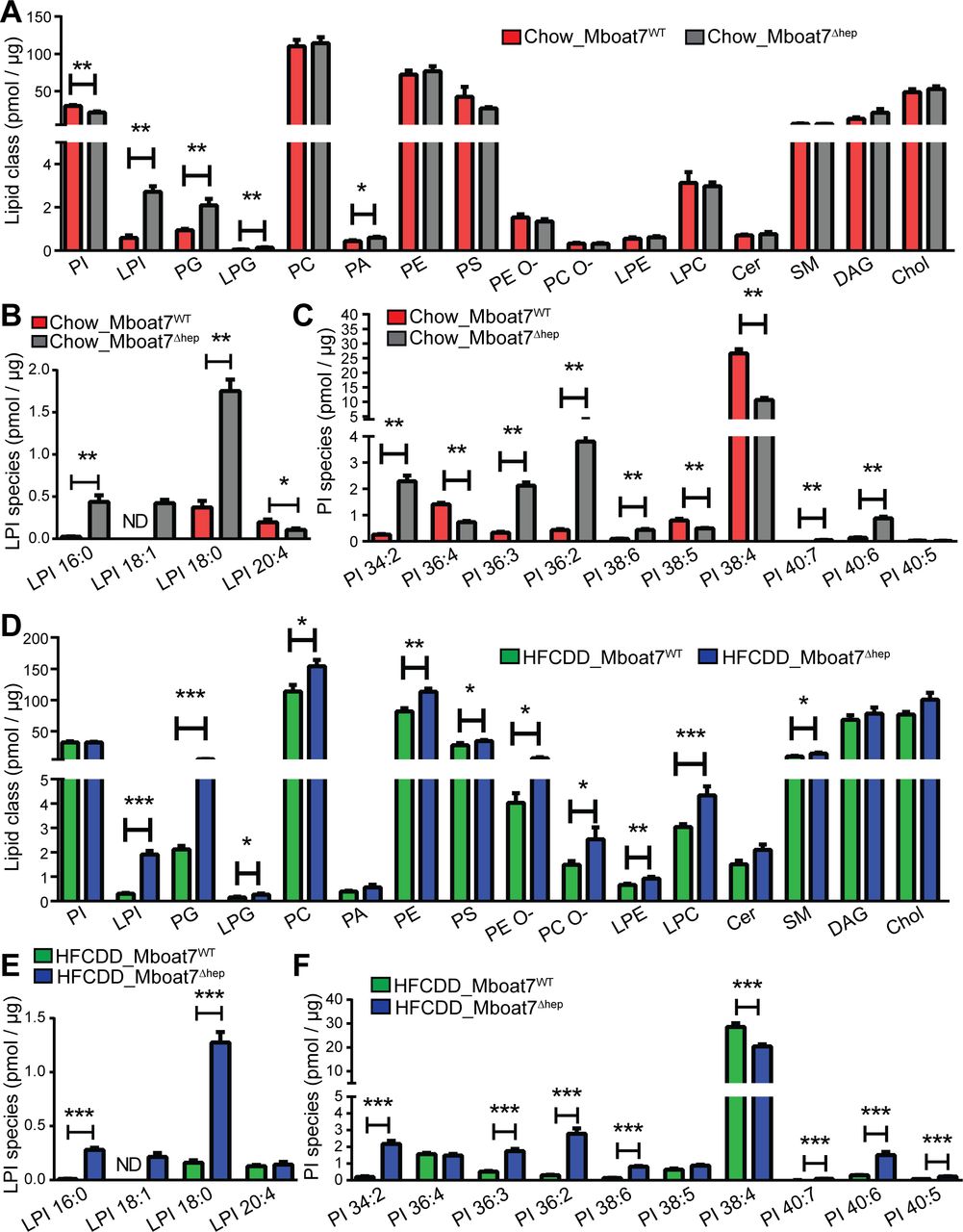
Lipidomic analysis of mouse liver on normal and HFCDD diet. (A–F) data are presented as mean±SEM. (Mann-Whitney U test). *P<0.05, **P<0.01, ***P<0.001. lipidomic analysis of livers on a normal diet (A–C, n=5 mice) and HFCDD diet (D–F, n=7–8 mice). Cer, ceramides; Chol, cholesterol; DAG, diacylglycerol; HFCDD, high-fat, methionine-low, choline-deficient diet; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; LPG, lysophosphatidylglycerol; LPI, lysophosphatidylinositol; MBOAT7, membrane-bound O-acyltransferase domain containing 7; ND, not detected; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; SEM, SE of the mean; SM, sphingomyelin.
