Article Text

Abstract
Objective Non-alcoholic fatty liver disease (NAFLD)-associated hepatocellular carcinoma (HCC) is an increasing healthcare burden worldwide. We examined the role of dietary cholesterol in driving NAFLD–HCC through modulating gut microbiota and its metabolites.
Design High-fat/high-cholesterol (HFHC), high-fat/low-cholesterol or normal chow diet was fed to C57BL/6 male littermates for 14 months. Cholesterol-lowering drug atorvastatin was administered to HFHC-fed mice. Germ-free mice were transplanted with stools from mice fed different diets to determine the direct role of cholesterol modulated-microbiota in NAFLD–HCC. Gut microbiota was analysed by 16S rRNA sequencing and serum metabolites by liquid chromatography–mass spectrometry (LC–MS) metabolomic analysis. Faecal microbial compositions were examined in 59 hypercholesterolemia patients and 39 healthy controls.
Results High dietary cholesterol led to the sequential progression of steatosis, steatohepatitis, fibrosis and eventually HCC in mice, concomitant with insulin resistance. Cholesterol-induced NAFLD–HCC formation was associated with gut microbiota dysbiosis. The microbiota composition clustered distinctly along stages of steatosis, steatohepatitis and HCC. Mucispirillum, Desulfovibrio, Anaerotruncus and Desulfovibrionaceae increased sequentially; while Bifidobacterium and Bacteroides were depleted in HFHC-fed mice, which was corroborated in human hypercholesteremia patients. Dietary cholesterol induced gut bacterial metabolites alteration including increased taurocholic acid and decreased 3-indolepropionic acid. Germ-free mice gavaged with stools from mice fed HFHC manifested hepatic lipid accumulation, inflammation and cell proliferation. Moreover, atorvastatin restored cholesterol-induced gut microbiota dysbiosis and completely prevented NAFLD–HCC development.
Conclusions Dietary cholesterol drives NAFLD–HCC formation by inducing alteration of gut microbiota and metabolites in mice. Cholesterol inhibitory therapy and gut microbiota manipulation may be effective strategies for NAFLD–HCC prevention.
- dietary factors
- fatty liver
- nonalcoholic steatohepatitis
- intestinal microbiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic fatty liver disease (NAFLD)-associated hepatocellular carcinoma (HCC) is an increasing healthcare burden worldwide.
Cholesterol is a major lipotoxic molecule.
What are the new findings?
High-fat high-cholesterol diet (HFHC) spontaneously and sequentially induced fatty liver, steatohepatitis, fibrosis and NAFLD–HCC development, while high-fat low-cholesterol diet induced only hepatic steatosis in male mice.
Dietary cholesterol-induced NAFLD–HCC formation was associated with gut microbiota dysbiosis. The microbiota composition changed along stages of NAFLD–HCC formation: Mucispirillum, Desulfovibrio, Anaerotruncus and Desulfovibrionaceae were sequentially increased; while Bifidobacterium and Bacteroides were depleted in HFHC-fed mice, which was corroborated in human hypercholesteremia patients.
Gut bacterial metabolites alteration including increased serum taurocholic acid (TCA) and depleted 3-indolepropionic acid (IPA) was found in NAFLD–HCC. IPA inhibited cholesterol-induced lipid accumulation and cell proliferation, while TCA aggravated cholesterol-induced triglyceride accumulation in human normal immortalised hepatocyte cell line.
Germ-free mice gavaged with stools from HFHC-fed mice manifested hepatic lipid accumulation, inflammation and cell proliferation, in consistence with donor mice phenomes.
Anticholesterol treatment restored dietary cholesterol-induced gut microbiota dysbiosis and completely prevented NAFLD–HCC formation.
How might it impact on clinical practice in the foreseeable future?
Our findings indicate that anticholesterol drug and manipulation of the gut microbiota might represent effective strategies in preventing NAFLD–HCC.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome, which encompasses a spectrum of liver pathologies that range from simple steatosis to non-alcoholic steatohepatitis (NASH).1 NASH can progress to hepatic cirrhosis, end-stage liver failure and hepatocellular carcinoma (HCC).2 Currently, NAFLD is a major cause of morbidity and a healthcare burden worldwide. Large population-based cohort studies demonstrate that the prevalence of NAFLD–HCC has increased by fourfold in the last decade compared with 2.5-fold for hepatitis, making it the most rapidly growing indication for liver transplantation.3 Lipotoxicity drives the progression of NASH, fibrosis/cirrhosis, and even HCC.4 Among hepatic lipid species, cholesterol is considered a major lipotoxic molecule in NASH development.4 The liver is central to the regulation of systemic cholesterol homeostasis. Abnormalities in hepatic cholesterol homeostasis have been demonstrated in both human and experimental models of NASH.5 6 We have revealed that squalene epoxidase, a rate-limiting enzyme in cholesterol biosynthesis, drives NAFLD–HCC development.7 Dietary cholesterol has an important impact on plasma and hepatic cholesterol homeostasis.5 Although the contribution of dietary cholesterol to NASH progression has been reported,5 8 the role and pathogenic basis of long-term cholesterol treatment on spontaneous and progressive NAFLD–HCC development are unknown.
The intestinal microbiota has a symbiotic relationship with its host and contributes nutrients and energy by metabolising dietary components including cholesterol in the large intestine.9 Several studies have suggested that the gut microbiome represents an environmental factor contributing to the development of NAFLD and its progression to NAFLD–HCC.10 ,11 Microbe-derived metabolites, such as bile acid, short-chain fatty acids and trimethylamine and the signalling pathways they affect may contribute to NAFLD development.12 13 In particular, 3-(4-hydroxyphenyl) lactate, a metabolite derived from gut microbiome, has a shared gene‐effect with both hepatic steatosis and fibrosis.14 Moreover, the alteration of gut microbiota profile by dietary cholesterol has been reported.15 However, whether gut microbiota dysbiosis is the cause or effect of dietary cholesterol-induced NASH and NAFLD–HCC progression remains unclear.
The present study was performed to determine the role and the associated molecular mechanisms of dietary cholesterol in the development of NAFLD–HCC. We found that dietary cholesterol caused spontaneous NAFLD–HCC formation by cholesterol-induced gut microbiota changes and metabolomic alterations. Cholesterol inhibition restored gut microbiota and completely prevented NAFLD–HCC development.
Materials and methods
Animals and diets
Male C57BL/6 wild-type littermates (8 weeks old) were fed with normal chow (NC, 18% fat, 58% carbohydrate, 24% protein, 0% cholesterol), high-fat/low-cholesterol diet (HFLC, 43.7% fat, 36.6% carbohydrate, 19.7% protein, 0.013% cholesterol) or high-fat/high-cholesterol diet (HFHC, 43.7% fat, 36.6% carbohydrate, 19.7% protein, 0.203% cholesterol) (Specialty Feeds, Glen Forrest, WA) ad libitum for 14 months. Mice were also fed with HFLC or HFHC for 3, 8,10 and 12 months (n=8–19 per group). In additional experiments, atorvastatin (20 mg/kg) was administered to HFHC-fed mice after 7 months of diet initiation and continued for additional 7 months (n=10 per group). Mice were kept on a 12/12 hour light/dark cycle. At experimental end points, mice were fasted and serum/tissues were harvested.16 Body weights and visceral fat weights were recorded. Livers were rapidly excised and weighed. The presence and dimensions of surface nodules were evaluated. HCCs were confirmed histologically from both grossly and histologically evident nodules. Liver tumours were isolated, snap frozen in liquid nitrogen and stored at −80°C for further experiments. Mice stool samples were collected for bacterial 16S rRNA gene sequencing and oral gavage.
