Article Text

Abstract
Complex rearrangement patterns and mitotic errors are hallmarks of most pancreatic ductal adenocarcinomas (PDAC), a disease with dismal prognosis despite some therapeutic advances in recent years. DNA double-strand breaks (DSB) bear the greatest risk of provoking genomic instability, and DNA damage repair (DDR) pathways are crucial in preserving genomic integrity following a plethora of damage types. Two major repair pathways dominate DSB repair for safeguarding the genome integrity: non-homologous end joining and homologous recombination (HR). Defective HR, but also alterations in other DDR pathways, such as BRCA1, BRCA2, ATM and PALB2, occur frequently in both inherited and sporadic PDAC. Personalised treatment of pancreatic cancer is still in its infancy and predictive biomarkers are lacking. DDR deficiency might render a PDAC vulnerable to a potential new therapeutic intervention that increases the DNA damage load beyond a tolerable threshold, as for example, induced by poly (ADP-ribose) polymerase inhibitors. The Pancreas Cancer Olaparib Ongoing (POLO) trial, in which olaparib as a maintenance treatment improved progression-free survival compared with placebo after platinum-based induction chemotherapy in patients with PDAC and germline BRCA1/2 mutations, raised great hopes of a substantially improved outcome for this patient subgroup. This review summarises the relationship between DDR and PDAC, the prevalence and characteristics of DNA repair mutations and options for the clinical management of patients with PDAC and DNA repair deficiency.
- pancreatic cancer
- DNA damage
- chemotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an exceptional malignancy with a distinct biology and epidemiology. It is nowadays the fourth leading cause of cancer-related deaths in the Western world and one of the few cancers with a rising incidence. PDAC has shown the least therapeutic progress out of all major GI cancers over the last decades.1 Various routes involving distinct precursor lesions guide PDAC development: (i) acinar cells undergo ductal reprogramming, forming metaplastic ducts which give rise to pancreatic intraepithelial neoplasia (PanIN1–3) with an increasing degree of dysplasia; (ii) pancreatic duct cells acquire oncogenic events that accelerate an aggressive PDAC formation2; (iii) mutations in pancreatic ducts, depending on the mutational make-up, lead to cystic precursor tumours: the mucinous cystic neoplasms and the intraductal papillary mucinous neoplasms (IPMN), each following separate genetic routes towards PDAC. The dismal prognosis of PDAC is mainly caused by a unique molecular complexity including (i) a desmoplastic, immunosuppressive and stroma-enriched environment; (ii) a high level of intratumoural and intertumoural heterogeneity3; (iii) a condition ultimately leading to early metastasis and high chemoresistance. Numerous, but not all clinical trials on PDAC have failed over the last decade.4 Single gemcitabine, gemcitabine/capecitabine and modified folinic acid, fluouracil, irinotecan, oxaliplatin (FOLFIRINOX) are now consolidated adjuvant treatment options.5 6 Neoadjuvant and perioperative treatment concepts are currently being investigated.7 In advanced PDAC, FOLFIRINOX and the combination of nanosized albumin-bound paclitaxel (nab-PTX)/gemcitabine are superior to gemcitabine alone in first-line therapy and are thus considered the standard of care.8 9 However, median overall survival (mOS) in advanced disease rarely exceeds 1 year, resulting in a 5-year OS of <10% for all stages.3 This underpins the necessity for more innovative, effective and targeted