In mice fed the HFCDD demonstrating the fibrotic liver phenotype, key phospholipids were also altered (figure 4D–F): Total LPI levels and LPI species LPI 16:0, LPI 18:1 and LPI 18:0 were increased in Mboat7Δhep livers (figure 4D–E). LPI 18:1 was not detected in Mboat7WT livers but was upregulated following hepatocyte-specific Mboat7 deletion (figure 4E). Other phospholipids and lysophospholipids such as PG, LPG, PC, PE, PS, PE O-, PC O-, LPE, LPC and SM were also increased in Mboat7Δhep livers during NAFLD (figure 4D). PI 38:4 was significantly reduced, whereas PI 34:2, PI 36:3, PI 36:2, PI 38:6, PI 40:7, PI 40:6 and PI 40:5 were increased in Mboat7Δhep livers as compared with Mboat7WT livers (figure 4F).
Lipidomic analysis of human liver biopsies
We performed a lipidomic analysis in a cross-sectional liver biopsy cohort (table 1) stratified by the rs641738C>T genotype. Except for cardiolipin, we found no differences in total amounts of lipid classes between CC, CT and TT groups (figure 5A–C). In individuals carrying the rs641738TT risk genotype, profound remodelling of PI species was observed, specifically PI 32:0, PI 32:1, PI 34:1, PI 34:2, PI 36:1, PI 36:2, PI 38:6, PI 40:5 and PI 40:6 were elevated in the TT group compared with the CC group, whereas PI 40:4, PI 36:4 and PI 38:4 were reduced in the TT group (figure 5D,E). The decrease of PI 38:4 in rs641738TT samples was also observed when stratified for disease condition, specifically in NAFL, early NASH and NASH samples (online supplementary figure 9A). Furthermore, LPI 18:0 levels were increased (p=0.002) and LPI 20:4 levels were decreased (p=0.009) in the TT group as compared with the CC group (figure 5F). By stratifying for presence or absence of fibrosis, we found that total LPI levels were positively correlated with rs641738TT genotype only in the presence of fibrosis (figure 5G). Additionally, LPA levels were also correlated with the rs641738TT genotype only in the presence of fibrosis, whereas other lipid classes were unaltered (online supplementary figure 9B–D). In the absence of fibrosis, no alterations were found in the CC as compared with the TT groups (online supplementary figure 9E–G). Changes in molecular composition of PI and LPI species were remarkably similar between humans homozygous for the MBOAT7 rs641738TT risk variant and Mboat7 deficient mice under normal diet and after HFCDD feeding (figure 5H–M). On stratifying for both, the MBOAT7 rs641738C>T genotype and disease condition, we observed significant first broad changes between phenotypic groups in lipid class compositions (online supplementary figure 10A–O). Alterations in lipid classes in the MBOAT7 genotype-specific analysis were observed in the normal control, NAFL, early NASH and NASH groups, whereas no differences were found in the healthy obese group. Consistent with the highest proportion of samples with histological fibrosis, genotype-specific lipid class alterations were most pronounced in the NASH group (online supplementary figure 10M–O). Lipidomic comparison of fibrotic and non-fibrotic NAFL revealed significant genotype-specific alterations in lysolipids LPI and LPA (online supplementary figure 9B–G). Results were robust to exclusion of patients on statin medication (n=14) as shown in online supplementary figure 11A–C.