C57BL/6 male germ-free mice (7 weeks) were bred at the Department of Laboratory Animal Science at the Third Military Medical University in Chongqing, China. Adult mice (8 weeks old) were divided into three groups (11–28 mice per group), and gavaged twice at 0 and 7 months with stools obtained from conventional mice fed with NC, HFLC, HFHC or HFHC mice treated with atorvastatin for 14 months. Briefly, 1 g of stool samples were homogenised in 5 mL of phosphate buffered saline (PBS). Recipient mice were then transplanted with 200 μL of the suspension by gastric gavage. Mice from each group were randomly selected and sacrificed at 8, 10 or 14 months following transplantation.
Additional methods are provided in online supplementary file 1.
Supplemental material
Results
Dietary cholesterol drives the development of NAFLD–HCC spontaneously
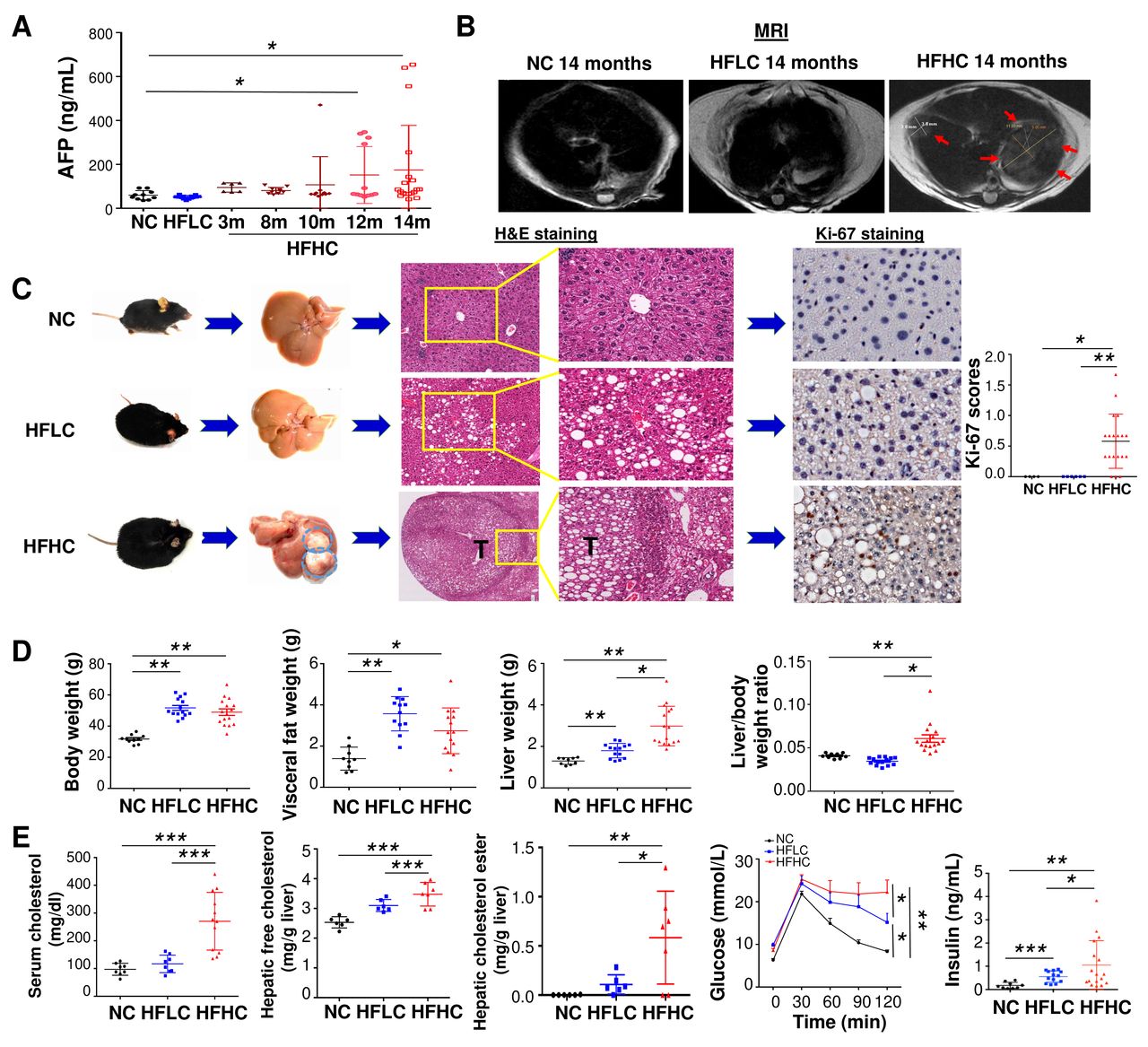
To examine the role of dietary cholesterol in the evolution of steatosis, NASH, fibrosis and subsequent NAFLD–HCC, mice were fed with HFHC,17 HFLC or NC. Serum level of alpha-fetoprotein (AFP, marker for liver cancer) was monitored at different time points (3, 8, 10, 12 and 14 months). An elevated AFP level was observed at month 10 (107.1±127.9 ng/mL), which was further increased at month 12 (151.6±129.3 ng/mL) and month 14 (174.2±203.1 ng/mL) in mice fed with HFHC compared with HFLC (50.9±7.5 ng/mL in HFLC) or NC (59.7±20.9 ng/mL) at month 14 (figure 1A). MRI scan showed liver tumours in HFHC-fed mice but not in HFLC-fed or NC-fed mice at month 14 (figure 1B). Mice were then harvested at month 14. Liver tumours were identified in 68% (13/19) of mice fed with HFHC but not in mice fed with HFLC or NC diet (figure 1C). Histological examination of liver sections confirmed that all liver tumours were HCCs (figure 1C) with an average number of 2.7±2.6 HCCs per mouse and maximal tumour diameter of 4.1±5.0 mm. The liver sections of HFHC-fed mice have significantly more Ki-67 positive cells compared with HFLC-fed mice, indicating increased cell proliferation in HFHC-fed mice (figure 1C). These results demonstrate that dietary cholesterol can spontaneously induce NAFLD–HCC formation.

Cholesterol induces spontaneous HCC formation in C57BL/6 mice. (A) Serum AFP levels in mice fed with HFHC diet for 3, 8, 10, 12 and 14 months as well as mice fed with NC or HFLC diet for 14 months; (B) liver MRI, (C) representative gross morphology, representative microscopic features and immunohistochemistry pictures of Ki67 staining in liver of mice fed with NC, HFLC and HFHC for 14 months, Ki-67 was scored according to the following criteria: 0 (<10% of cells staining), 1 (11%–30% of cells staining), 2 (30%–50% of cells staining) or 3 (>50% of cells staining); (D) body weight, visceral fat, liver weight, liver-to-body weight ratio, (E) serum cholesterol level, hepatic free cholesterol, hepatic cholesterol ester content, glucose tolerance test and fasting insulin levels in mice fed with NC, HFLC and HFHC for 14 months. *p<0.05, **p<0.01, ***p<0.001. AFP, alpha-fetoprotein; NC, normal chow; HFLC, high-fat/low-cholesterol diet; HFHC, high-fat/high-cholesterol diet; H&E, hematoxylin and eosin.