regimens. However, in contrast to various other cancers, tailored approaches have been largely disappointing in PDAC.10–13 Activating KRAS mutations are major drivers of malignant growth in PDAC, and although yet undruggable, first promising developments have been made.14 The oncogenic KRAS dosage also regulates diverse phenotypes in PDAC, leading to different routes of tumourigenesis permitted by the loss of distinct tumour suppressor gene pathways (eg, TP53, CDKN2A, SMAD4).15 Accordingly, oncogenic KRAS may synergise with additional tumour suppressor mutations to deregulate double-strand break (DSB) repair and subsequently induce genomic instability.16 ,17 Besides, a tremendously high number of passenger mutations establishes the high intratumoural and intertumoural heterogeneity. To approach this heterogeneity, transcriptional profiling of purified pancreatic cancer epithelial cells allowed a certain degree of subgrouping, having led to various partly overlapping classifications according to Collisson et al,18 Moffitt et al 19 and Bailey et al.20 The basal-like and classical subtypes have meanwhile evolved as the most robust transcriptional classifier. However, transcriptional phenotypes could be recently integrated with genomic alterations by using whole-genome analysis from purified epithelium of primary and metastatic human PDAC together with single-cell RNA sequencing. Thereby, the Notta group revealed that molecular subtypes are specifically linked to copy number aberrations, for example, in the KRAS gene, which in turn additionally drive genomic instability.21 These observations indicate that genomic aberrations can define the molecular subtype, and PDAC heterogeneity as well as evolution during disease progression is caused by certain mutational triggers.21 To genomically classify PDAC according to patterns of variation in chromosomal structure, four subtypes have been defined and also allowed predictions in terms of a given treatment. Those subtypes have been termed (i) ‘stable’, (ii) ‘locally rearranged’, (iii) ‘scattered’ and (iv) ‘unstable’.16 The latter accounts for around 14% of human PDACs and harbours mutations in the genes responsible for DNA repair that are involved in the so-called DNA damage repair (DDR), such as BRCA1/2, PALB2 and ATM. Mutations in these genes also cluster in inherited forms of PDAC.22 Thus, a significant proportion of human PDACs with either somatic or germline mutations in DDR genes might benefit from tailored, targeted therapies. These aspects form the focus of the current review.
DNA damage repair and cancer
Preserving the integrity of our DNA is eminent in preventing genomic instability, a hallmark of cancer, various chronic diseases and the normal process of ageing.23 DNA damage is a common event and must undergo immediate repair in order to ensure the exact transfer of genetic information during cell division. The inability of the DDR to repair following endogenous and exogenous insults can lead to (i) an accumulation of genomic defects, (ii) subsequent malignant transformation, (iii) cancer progression and (iv) further impairment of the DNA repair capacity. By contrast, during tumour progression or on therapy-induced tumour evolution, the DDR machinery can be reconstituted to assign new growth advantages to outgrowing tumour clones with disturbed genomic integrity. Repair mechanisms include the detection and excision of the defect, the rejoining of DNA ends and the reconstruction of the sequence based on a DNA matrix (figure 1). Basically, DNA lesions can occur in two major ways, affecting either a single-strand break (SSB) or DSB or mono-adducts and interstrand