Supplemental material
Supplemental material
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lipidomic analysis of human liver biopsies stratified by MBOAT7 rs641738C>T genotype. (A–E) lipidomic analysis of human livers (lipidomic cohort) stratified by rs641738C>T genotype (not grouped for a disease condition, age, sex, and BMI). Data are presented as mean±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Patient samples in which a specific lipid class was not detected were excluded from analysis. Alterations in PI and LPI species are presented as mol% (% of total PI and % of total LPI). (A) alterations in hepatic triglyceride (TAG) levels. CC, n=94; CT, n=136; TT, n=50. (B) alterations in hepatic lipid classes. CC, n=94; CT, n=136; TT, n=50. (C) alterations in hepatic lipid classes. CER, ceramide (CC, n=94; CT, n=136; TT, n=50); CL*, cardiolipin (CC, n=55; CT, n=87; TT, n=24); LPA*, lyso-phosphatidate (CC, n=45; CT, n=56; TT, n=11); LPC, lysophosphatidylcholine (CC, n=94; CT, n=136; TT, n=50); LPE, lysophosphatidylethanolamine (CC, n=94; CT, n=136; TT, n=50); LPG*, lysophosphatidylglycerol (CC, n=87; CT, n=123; TT, n=48); LPI*, lysophosphatidylinositol (CC, n=85; CT, n=120; TT, n=48); LPS*, lysophosphatidylserine (CC, n=91; CT, n=132; TT, n=48). *Patient samples in which a specific lipid class was not detected were excluded from analysis. (D) Alterations in PI species presented as mol% (% of total PI). CC, n=94; CT, n=136; TT, n=50. (E) Alterations in PI species presented as mol% (% of total PI). *PI 32:0 (CC, n=68; CT, n=103; TT, n=37) *PI 32:1 (CC, n=54; CT, n=59; TT, n=30) *PI 34:0 (CC, n=87; CT, n=116; TT, n=37); *PI 34:1 (CC, n=92; CT, n=128; TT, n=46); *PI 34:2 (CC, n=93; CT, n=130; TT, n=46); *PI 36:1 (CC, n=93; CT, n=136; TT, n=50); *PI 38:3 (CC, n=3; CT, n=7; TT, n=3); *PI 38:6 (CC, n=92; CT, n=128; TT, n=50); *PI 40:4 (CC, n=34; CT, n=41; TT, n=11); *PI 40:7 (CC, n=27; CT, n=32; TT, n=19). *Patient samples in which a specific lipid species was not detected were excluded from analysis. (F) Alterations in LPI species presented as mol% (% of total LPI). CC, n=10; TT, n=7. (analysis was done on patient liver samples in which 4 LPI species were detected). (G) comparison of total LPI level in NAFLD (include NAFL, early NASH and NASH samples) patient livers with and without the presence of fibrosis. with fibrosis, CC, n=25; with fibrosis, TT, n=17; without fibrosis, CC, n=39; without fibrosis, TT, n=20. NS, not significant. (Patient samples in which no LPI species was detected were excluded). (H–M) Difference of means of common PI and LPI species (mol%) in mice and humans. (H, K) Lipidomic analysis of human livers obtained from people with either the CC or TT genotype (not grouped for a disease condition, age, sex, and BMI), (I, L) livers from Mboat7WT and Mboat7Δhep mice fed a normal diet and (J, M) livers from Mboat7WT and Mboat7Δhep mice fed an HFCDD for 6 weeks. (H, K) Difference of means (mol% values, TT-CC) of PI and LPI species in human livers. (I, L) Difference of means (mol% values) of PI and LPI species in Mboat7WT and Mboat7Δhep mice fed a normal diet (Mboat7Δhep - Mboat7WT). n=5 mice. (J, M) Difference of mean (mol% values) of PI and LPI species in Mboat7WT and Mboat7Δhep mice fed an HFCDD (Mboat7Δhep - Mboat7WT). n=7–8 mice. BMI, body mass index; DAG, diacylglycerol; NAFL, non-alcoholic fatty liver; MBOAT7, membrane-bound O-acyltransferase domain containing 7; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Discussion
NAFLD encompasses a broad phenotypic spectrum from benign steatosis to liver cirrhosis and liver-related death.2 3 The strongest and ultimately sole prognostic factor for liver-related mortality is the presence and progression of fibrosis. Interestingly, fibrosis can occur in up to 30% of patients without relevant inflammation.5 In contrast to its clinical and prognostic relevance, the process of fibrogenesis in the context of NAFLD is poorly understood. Discovery of the MBOAT7 locus, which is associated with a strong fibrotic signature in human NAFLD, alcoholic liver disease and infectious hepatitis,13 14 16 17 raised expectations for uncovering novel mechanisms underlying hepatic fibrogenesis of broad relevance. Here, we provide a combined mouse and human dataset that advances our knowledge pertinent to NAFLD-related fibrosis and at the same time challenges several central paradigms of current NAFLD research.