Along with HCC formation, mice fed with HFHC showed significantly increased body weight, visceral fat, liver weight and liver-to-body weight ratio compared with NC-fed mice at month 14 (figure 1D). HFLC-fed mice also displayed enhanced body weight, visceral fat and liver weight (figure 1D). Significantly increased serum cholesterol, hepatic free cholesterol and cholesterol ester, glucose intolerance and fasting insulin were demonstrated in HFHC-fed mice compared with HFLC-fed or NC-fed mice (figure 1E). Enhanced glucose intolerance and fasting insulin were also observed in mice fed with HFLC compared with NC (figure 1E).
NASH and fibrosis are present in mice fed with high-cholesterol diet for 14 months
Further examination of liver sections revealed the presence of steatohepatitis characterised by steatosis and lobular inflammation in HCC adjacent liver tissues and non-HCC liver tissues of mice fed with HFHC for 14 months, while only steatosis was observed in HFLC-fed mice (figure 2A, online supplementary figure S1). Consistent with histological inflammation, serum alanine aminotransferase (ALT) (p<0.01) and aspartate aminotransferase (AST) (p<0.01) levels were significantly increased in HFHC-fed mice compared with mice fed with HFLC or NC (figure 2B). Serum and hepatic proinflammatory cytokines including IL-6, IL-1α and IL-1β (figure 2C,D) and proinflammatory factors (online supplementary figure S2A-B) are increased in HFHC-fed mice as measured by cytokine profiling assay and ELISA. The crucial NASH-related proinflammatory cytokines, including Cx3cl1, Mcp1, Cxcl10, Mip1β, Mip1α, Ccl5, Cxcl16 and Tnfα, are significantly upregulated in the liver tissues of 14 months HFHC-fed mice compared with HFLC-fed mice as indicated by RNA sequencing analysis (figure 2D). HFHC-fed mice presented severe fibrotic injury with significantly more collagen distribution areas (figure 2E), collagen content by hepatic hydroxyproline assay (figure 2F) and hepatic stellate cells activation as evidenced by increased alpha-smooth muscle actin (α-SMA) mRNA and protein levels (figure 2E). Examination of hepatic oxidative stress revealed that oxidised nicotinamide adenine dinucleotide (NAD+) to NADH (reduced form of NAD)ratio and antioxidant superoxide dismutase (SOD) activity were significantly decreased in HFHC-fed mice (figure 2G), suggesting the induction of hepatic oxidative stress by dietary cholesterol. Collectively, these findings indicate that NASH and fibrosis were formed in non-HCC liver tissues of HFHC-fed mice.
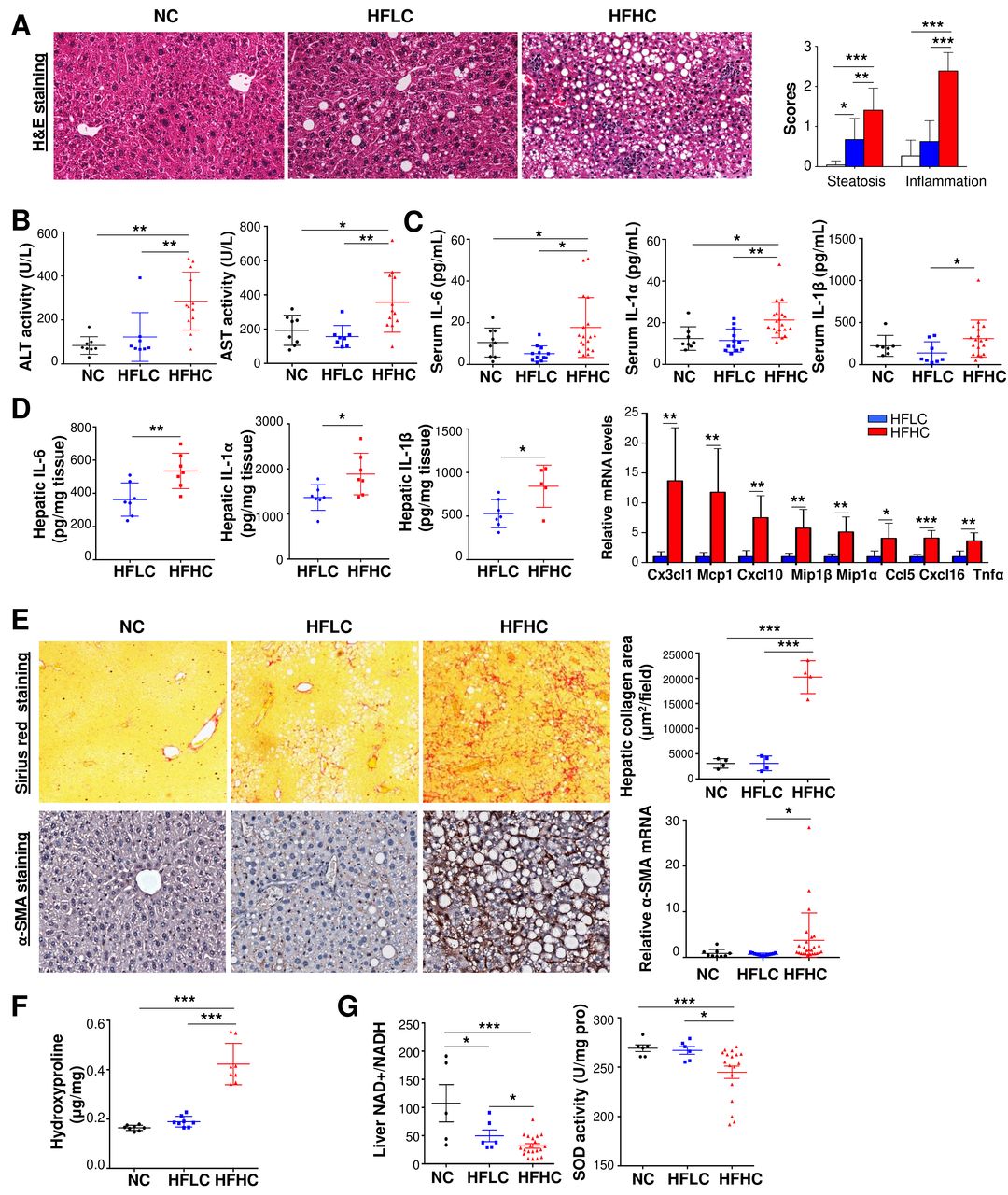
Cholesterol causes NASH and fibrosis in non-HCC liver tissues of mice fed with HFHC diet. (A) Representative H&E staining, histological scores of liver sections; (B) serum ALT and AST levels; (C) serum IL-6, IL-1α and IL-1β protein levels by cytokine profiling assay in mice fed with NC, HFLC and HFHC for 14 months; (D) hepatic proinflammatory cytokines IL-6, IL-1α and IL-1β protein levels by ELISA and Cx3cl1, Mcp1, Cxcl10, Mip1β, Mip1α, Ccl5, Cxcl16, Tnfα mRNA levels by RNA sequencing in mice fed with HFLC and HFHC for 14 months; (E) collagen deposition by Sirius Red staining, α-SMA protein and mRNA levels by immunohistochemistry staining and RT-PCR, respectively; (F) hepatic hydroxyproline content, (G) liver NAD+ to NADH ratio and SOD activity in mice fed with NC, HFLC and HFHC for 14 months. *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; α-SMA, alpha-smooth muscle actin; HCC, hepatocellular carcinoma; HFHC, high-fat/high-cholesterol diet; HFLC, high-fat/low-cholesterol diet; H&E, Hematoxylin and eosin; NAD, nicotinamide adenine dinucleotide; NASH, non-alcoholic steatohepatitis; NC, normal chow; RT-PCR, reverse transcription polymerase chain reaction; SOD, superoxide dismutase.