crosslinks, respectively. Damage affecting a single DNA strand can be removed either by base excision, mismatch (MMR)24 or nucleotide excision repair (NER),25 all of which use the sister strand as a template (figure 1). Conversely, DSBs or crosslinked DNA strands (eg, caused by platinum agents or irradiation) require partial strand substitution, a process that is much more complex and susceptible to errors.26 It is noteworthy that the accumulation of unresolved SSBs inexorably induces DSB formation, namely when encountering a replication fork. Two main mechanisms affect DSB repair, namely non-homologous end joining (NHEJ) and homologous recombination (HR) (figure 1). While NHEJ is a more error-prone form of DSB repair, in which a DNA segment is removed and both ends are adjoined without consideration of homology, HR is an accurate process that uses the sister chromatid as a repair template.27 Herein, the MRN (Mre11, Rad50 and Nbs1 proteins) complex makes up the core of the initial DSB repair machinery as an upstream effector of HR and partially NHEJ. This complex crucially participates in all DSB repair events such as (i) DNA damage sensing, (ii) DDR protein recruitment to the damaged site, (iii) cell cycle checkpoints activation and (iv) damage repair.28 29 Accordingly, DSBs detected by the MRN complex activate the cell cycle regulatory serine/threonine kinases ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein (ATR) to allow the formation of protruding 3’ ends at both sides of the break.24 Subsequently, ATM activates CHK2, which arrests cell cycle progression, interacts with TP53 that is responsible for cell cycle and apoptosis control and contributes to regulating BRCA1 in DNA repair.30 The MRN complex also attracts BRCA1 to the DNA damage site, supporting DNA resection, forming the adjoining 3’ ends and recruiting PALB2 and BRCA2.31 This newly formed complex of BRCA1, PALB2 and BRCA2 finally activates RAD51, that is responsible for binding single-stranded DNA segments and invading the homologous sequences in the sister chromatid.31 Alternatively, ATR is activated by the presence of a DNA crosslinking adduct phosphorylating the Fanconi anaemia (FA) core complex, which helps to excise the defect.32 During this DNA crosslink repair process involving NER, DSBs are generated in the proximity of the incised oligonucleotide.32 Subsequently, this accumulation of DSBs requires repair, particularly by HR and not NHEJ.33 34 Consequently, mutations in HR genes (such as BRCA1, BRCA2, XRCC2 and XRCC3) can further exhibit hypersensitivity to crosslinking agents (figure 1).32
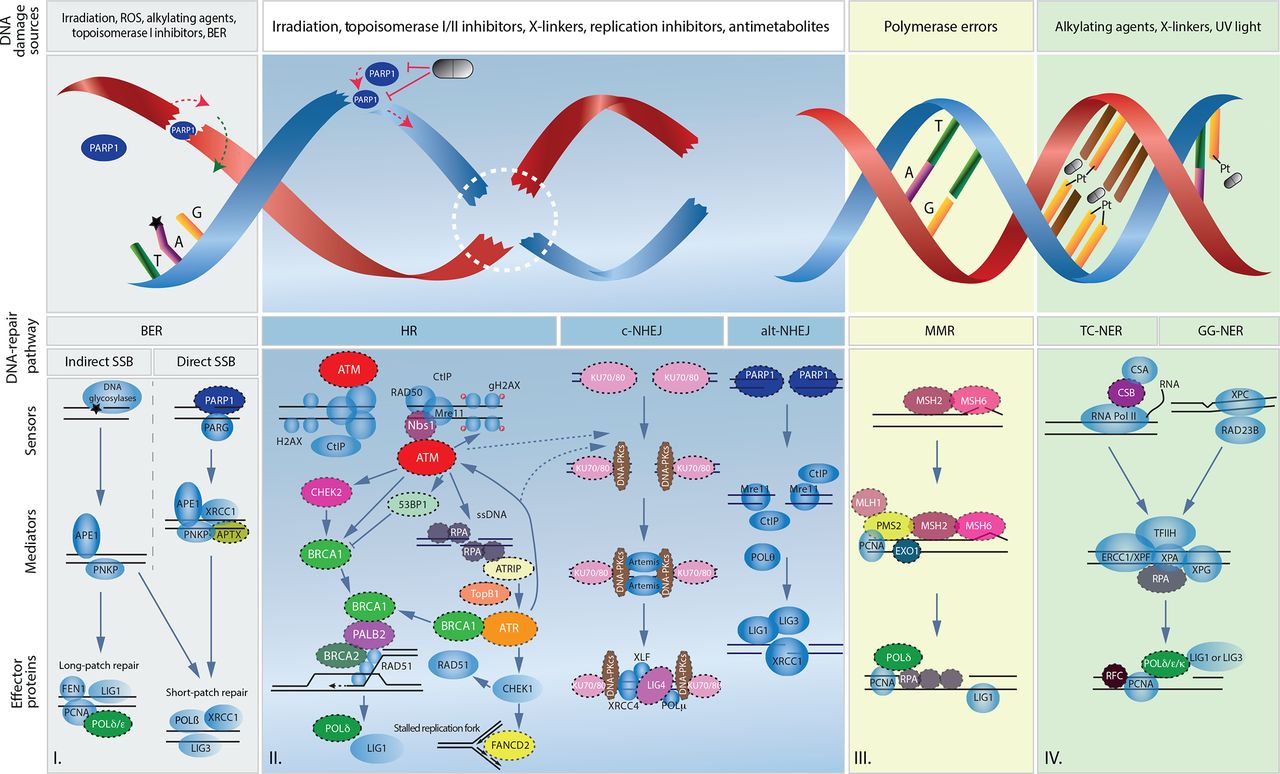
Overview of main DNA lesions and their related DNA damage repair pathways. Schematic representation of I. single-strand break repair by direct and indirect base excision repair, II. double-strand break repair by homologous recombination and non-homologous end joining, III. replication error repair by mismatch repair and IV. DNA adducts repair by either transcription-coupled nucleotide excision repair (TC-NER) or global genomic nucleotide excision repair (GG-NER). Symbolic pills show potential targeted therapeutic interventions by PARP inhibition and platinum agents (Pt). Black stars represent an indirect single-strand break. Dashed circles emphasize proteins found to be mutated in human pancreatic ductal adenocarcinoma. Dashed lines represent the regulatory role of ATM and ATR on non-homologous end joining. alt-NHEJ, alternative non-homologous end joining; BER, base excision repair; c-NHEJ, canonical non-homologous end joining; MMR, mismatch repair; ROS, reactive oxygen species; SSB, single-strand break.