We demonstrate that a disrupted PI remodelling pathway in hepatocytes is associated with phenotypes on standard and a NASH-inducing diet: Mboat7Δhep mice developed spontaneous steatosis on normal diet. Steatosis was characterised by increased Chol esters while TAGs were unchanged. The lipogenic enzyme, Scd1 (SCD1, stearoyl-CoA desaturase) was upregulated and several fatty acid oxidation genes were downregulated in Mboat7 deficient livers at steady state. The major product of SCD1 is oleic acid (18:1) which is the preferred substrate for acyl-CoA:cholesterol acyltransferase, the Chol esterification enzyme.34 35 The reduced fatty-acid oxidation and increased SCD1 may at least partly explain the increased CE levels and spontaneous steatosis seen in Mboat7-deficient livers, however, further investigation is required to completely understand the mechanisms involved. Similar to the current study, it was shown recently that silencing Mboat7 in mice using antisense oligonucleotide conjugated with morpholinos results in spontaneous lipid accumulation in the liver; however, the study concludes that this was due to increased TAG accumulation although CE levels were not measured in their samples.36 Another study, in which antisense oligonucleotides were used to knock down Mboat7 in adipose tissue, liver and cells of the reticuloendothelial system reported no spontaneous phenotype.20 However, following high-fat diet (HFD) feeding, these mice exhibited elevated levels of TAGs and CEs in the liver.20 The phenotypic differences seen between these previous studies and the current study might be explained by the broader knock-down of Mboat7 using the antisense oligonucleotide approach20 36
Disturbed lipid metabolism in hepatocytes is sufficient to drive liver fibrosis
After HFCDD feeding, Mboat7Δhep mice developed increased hepatic-fibrosis as measured by picrosirius staining and relative hepatic hydroxyproline content and transcriptomic analysis revealed a broad upregulation of ECM genes. Thus, disturbance of the PI side chain remodelling pathway in hepatocytes elicits fibrosis on an induction diet—that is, hepatocytes themselves may serve as a pathogenic driver of fibrosis.
Mboat7-deficiency elicits liver fibrosis in the absence of inflammation
We found that, the MBOAT7 risk variant was associated with hepatic fibrosis independent of the presence of inflammation. This represents an intriguing and novel mechanism for liver fibrosis, suggesting that hepatocyte signalling in the fibrogenic mesenchymal cell compartment, that is, primarily myofibroblasts/hepatic stellate cells, is sufficient to induce fibrosis without an increased inflammatory infiltrate. Mancina et al have shown that MBOAT7 rs641738C>T is associated with more necroinflammation.14 In mice, Helsley et al have shown that HFD feeding of Mboat7 deficient mice results in reduced hepatic-accumulation of total macrophages and M2 macrophages, whereas, CD8+T cells and M1 macrophages were increased.20 Differences in the inflammatory cell accumulation seen between the current study and the study by Helsley et al may be due to the strategy they used for Mboat7 deletion (antisense oligonucleotide) which is known to silence Mboat7 in adipose tissue, liver and cells of the reticuloendothelial system, which includes macrophages; therefore, these changes in inflammatory cells could derive from the loss of macrophage-specific Mboat7 function.20 Moreover, in the study by Helsley et al, mice were treated with the antisense oligonucleotide to silence Mboat7 concurrently during HFD feeding, therefore, Mboat7 was not silenced before the start of the feeding.
FFA and oxylipins are unchanged in the fibrotic mouse phenotype
Since Mboat7 exhibits LPI acyltransferase activity towards arachidonoyl-CoA,8 deletion of Mboat7 could lead to elevated levels of free arachidonic acid and the generation of its proinflammatory lipid metabolites. However, we found no significant increase in FFA and oxylipins in Mboat7Δhep mice, suggesting that these lipid mediators and arachidonic acid-derived metabolites do not contribute to fibrosis development in this model. These findings are concordant to a recent study that has shown that arachidonic acid-derived proinflammatory lipid mediators were not altered in the livers of Mboat7 deficient mice.20 37 Because FFA and fatty acid metabolites are known proinflammatory mediators38–40 this finding is consistent with the lack of changes in the lobular inflammatory compartment.