High-cholesterol diet causes fatty liver, steatohepatitis and fibrosis sequentially in mice
To clarify the progression of NAFLD prior to HCC formation by dietary cholesterol, we monitored serum ALT, AST, cholesterol, AFP and liver histological changes in mice fed with HFHC. Mice were harvested at 3, 8, 10 and 12 months following diet feeding (figure 3A). We observed increases in body weight and visceral fat weight (online supplementary figure S3A), accompanied with increased liver weight and liver-to-body weight ratio in mice fed with HFHC compared with mice fed with HFLC at 3 and 8 months (online supplementary figure S3B). The increased profiles of serum ALT and AST (online supplementary figure S3C) were consistent with enhanced serum cholesterol levels in HFHC-fed mice (online supplementary figure S3D). Liver histology of HFHC-fed mice showed steatosis with mild inflammation at 3 months, steatohepatitis with fibrosis at 8 months and HCC formation at 10, 12 and 14 months, while HFLC-fed mice showed only steatosis without further HCC development at 3, 8, 10 and 14 months (figure 3B and online supplementary figure S4A-B). We also performed immunohistochemistry for two HCC markers AFP and Golgi protein 73 (GP73). Positive staining was observed on liver tissues from HFHC-fed mice but not from HFLC-fed mice (figure 3B and online supplementary figure S4A). Scores of hepatic steatosis, and lobular inflammation and hepatic collagen areas confirmed the increased severity of liver histology across the disease stages (figure 3B). Moreover, 10% (1/10) of HFHC-fed mice developed HCC at 10 months (tumour number=0.1 ± 0.32, maximum tumour diameter=0.55 ± 1.74 mm), 25% (3/12) developed HCC at 12 months (tumour number=0.25 ± 0.45, maximum tumour diameter=1.57 ± 3.10 mm) and 68% (13/19) developed HCC at 14 months (figure 3C), indicating that HFHC-fed mice progressively developed steatosis, steatohepatitis, fibrosis and NAFLD–HCC.

HFHC-fed mice sequentially develop a fatty liver, steatohepatitis, fibrosis and HCC. (A) Schematic illustration of the treatment of C57BL/6 mice with HFHC diet. Mice were sacrificed at 3, 8, 10, and 12 months of age. (B) Representative gross morphology, H&E staining and IHC staining of tumour markers AFP and GP73 in the liver of mice fed with HFLC for 14 months and HFHC for 3, 8, 10, 12 and 14 months; histological scoring of steatosis and inflammation and the quantitation of Sirius Red staining were calculated. (C) Tumour incidence, tumour number and maximum tumour diameter of the largest tumour from mice fed with HFHC at different time points. *p<0.05, **p<0.01, ***p<0.001. AFP, alpha-fetoprotein; HCC, hepatocellular carcinoma; HFHC, high-fat/high-cholesterol diet; HFLC, high-fat/low-cholesterol diet; H&E, hematoxylin and eosin; IHC, immunohistochemistry.
Dietary cholesterol-induced gut microbiota dysbiosis in the initiation and progression of NAFLD–HCC
In order to explore the potential involvement of the gut microbiota in mediating dietary cholesterol-induced NAFLD–HCC, we performed 16S rRNA gene sequencing on stools from HFLC-fed and HFHC-fed mice at 14 months. Rarefaction analysis shows that observed number of operational taxonomic units (OTUs) reached saturation (online supplementary figure S5A-B). Gut microbiota compositional discrimination, by weighted Unifrac principal component analysis (PCA), was observed between HFLC-fed and HFHC-fed mice at 14 months (p<0.001) (figure 4A). Additionally, lower bacterial diversity and increased bacterial richness were observed in HFHC-fed mice with HCC than HFLC diet-fed mice with only simple steatosis at 14 months (figure 4A). Cholesterol associated bacterial taxa were determined by differential abundance analysis. Several bacterial OTUs were differentially abundant in mice fed with HFHC compared with HFLC-fed mice (figure 4B, online supplementary tables S1-5). Principal component and redundancy analyses also showed that the microbiota composition clustered distinctly for mice fed with HFHC for 3 months (simple steatosis with mild inflammation), 8 months (steatohepatitis with fibrosis) and 14 months (HCC) (figure 4C) indicating compositional changes in gut microbiota along stages of NAFLD–HCC progression. Moreover, bacterial richness was sequentially increased with NAFLD–HCC progression (figure 4D). In particular, Mucispirillum schaedleri_Otu038, Desulfovibrio_Otu047, Anaerotruncus_Otu107 and Desulfovibrionaceae_Otu073 were observed to have sequentially increased from 3 to 8, and to 14 months of HFHC feeding (figure 4D). Also, among others, we observed the enrichment of Clostridium OTUs such as Clostridium celatum_Otu070, C. ruminantium_Otu059, C. cocelatum_Otu036 and C. methylpentosum_Otu053, and the depletion of Bifidobacterium_Otu026, Akkermansia municiphila_Otu034, Lactobacillus_Otu009, Bacteroides acidifaciens_Otu032, Bacteroides_Otu012, B. uniformis_Otu080 and B. eggerthii_Otu079 with high dietary cholesterol. Moreover, HFHC-enriched Helicobacter ganmanii_Otu031 was more abundant in HFHC-fed mice with tumour compared with HFHC-fed mice without tumour, while HFHC-depleted Bacteroides_Otu012 was reduced in HFHC-fed mice with tumour compared with HFHC-fed mice without tumour (online supplementary figure S5C). These suggest that further enrichment of Helicobacter ganmanii_Otu031 and depletion of Bacteroides_Otu012 may be important for the role of gut microbiota in NAFLD–HCC. In addition, gut microbiota associated tryptophan metabolising capacity was reduced in HFHC-fed mice compared with HFLC-fed mice (figure 4E). These, taken together, suggest that high-cholesterol diet-induced gut microbiota dysbiosis and impaired microbial tryptophan metabolism.