In summary, a plethora of distinct and tailored repair mechanisms work to preserve genomic integrity in healthy and in cancerous cells. Any failure of the DDR leads to a subsequent accumulation of mutations as well as structural aberrations, usually generating particularly aggressive tumours. It is of great importance that the sole dependence on the remaining DDR pathways has yielded several conceptual approaches such as synthetic lethality for discovering and characterising cancer-specific vulnerabilities in customised interventions.35
DNA damage repair defects and pancreatic cancer
The incidence of PDAC shows significant variations from a geographic perspective, with the highest in high-income countries.36 Although the cause of PDAC is complex and multifactorial, a variety of inherited and non-inherited risk factors have been described, some of which may explain these variations. Non-inherited risk factors include chronic pancreatitis, diabetes mellitus, smoking, alcohol consumption, obesity and possibly Helicobacter pylori infection.3 Apart from these, various genes have been associated with increased PDAC susceptibility.22
Sporadic pancreatic cancer with somatic DDR gene mutations
Tumour biology in PDAC is determined by the type of mutations and structural aberrations and not by their pathogenic or somatic occurrence,37 the latter dominating the most frequently occurring sporadic PDAC. In fact, biallelic inactivation, zygosity-dependent phenotype penetrance and sensitivity to poly (ADP-ribose) polymerase (PARP) inhibition, as shown for BRCA1/2 in PDAC, are frequently associated with genomic features such as dramatic chromosomal rearrangements, indicating a deficient HR profile.16 37 Particularly in pancreatic carcinogenesis, single mitotic errors as well as distinct genomic rearrangements such as chromothripsis (an unstable subtype enriched feature) can lead to a dramatic loss of genetic information accompanied by a simultaneous loss of function of several tumour suppressors. In turn, rapid and synchronous clonogenic escape is thought to establish the metastatic disease.38 Large-scale genomic analysis revealed 63 genetic alterations per single PDAC, with ‘DNA damage control’ being one of the most prominent terms (figure 2).39 Another study reported either an unstable genome or a BRCAness mutational signature in 24% of analysed PDACs, with 17% germline and 29% somatic mutations in BRCA1/2, ATM or PALB2.16 Basically, BRCAness encompasses defects in the HR pathway, mimicking the consequences of BRCA1 or BRCA2 loss.40 Overall, ATM appears to be the most frequently mutated DDR gene in somatically mutated sporadic PDAC, with an overall mutational frequency of approximately 4%, followed by BRCA2, STK11 and BRCA1 (figure 2). Loss of ATM occurs in precancerous lesions such as PanINs or IPMNs and in primary tumours, underpinning its crucial role in genomic integrity.41 42 The ultimate therapeutic strategy for PDAC is the delineation of patient subgroups who might be susceptible to an interference with the DDR due to the intrinsically high DNA damage load, leading to a further increase beyond a tolerable threshold.