Resemblance of human NAFLD and the mouse model
Interestingly, the lipidomic changes associated with hepatocyte-specific Mboat7 deficiency closely resemble the lipidomic alterations in humans with the MBOAT7 risk variant and may thus represent a promising model to gain further insights into the patients with NAFLD fibrosis, although all NALFD patients in this study were recruited from bariatric surgery. Furthermore, murine and human data are in agreement with regard to the genetic association of MBOAT7 genotype with fibrosis and inflammation: We show that the correlation between the rs641738C>T variant and hepatic-fibrosis was indeed independent of inflammation in our biopsied NAFLD patients with a BMI≤35.
Novel lipid signalling targets in NAFLD
Mboat7 functions in the Lands’ cycle, where it adds arachidonic acid (20:4) to 1-stearoyl (18:0) LPI thereby generating sn-1-stearoyl-sn-2-arachidonoyl PI species,9 which in turn might lead to increased LPI levels and an altered PI spectrum if Mboat7 is absent in liver. In the absence of Mboat7, alterations in PI species have been reported in mice, humans and in cultured hepatocytes, previously.14 18 19 36 We confirm and extend these findings through broader and liver-specific lipidomics in mice and humans and observe characteristic genotype-specific alterations in lysolipid patterns between fibrotic and non-fibrotic NAFL (online supplementary figure 9B–G, figure 5G). Intriguingly, the lipidomic patterns in mouse Mboat7Δhep livers and humans carrying the rs641738TT variant showed striking similarities. We demonstrate a profound remodelling of PI with a reduction of PI species containing arachidonoyl side chains and increase of other PI species particularly with monounsaturated fatty acids. Increased LPI levels and particularly LPI 18:0 correlated with the presence of fibrosis in the human rs641738TT genotype and in the murine fibrotic phenotype. LPI is exported from cells via ABCC1,41–43 thereby rendering it a candidate profibrotic lipid mediator, which might directly interact with the stellate cell compartment. Circulating LPI levels are significantly increased in patients with liver fibrosis as compared with healthy people20 and treating obese mice with exogenous LPI 18:1 promotes pro-fibrotic gene expression in the liver.20 LPI might thus play a signalling role in fibrosis development.
In summary, our combined murine and human data provide novel insights into NAFLD pathogenesis (online supplementary figure 12). Further mechanistic studies of the new candidate lipid mediators and their effects on other hepatic cell populations are required to provide a deeper understanding of these pathways and to transform them into viable target molecules.
Supplemental material
Data availability statement
Data are available on reasonable request. Not applicable.
Ethics statements
Ethics approval
Subjects recruited for the genetic association study came from different sites, at which the study protocol was approved by the ethics committees of the participating institutions.
Acknowledgments
The authors wish to thank all study participants, researchers, clinicians, technicians and administrative staff who contributed to this study.
References
Footnotes
JH and PS are joint senior authors.
Contributors Project was conceived and experiments were conceptualised by: VRT, PS, MB, EK, TC, FS, MMS, and JH. Experiments were performed by VRT, PS, OK, EP, OV, KH, MN, SN, AD, DRR and MVB. Patient sample collection and coordination was done by CS, WVS, SH, AH, TB, EA, CD and CR. Data analysis was done by VRT, PS, SB, AH, JAHW, JKP and JMA. Manuscript was written and edited by VRT, PS, JH, TC, AS, SZ and FS.
Funding This study was supported by the German Ministry of Research and Education (BmBF) through the Liver Systems Medicine (LiSyM) network grant (to JH and AS) and ERACOSysMed (Dynaflow) grant (to JH), an ERC grant (DEMETINL to TC) and Lipidomics and Informatics for Life Sciences (LIFS) grant Unit of the de. NBI Consortium (AS). Additional support came from the Swiss National Funds (grants 310030_138747/1 and 310030_169196 to F.S.) Contributions by JP were partly funded by the Bavarian State Ministry of Science and the Arts in the framework of the Centre Digitisation. Bavaria (ZDB).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note The transciptomic data is available at GEO under id GSE139992