Dietary cholesterol-induced gut microbiota dysbiosis. (A) Principal component ordination analysis (PcoA), Shannon diversity and the richness of gut microbiota between 14 months HFLC and HFHC-fed mice. (B) Heatmap plot of the bacteria in stool of mice fed with HFLC or HFHC for 14 months. (C) PCoA and redundancy analysis of gut microbiota in fed with HFHC for 3 months, 8 months and 14 months, respectively. (D) The microbiota richness measured by chao1 index and sequentially increased bacteria from 3, 8 to 14 months of HFHC feeding. (E) Gut microbiota tryptophan metabolism capacity in HFLC-fed and HFHC-fed mice. (F) Correlation of bacterial abundance with mice phenomes. (G) The association of bacteria by metagenomic sequencing and serum total cholesterol, triglyceride, LDL-cholesterol and HDL cholesterol in 59 human cases of hypercholesterolemia and 39 healthy subjects. *p<0.05, **p<0.01, ***p<0.001. AFP, alpha-fetoprotein; ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL, high-density lipoprotein; HFHC, high-fat/high-cholesterol diet; HFLC, high-fat/low-cholesterol diet; LDL, low-density lipoprotein; TCHO, total cholesterol; TG, triglyceride.
Correlation analysis was performed to determine the potential association of bacterial abundance with mice phenomes. We observed that M. schaedleri_Otu038, Desulfovibrio_Otu047, Anaerotruncus_Otu107, C celatum_Otu070, C. cocelatum_Otu036 and C. methylpentosum_Otu053 which were enriched in faecal samples of HFHC-fed mice were positively correlated (figure 4F) while Bifidobacterium_Otu026, B. acidifaciens_Otu032, B. uniformis_Otu080, A. municiphila_Otu034 and Lactobacillus_Otu009, which were depleted in HFHC-fed mice, were negatively correlated with high-cholesterol diet, serum and liver cholesterol levels (figure 4F). These results suggest that gut microbiota dysbiosis in NAFLD–HCC correlates with cholesterol levels.
To corroborate our findings from animal experiments in human patients, we analysed the correlation of serum cholesterol and gut microbiota in 59 cases of hypercholesterolemia and 39 healthy subjects. The characteristics of these 98 subjects are shown in online supplementary table S6. Bifidobacterium and Bacteroides were negatively correlated with serum total cholesterol and low-density lipoprotein (LDL)-cholesterol but positively correlated with high-density lipoprotein (HDL)-cholesterol (figure 4G). These results were consistent with observations in HFHC-fed mice (online supplementary tables S1-5), further inferring the involvement of the gut microbiota in contributing to cholesterol-induced disorder.
Dietary cholesterol promotes NASH–HCC progression by inducing metabolites alteration
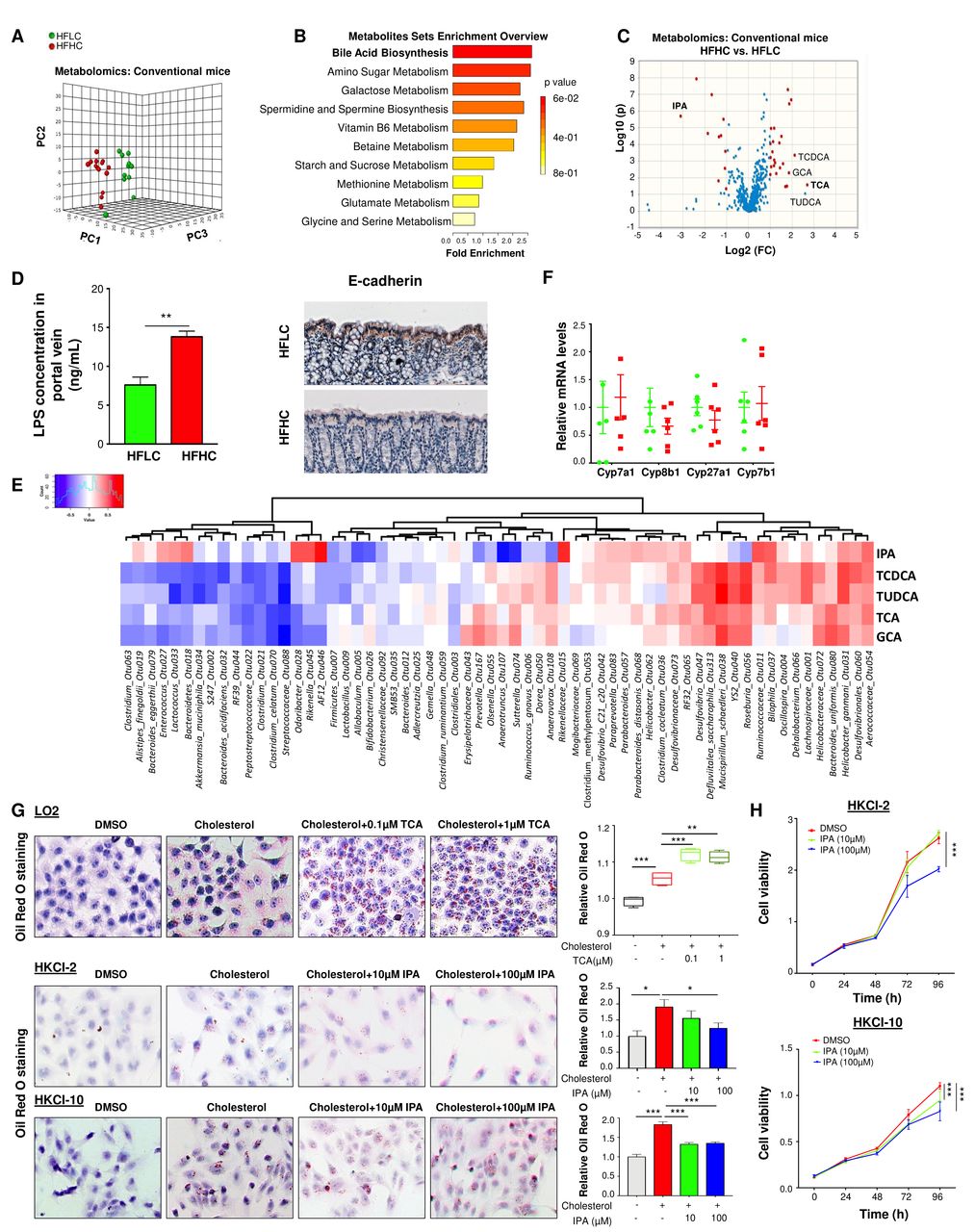
To reveal metabolic phenotypes, related to the gut microbiome, that are potentially involved in cholesterol-induced NAFLD–HCC, we performed metabolic profiling of the serum from HFHC-fed and HFLC-fed mice. Serum metabolites were significantly different according to dietary cholesterol content by PCA (figure 5A and online supplementary table S7). Bile acid biosynthesis was a key pathway altered in mice fed with high-cholesterol diet (figure 5B). Primary bile acids including taurocholic acid (TCA), tauroursodeoxycholic acid (TUDCA), glycocholic acid (GCA) and taurochenodeoxycholic acid (TCDCA) were outlier upregulated metabolites in HFHC-fed mice (figure 5C). On the other hand, 3-indolepropionic acid (IPA), which is a product of microbial tryptophan metabolism,18 was an outlier down-regulated metabolite in HFHC-fed mice (figure 5C). Moreover, serum lipopolysaccharides (LPS) concentration in portal vein was elevated and loss of colonic E-cadherin was identified in HFHC-fed mice compared with HFLC-fed mice (figure 5D), suggesting that dietary cholesterol could impair intestinal barrier function. Correlation analysis was performed to determine the potential association of the HFHC-altered microbes and metabolites. Consistently, correlation analysis revealed that HFHC-enriched Mucispirillum schaedleri_Otu038 is positively correlated with TUDCA, TCDCA, TCA and GCA. HFHC-enriched Roseburia_Otu056 and Helicobacter_ganmanii_Otu031 are also positively associated with TUDCA and TCDCA. Moreover, HFHC-depleted Akkermancia_muciniphila_Otu034 is negatively associated with TCDCA and TUDCA while HFHC-enriched Anaerotruncus_Otu107 is negatively correlated with IPA (figure 5E).