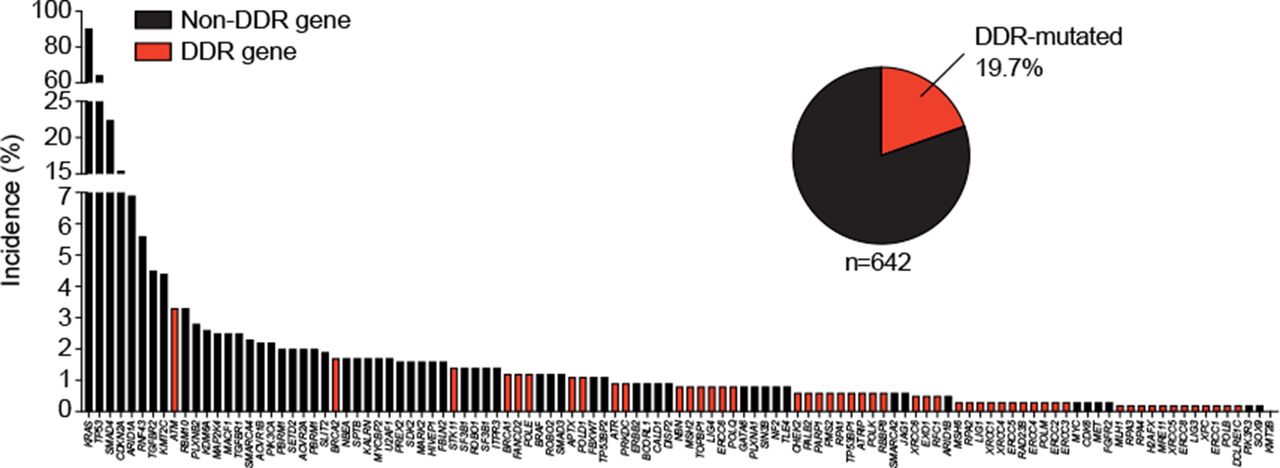
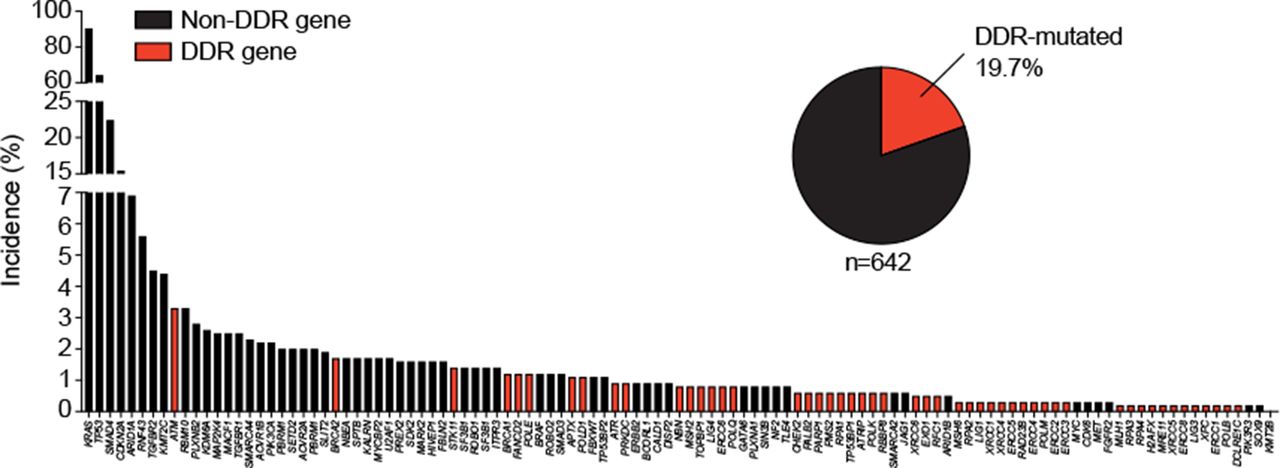
Frequency of gene alterations in primary pancreatic ductal adenocarcinomas. Three available pancreatic cancer sequencing data sets123 ,124 ,125 (n=751) were assessed for somatic gene mutations (panel of 118 genes from 16 ,123 and additionally extended in more detail for DNA damage repair genes from cBioPortal). DDR, DNA damage repair.
DDR gene mutations in the germline of patients with pancreatic cancer
Familial pancreatic cancer and hereditary pancreatic cancer syndromes
Unlike sporadic PDACs, up to 10% of the cases cluster in families, with at least two first-degree relatives (FDR) being affected. These cases are designated as familial pancreatic cancers (FPC). The risk of developing PDAC in relatives fulfilling the FPC criteria increases with the number of affected FDRs by 4.6, 6.4 and up to 32-fold for 1, 2 and ≥3 FDRs, respectively, compared with the whole population.43 Nevertheless, 80%–90% of the genetic events leading to FPC remain unknown, leaving as few as 10%–20% with a clearly identifiable germline mutation. This makes it difficult to properly distinguish FPCs from apparently sporadic PDACs. Specifically, inheritance can be frequently attributed to germline DDR gene mutations (ATM, BRCA1, BRCA2, MLH1, MSH2, PALB2, PMS2 and STK11) and to mutations in classical cancer susceptibility genes such as CDKN2A or TP53. Table 1 summarises the frequencies of the most relevant FPC-causing genes.22 44 45
Frequency of the most common deleteriously mutated genes in familial pancreatic cancer
Moreover, several inherited cancer predisposition syndromes are characterised by usually monoallelic, dominantly inherited autosomal mutations that predispose to PDAC and other cancers such as prostate, breast and ovarian cancer (eg, BRCA1/2 mutations).46 ,47 The majority of the underlying germline mutations are well-characterised, such as Peutz-Jeghers caused by STK11 mutations or Lynch syndrome caused by MLH1 and MSH2 mutations. Furthermore, the risk of PDAC is increased in familial adenomatous polyposis (usually associated with APC mutations), familial atypical multiple-mole melanoma syndrome (FAMMM; CDK2NA mutations), hereditary pancreatitis (PRSS1 and SPINK1 mutations) and Li-Fraumeni syndrome (TP53 mutations).48–50 It is to be noted that Peutz-Jeghers syndrome-related PDACs are mostly, but not exclusively, associated with the IPMN precursor lesion pathway.51 Finally, chronic pancreatitis in patients with cystic fibrosis related to the CFTR gene has been associated with a moderately increased incidence of PDAC in mutation carriers.52
Sporadic pancreatic cancer with germline mutations
Interestingly, in about 3.9%–13.5% of patients with apparently sporadic PDAC, DDR gene mutations can be detected in the germline, although negative family history of cancer.53 ,54 ,55 ,55 ,56 ,57 This mirrors an incomplete penetration, rather than a de novo mutation in the germline. With a frequency up to 3.4%,56 ATM serine/threonine kinase is the most frequently mutated DDR gene in this group (table 2). Biallelic mutations in ATM cause a syndrome known as ataxia telangiectasia that predisposes to various cancers, including PDAC (twofold to threefold increased risk).58 59 BRCA1/2 mutations are also frequent in this subgroup and have the highest prevalence (12.1%) in Ashkenazi Jews.60 A recent study also identified germline mutations in 7.3% of patients with IPMN and associated them with an increased risk of developing PDAC,42 for instance, PALB2, which codes for a BRCA2-interacting protein essential for the DSB repair function. Monoallelic germline mutations in PALB2 are also associated with breast cancer.61 Biallelic germline mutations result in a rare subtype of FA (FA-N) associated with a severe predisposition to paediatric malignancies.62
Frequency of germline mutations in DNA damage repair and cell cycle control genes in sporadic pancreatic ductal adenocarcinoma and frequency of loss-of-function variants in gnomAD controls
BRCAness revisited
Most of the DDR genes mutated in PDAC are crucial for the proper functioning of the HR pathway. Alterations in these genes lead to a homologous recombination-deficient (HRD) phenotype within a given tumour defining the term BRCAness. This defect can arise following certain genomic, epigenetic or post-translational alterations in certain DDR genes. Recently, it has been shown that loss-of-function alterations in BRCA1/2 are an indispensable initiating event in so-called BRCA-associated cancer types (pancreatic, prostate, breast or ovarian cancer). Thus, the tumour lineage, together with a given mutation, determines the tumour biology, and, subsequently, therapeutic decision-making in non-BRCA-associated cancers, for example, BRCA1/2 mutations, is carried out in a biologically neutral manner.37 Molecularly, the BRCA1/PALB2/BRCA2 complex activates RAD51 to allow the incursion of the sister chromatid homologous sequences, a crucial step during HR (as described before). Similarly, ATM or ATR loss-driven DSB sensor defects lead to failure of this repair complex (figure 1). Therefore, any disruption of HR influences cells to rely on error-prone DNA repair pathways (such as NHEJ) for DSB repair, cumulating in augmented genomic instability. The term HRDness extends this phenotypic spectrum to other somatic or germline mutations causing HR defects, including non-BRCA-related phenotypes.63 Along this line, a highly promising concept could be the chemical induction of HRDness using certain therapeutics (eg, ATM or ATR inhibitors) and subsequently targeting the respective tumour with classical DNA-damaging agents. However, testing such concepts requires appropriate preclinical data in models that faithfully recapitulate human disease.