Dietary cholesterol-induced alteration of metabolic profiles in mice serum. (A) Serum metabolites were significantly different between mice fed with HFLC and HFHC diet by principal component ordination analysis. (B) Pathway analysis of differentially enriched metabolites in mice fed with HFHC diet. (C) Volcano plot of serum metabolomics of mice fed with HFLC and HFHC diet, the outliers of metabolites are indicated. (D) LPS concentration in portal vein blood of mice fed with HFLC and HFHC diet for 3 months and E-cadherin expression in the colon tissues of 14 months HFLC and HFHC diet fed mice. (E) Correlation analysis of the association of the HFHC-altered microbes and metabolites. (F) mRNA levels of Cyp7a1, Cyp8b1, Cyp27a1 and Cyp7b1 in the liver tissues of mice fed with HFLC and HFHC diet. (G) TCA aggravated cholesterol-induced triglyceride accumulation in human LO2 cell line while IPA inhibited cholesterol-induced triglyceride accumulation in NASH–HCC cell lines, HKCI-2 and HKCI-10 by Oil Red O staining. Cholesterol, 200 μg/mL; (H) IPA inhibited cell proliferation in NASH–HCC cell lines. *p<0.05, **p<0.01, ***p<0.001. DMSO, dimethyl sulfoxide; GCA, glycocholic acid; HCC, hepatocellular carcinoma; HFHC, high-fat/high-cholesterol diet; HFLC, high-fat/low-cholesterol diet; IPA, 3-indolepropionic acid; NASH, non-alcoholic steatohepatitis; LPS, lipopolysaccharides; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; TUDCA, tauroursodeoxycholic acid.
Further experiments revealed that high dietary cholesterol could not change the mRNA expression of bile acid synthesis enzymes, including cytochrome P450 (Cyp)7a1, Cyp8b1, Cyp27a1 and Cyp7b1 in the liver (figure 5F). Additional in-vitro experiments showed that TCA aggravated cholesterol-induced triglyceride accumulation in human normal immortalised hepatocyte cell line LO2 (figure 5G), while IPA suppressed cholesterol-induced lipid accumulation (figure 5G), and cell proliferation (figure 5H) in NASH–HCC cell lines, HKCI-2 and HKCI-10. These results indicated that cholesterol promoted NASH–HCC progression through the modulation of host serum metabolites and, at least in part, through increased TCA and decreased IPA.
High cholesterol-modulated gut microbiota promotes steatohepatitis and hepatocyte proliferation in germ-free mice
We evaluated the contribution of cholesterol-modulated microbiota in NAFLD–HCC by faecal microbiota transplantation (FMT). Stools of NC-fed, HFLC-fed and HFHC-fed mice (14 months) were gavaged to germ-free mice (G-NC, G-HFLC and G-HFHC) under NC diet (figure 6A). The liver-to-body weight ratio was significantly higher in G-HFHC mice compared with G-NC mice (online supplementary figure S6A). Moreover, hepatic lipid accumulation by triglyceride assay and Oil Red O staining (figure 6B) and peroxidation by thiobarbituric acid reactive substances assay (figure 6B) were significantly increased accompanied with impaired liver histology by H&E staining (figure 6B) in G-HFHC mice at 8, 10 and 14 months following FMT. Increased inflammation was evidenced by enhanced hepatic cytokines and chemokines including IL-6 (figure 6C) by cytokine profiling assay, Fos, Ccl12, Cxcr1, Ccl1, Myd88, Il-1β, Cxcl10 and C3ar1 (online supplementary table S8) by cDNA expression assay and enhanced CD45+ lymphocytes accumulation (online supplementary figure S6B) by flow cytometry in G-HFHC mice compared with G-HFLC mice.

High cholesterol-modulated microbiota promotes hepatocyte proliferation in germ-free mouse model. (A) Stools were transplanted from NC-fed, HFLC-fed and HFHC-fed mice (14 months) to germ-free mice (G-NC, G-HFLC and G-HFHC) under NC. Gross morphology, histological examination and Ki-67 staining of livers in stool-gavaged germ-free mice in the G-NC, G-HFLC or GHFHC groups were shown. (B) Liver triglyceride content, lipid peroxidation and liver histology in recipient germ-free mice of the G-NC, G-HFLC or G-HFHC groups at 8, 10 and 14 months. (C) Hepatic IL-6 protein levels and (D) Ki-67 staining of liver sections in the G-NC, G-HFLC or G-HFHC groups at 14 months. (E1) Mouse cancer pathway Finder PCR array in the liver tissues of G-NC, G-HFLC or G-HFHC mice. (E2) CDC20 protein level was validated. (F) Principal component ordination analysis and histogram of the linear discriminant analysis (LDA) scores of gut microbiota among G-NC, G-HFLC or G-HFHC mice. (G) Serum metabolomic analysis of G-HFLC and G-HFHC mice. *p<0.05, **p<0.01, ***p<0.001. HFHC, high-fat/high-cholesterol diet; HFLC, high-fat/low-cholesterol diet; NC, normal chow; PCA, principal component analysis.
Increased hepatocyte proliferation was observed in the liver of the G-HFHC mice at 14 months (p<0.05) (figure 6D) but not at 8 and 10 months after FMT. Cancer pathways PCR array revealed the upregulation of genes involved in oncogenic pathways including cell proliferation (Cdc20), angiogenesis (Pgf), invasion/metastasis (Serpinb2, Snai3) and downregulation of genes involved in apoptosis (Fasl and Lpl) in liver tissues of G-HFHC mice compared with G-NC and G-HFLC mice (figure 6E1). Upregulation of CDC20 was validated by Western blot (figure 6E2). One liver nodule was observed in G-HFHC mice at 14 months, but not at 8 and 10 months (figure 6A). Histology examination confirmed the nodule as dysplasia with globules and increased cell proliferation (figure 6A). We evaluated the composition of gut microbiota in recipient germ-free mice and their corresponding donor conventional mice. We found that gut microbiota was significantly different among the recipient germ-free mice in relation to donors’ diet (figure 6F). The microbial ecosystem after FMT remained stable over time in all groups of mice as shown by β-diversity and composition analysis (online supplementary figure S7A-D). Furthermore, analysis of serum metabolites from germ-free mice revealed decreased IPA in G-HFHC mice compared with G-HFLC mice in consistence with conventional mice fed with HFHC (figure 6G). Taken together, these data indicated that dietary cholesterol-modulated microbiota promote NAFLD and hepatocyte proliferation by inducing metabolite alteration, in contributing to cholesterol-induced NAFLD–HCC formation.
Anticholesterol treatment completely prevents NAFLD–HCC formation in HFHC-fed mice
As dietary cholesterol drives the progression of NAFLD–HCC, we evaluated if anticholesterol drug could dampen NAFLD and its progression to HCC. Atorvastatin (20 mg/kg), a cholesterol-lowering drug, was administered to mice that had been fed with HFHC for 7 months, and continued for additional 7 months with HFHC and atorvastatin (figure 7A). At the end of experiment (14 months), atorvastatin completely prevented NAFLD–HCC formation induced by HFHC diet and improved severity of NASH (figure 7B). This was concomitant with significantly reduced serum cholesterol, hepatic free cholesterol, serum AFP (figure 7C), ALT, lowered serum pro-inflammatory cytokines (IL-6, IL-1α, IL-1β, MCP-1, MIP-1α and MIP-1β) and oxidative stress (increased NAD+ to NADH ratio and SOD activity) (figure 7D). Atorvastatin also ameliorated liver fibrosis as supported by significant reduction of hepatic collagen deposition and hydroxyproline content (figure 7E). Microbiota analysis by 16S rRNA gene sequencing was performed on the stools of mice fed with HFHC under atorvastatin treatment (HFHC+At) and compared with mice fed with NC, HFLC and HFHC. Bacterial richness which increased in HFHC-fed mice was significantly restored by anticholesterol atorvastatin treatment (figure 7F). Furthermore, among the dysregulated OTUs in HFHC-fed mice (figure 4B and D), the abundance of Mucispirillum schaedleri_Otu038, Desulfovibrio_Otu047, Anaerotruncus_Otu107 and Desulfovibrionaceae_Otu073 were found to be reversed by anticholesterol treatment (figure 7F). To study the direct effect of the microbiota in anticholesterol drug-prevented NAFLD–HCC, we gavaged stool from HFHC+At mice to germ-free mice (G-HFHCAt, n=10). G-HFHCAt mice showed improved liver histology with decreased liver triglyceride and lipid peroxidation compared with G-HFHC group at 14 months after stool gavage (figure 7G). An induction of IPA and a reduction of TCA were observed in HFHC mice treated with atorvastatin (online supplementary figure S8). These results further show that gut microbiota plays an active role in mediating cholesterol-induced NAFLD–HCC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cholesterol inhibition abrogated NAFLD–HCC progression in HFHC-fed mice. (A) Schematic illustration of the atorvastatin treatment of C57BL/6 mice fed with HFHC diet. (B) Representative gross morphology of liver and microscopic features in H&E-stained HFHC-fed mice with or without the treatment of atorvastatin, histological scores of steatosis, inflammation and collagen were calculated. (C) Serum cholesterol levels, hepatic free cholesterol levels, serum AFP levels, (D) serum ALT levels, liver NAD+ to NADH ratio, liver SOD activity, serum IL-6, IL-1α, IL-1β, MCP-1, MIP-1α, MCP-1β protein levels, (E) collagen deposition and hydroxyproline content in HFHC-fed mice with or without the treatment of atorvastatin. (F) Bacterial richness and heatmap plot of the bacteria in stool of mice fed with NC, HFLC and HFHC with or without atorvastatin treatment. (G) Gross morphology, histological examination, triglyceride and lipid peroxidation of livers in HFHCAt stool gavaged germ-free mice (G-HFHCAt). (H) Schematic diagram of the mechanism of cholesterol-induced NAFLD–HCC development. *p<0.05, **p<0.01, ***p<0.001. AFP, alpha-fetoprotein; At, atorvastatin, ALT, alanine aminotransferase; HCC, hepatocellular carcinoma; HFHC, high-fat/high-cholesterol diet; IPA, 3-indolepropionic acid; NAD, nicotinamide adenine dinucleotide; NAFLD, non-alcoholic fatty liver disease; ROS, reactive oxygen species; SOD, superoxide dismutase; TCA, taurocholic acid.
Discussion
Despite cholesterol being a known cytotoxic lipid in NASH,5 8 information about the role and the underline mechanisms of cholesterol in HCC development from NASH is limited. In this study, we demonstrated for the first time that prolonged high dietary cholesterol feeding caused spontaneous NAFLD–HCC development in mice. High-fat diet induces obesity, insulin resistance, glucose intolerance and steatosis. The progression of steatohepatitis to fibrosis and HCC was, however, predominantly associated with high dietary cholesterol. Germ-free mice transplanted with faecal microbiota from high-cholesterol diet-fed mice substantially phenocopied the donor mice in lipid accumulation, inflammation and cell proliferation, suggesting that gut microbiota dysbiosis contributes to dietary cholesterol-induced NASH and potential development of NAFLD–HCC.
We investigated the underline mechanisms of HCC development from NAFLD by high dietary cholesterol. We found enhanced ROS accumulation in NAFLD–HCC in mice fed with high-cholesterol diet. The accumulated ROS is a toxic mediator, which can induce inflammatory response, insulin resistance and oxidative damage.19 Indeed, our analysis revealed that dietary cholesterol induced both ROS and proinflammatory cytokines, thereby promoting NASH and HCC development. In line with our findings, Wolf et al have shown that long-term choline-deficient high-fat diet feeding in mice-induced hepatic ROS, intrahepatic CD8+ T cells, NKT cells and their secreted inflammatory cytokines to promote NASH and HCC transition.20
Accumulating evidence demonstrates that the human gut microbiota can influence various pathologic conditions including liver cancer.21 We therefore explored the potential involvement of the gut microbiota in mediating dietary cholesterol-induced NAFLD–HCC. Along with sequentially increased bacterial richness, M. schaedleri_Otu038, Desulfovibrio_Otu047, Anaerotruncus_Otu107 and Desulfovibrionaceae_Otu073 increased along NAFLD–HCC progression. This is consistent with the report that M. schaedleri and Desulfovibrionaceae are strongly correlated with obesity, metabolic syndrome and inflammation in mouse models.22–24 Desulfovibrionaceae and Desulfovibrio were described to be abundant in a swine NASH model.25 Enriched Anaerotruncus was also reported in MCD diet-induced experimental NASH mouse model.26 In addition, we observed that several Clostridium OTUs were enriched with high-cholesterol diet and positively associated with high serum cholesterol. Previous reports have shown increased prevalence of Clostridia in NASH patients,27 supporting our observation in this study. Moreover, the depletion of OTUs of Akkermansia, Lactobacillus, Bifidobacterium and Bacteroides were demonstrated in NAFLD–HCC mice. These OTUs including A. municiphila_Otu034, Lactobacillus_Otu009, Bifidobacterium_Otu026, B. uniformis_Otu080 and B. acidifaciens_Otu032 consistently decreased with high serum cholesterol. Bifidobacterium and Bacteroides were further shown to be negatively associated with total serum cholesterol in human hypercholesteremia patients in this study. Depletion of Bifidobacterium and Bacteroides has been previously demonstrated in human NASH patients.27 28 There are evidences that Bifidobacterium has cholesterol-lowering activity through gut microbiota modulation. Additionally, B. acidifaciens is a potential probiotic bacteria as it possess the ability to prevent obesity and improve insulin sensitivity in mice.29 A strain of B. uniformis, namely B. uniformis CECT 7771 reportedly ameliorated metabolic and immunological dysfunction induced by high-fat diet in mice.30 Strains of Lactobacillus has also been reported to be protective against NASH31 and beneficially influence liver cholesterol metabolism in a pattern similar to that resulting from statin treatment in murine model.32 Moreover, decrease in A. muciniphila reportedly promotes NAFLD through thinning of the intestinal mucus layer, thereby impairing gut permeability barrier.33 These results support our observations in this study and further indicate that the suppression of protective gut microbes might represent a means through which cholesterol exerts its procarcinogenic effect in the liver. Our results provide new insights into the potential roles of gut dysbiosis in cholesterol-induced NAFLD–HCC progression.