Translational models of DDR in pancreatic cancer
Nowadays, the prediction of tumour biology and treatment response in patients is mainly based on DNA and RNA-sequencing of the tumour. Here, subclonal genetic events require high coverage, and an incomplete characterisation of the mutational spectrum, for example, due to simplified panel sequencing, limits the interpretation of the results. To study the effects of DDR deficiency on pancreatic carcinogenesis and to test novel therapeutic strategies, in vivo and in vitro model systems with a high translational capacity have been developed.64 Thus, the greatest challenge is the recapitulation of the exceptionally high intertumoural and intratumoural heterogeneity in PDAC.
Human PDAC cells harbouring BRCA2, FANCC, FANCG gene mutations display in vitro hypersensitivity to DNA interstrand crosslinking agents (ICA) such as mitomycin C or cisplatin, which do, in fact, result in structural and chromosomal alterations following xenograft transplantation.65 These two-dimensional cell monolayers have the advantage of easy propagation, access and replicability. However, artefacts due to culture methods and clonal selection, as well as the lack of microenvironmental tumour interactions, limit their value and can only be a basis for more complex models.
Accordingly, in a genetically engineered mouse model the loss of Brca2 does not propagate PDAC development but needs further drivers such as Trp53. However, in both situations more chromosomal instability is generated that sensitises to DNA-damaging agents.17 66 Interestingly, oncogenic KRASG12D in a BRCA2-deficient background promotes chromosomal instability and apoptosis, but inhibits tumour growth. However, loss of Palb2 and Brca1 in a Trp53, KrasG12D -mutated context (KPC) enhances genomic instability, decreases survival, but generates vulnerability to ICA and PARP inhibitors.67 Interestingly, these tumours also showed a genotype-specific immune cell infiltration pattern with CD3+ T cells, resulting in decreased tumour progression and improved survival, after treatment with an interleukin (IL)-6 and programmed death-ligand 1 (PD-L1) antibody.68
Atm-deficient mice expressing oncogenic KRASG12D (AKC) showed accelerated dysplastic growth in the pancreas, epithelial-to-mesenchymal transition and a highly metastatic phenotype (figure 3).69 AKC mice exhibit features of the unstable human PDAC subtype including chromosomal instability, deregulated DNA integrity checkpoints and aneuploidy.41 70 Single targeting of either PARP1 or ATR in this model yielded moderate efficacy comparable to gemcitabine.70 Moreover, targeting of the remaining HR-associated ATR protein and NHEJ by DNA-PK inhibition led to a lethal accumulation of DNA damage. Likewise, adding an ATM inhibitor to PARP, ATR and DNA-PK inhibition in ATM-proficient PDAC cells virtually removes DDR. Preclinically, maintenance therapy with single PARP inhibitors can force the selection of aneuploid, highly aggressive, multidrug-resistant subclones.71

{kind=link}
{kind=link}
{kind=link}
Schematic representation of pancreatic cancer progression in a homologous recombination-deficient context. HRDness resulting from ATM deficiency sensitises pancreatic cells to exogenous and endogenous DNA damaging factors enabling an oncogenic cascade through acinar-to-ductal metaplasia. Sustained impaired double-strand break repair results in accelerated genomic instability driving the progression of preneoplastic PanIN stages to HRD pancreatic ductal adenocarcinoma (PDAC). Persistent HRD or therapeutically induced HRD by eg, ATMi renders cancer cells vulnerable to therapeutic interventions promoting DNA damage. Monotherapeutic approaches as PARPi subsequently tend to complex multidrug resistance (MDR) involving epithelial-to-mesenchymal-transition (EMT) and alternative DNA repair. Smart tailored use of synergistically druggable vulnerabilities within the DNA damage repair machinery could be exploited to hit HRD tumors “hard and early” and prevent further MDR acquisition, as eg, recently shown upon inhibition of PARP, ATR and DNA-PKcs.71 ADM, acinar-to-ductal metaplasia; ADR, acinar-to-ductal reprogramming; DSB, double-strand break; HRD, homologous recombination-deficient; NHEJ, non-homologous end joining; PanIN, pancreatic intraepithelial neoplasia.