In addition to gut microbiota dysbiosis, small molecule metabolites produced by commensal bacteria could contribute to the pathogenesis of NAFLD.13 Gut microbiota-related metabolites, including short-chain fatty acids, amino acid catabolites and bile acid, can both agonise and antagonise their cognate receptors to reduce or exacerbate liver steatosis and inflammation.34 Recently, a new gut flora generated metabolite, N,N,N-trimethyl-5-aminovaleric acid (TMAVA), is found to be elevated in plasma of human liver steatosis and exacerbates fatty liver progression in mice.35 We therefore attempted to unravel metabolites that potentially mediate the NAFLD–HCC promoting effect of high dietary cholesterol. Metabolomics profiling revealed that bile acid metabolism was impaired with high-cholesterol diet. Primary bile acids including TCA, GCA, TCDCA and TUDCA were enriched in HFHC-fed mice, consistent with high plasma TCA, GCA and TCDCA previously reported in human NASH patients compared with healthy subjects.36 Disruption of bile acid homeostasis alters metabolic homeostasis in the liver and can lead to hepatic inflammation and metabolic diseases including diabetes and NAFLD.37 38 TCA, GCA, TCDCA and TUDCA are key signalling molecules linking the gut and the liver to impact hepatic lipid accumulation and inflammation.39 Bifidobacterium and Bacteroides are the main bacterial genera of the gut microbiota involved in bile acid metabolism.40 They can deconjugate taurine-conjugated and glycine-conjugated bile acids to their unconjugated free forms through bile acid hydrolase and convert the unconjugated primary bile acids into secondary bile acids.40 Therefore, the decreased Bifidobacterium and Bacteroides in the stool of HFHC-fed mice may account for the accumulated taurine-conjugated bile acids in HFHC-fed mice. TCA aggravated cholesterol-induced triglyceride accumulation in vitro in this study which further supports its contribution and potentially, those of other enriched bile acids to the pathogenesis of cholesterol-induced NAFLD–HCC. Additionally, IPA, which is exclusively produced by commensal gut microbes,41 42 was depleted in both HFHC-fed conventional mice and germ-free mice transplanted with stools from HFHC-fed mice. IPA is a specific metabolite whose production completely depends on tryptophan metabolism by the gut microbiota18 and characterised as being anti-inflammatory and protective of intestinal barrier integrity.42 We identified significant reduction in microbial tryptophan metabolism. Our in-vitro functional analysis showed that IPA could inhibit cell proliferation and suppress lipid accumulation in NASH–HCC cell lines. Collectively, our findings suggest that cholesterol could impair bile acid metabolism and microbial tryptophan metabolism, leading to enhanced serum TCA, and reduced IPA, thereby promoting NAFLD–HCC development (figure 7H).
Gut microbiota transfer through faecal transplantation have been used to demonstrate active roles of gut microbiota in metabolic diseases including obesity and NAFLD.27 43 Therefore, we sought to determine whether the gut microbiota can play a direct causative role in dietary cholesterol-induced liver steatohepatitis and tumourigenesis. Compared with control groups, cholesterol-modulated microbiota promoted steatohepatitis and hepatocyte proliferation/dysplasia in germ-free mice. Proinflammatory factor, and decreased IPA were associated with microbiota-induced liver steatohepatitis and tumourigenesis in germ-free mice, in consistence with observations in conventional mouse model. In addition to hepatic lipid accumulation, inflammation, cell proliferation and dysplasia, HCC was not observed in germ-free mice transplanted with faecal microbiota from high-cholesterol diet-fed mice for 14 months. This suggested that dietary cholesterol plays an important role in inducing NAFLD–HCC formation and that the change of gut microbiota by dietary cholesterol contributes to the disease progression.
If cholesterol plays a key role in the pathogenesis of NASH and HCC, it would be important to establish that its functional blockade ameliorates the severity of steatohepatitis and NAFLD–HCC development. To test this, we used atorvastatin, a 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitor widely used to treat hypercholesterolemia, to inhibit cholesterol biosynthesis in HFHC-fed mice. We found that no HCC was identified in atorvastatin-treated mice and the severities of steatohepatitis and fibrosis were largely blunted. Additionally, bacterial richness, Mucispirillum, Desulfovibrio, Anaerotruncus and Desulfovibrionaceae which increased along cholesterol-induced NAFLD–HCC progression were reversed with atorvastatin treatment. Restoration of the gut microbiota diversity could be one of the mechanisms through which atorvastatin elicits its protective effect against NAFLD–HCC as shown in this study. Moreover, unlike the mice gavaged with stools from mice fed with HFHC only, the stools from atorvastatin-treated HFHC-fed mice did not promote hepatic cell proliferation in recipient germ-free mice. These observations further highlight the potential use of atorvastatin in preventing the development of cholesterol-induced NAFLD–HCC.
In conclusion, this study shows for the first time, that prolonged high dietary cholesterol induces spontaneous and progressive development of NAFLD–HCC in male mice by modulating the gut microbiota. Cholesterol induces increased TCA and decreased IPA through gut microbiota alteration, thereby promoting lipid accumulation, cell proliferation in the liver, leading to NAFLD–HCC development (figure 7H). Anticholesterol treatment completely abrogated dietary cholesterol-induced NAFLD–HCC formation. This study highlights that cholesterol inhibition and manipulation of the gut microbiota and its related metabolites might represent effective strategies in preventing NAFLD–HCC.
References
Footnotes
XZ and OOC are co-first authors.
Contributors XZ and OOC were involved in study design, conducted the experiments and drafted the paper; EC, KF, HCHL and Y-XW performed the experiments; AWHC determined the histology; HW provided germ-free animal; XY and JJYS commented and revised the manuscript; JY designed, supervised the study and wrote the paper.
Funding This project was supported by research funds from Guangdong Natural Science Foundation (2018B030312009), RGC Collaborative Research Fund (C4041-17GF, C7026-18G, C7065-18G), RGC Theme-based Research Scheme Hong Kong (T12-703/19 R), CUHK direct grant for research, Vice-Chancellor's Discretionary Fund CUHK.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal studies were performed in accordance with guidelines approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and the Third Military Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.