Patient-derived xenografts (PDX) are today state of the art for the human translation of the aforementioned models72 and do, in fact, faithfully reflect phenotypic and therapeutic features.73 However, PDX are not suitable for either high-throughput drug screens or response prediction analysis in an individual patient due the long time required for their determination and propagation.
Therefore, three-dimensional cultured organoids can be easily determined from different sources including circulating tumour cells,74 metastasis or primary PDAC itself in a short time span.75 Patient-derived organoids (PDOs) represent PDAC heterogeneity and partly its microenvironment,75 76 providing a potentially better model system for testing/predicting drug response.77 78
Therapeutic interference with the DDR in pancreatic cancer
Platinum analogues
Platinum agents can crosslink purine bases on the DNA, thereby interrupting DNA transcription and stall replication which can consecutively lead to DSBs and the induction of apoptosis.79 The removal of monoadducts on one DNA strand in response to platinum-induced DNA damage primarily involves NER. The repair of interstrand crosslinks additionally uses the FA pathway, translesion synthesis and HR, making these agents interesting for patients with an HRDness phenotype (figure 1).80 81 It is of note that structural differences in platinum agents cause differences in DDR recognition as has been observed for cisplatin and oxaliplatin, although they form similar adducts at the same sites on the DNA. These differences in recognition, excision and processing affect the cytotoxicity and activity of the individual platinum adducts.82 Genomic instability in PDAC, at least in the case of BRCA1/2 or PALB2 mutations, co-segregates with (i) defective DNA maintenance, (ii) a mutational signature of DNA damage repair deficiency and (iii) an exceptional response to platinum. This may allow treatment stratification according to a given genetic event.16 Molecular subtyping is necessary for identifying these new target populations of unstable PDACs. The response of 820 patients with PDAC to a platinum-based regimen was recently evaluated in a large registry with a comprehensive genomic profiling programme. HRDness-causing germline or somatic mutations were grouped into three categories according to a known or suspected platinum responsiveness: (i) BRCA1/BRCA2/PALB2, (ii) ATM/ATR and (iii) FA core/MRN complex effectors. Overall, HR mutations were prevalent in 16.5% of patients. Interestingly, patients with advanced HR-defective (HRD) PDAC had a worse outcome than patients with HR-proficient (HRP) PDAC if they were not treated with platinum, underpinning their overall more aggressive tumour biology and particular platinum susceptibility (mOS: HRD 0.76 years vs HRP 1.13 years, p=0.1535). In line platinum treatment substantially prolonged mOS in the HRD patients (n=53) compared with the HRP patients (n=258) (mOS: HRD 2.37 years vs HRP 1.45 years; p=0.000072; HR, 0.44, 95% CI 0.29 to 0.66). The mOS positively correlated with an increasing number of HR-related mutations and was independent of the therapy line.83 Golan et al collected data from 43 patients with advanced BRCA1/2-mutated PDAC, showing a significant survival benefit for platinum treatment compared with platinum-naïve patients (22 vs 9 months; p<0.039).84 Interestingly, in patients with FPC, platinum efficacy increases with the number of affected FDRs.85 A recent phase II trial further determined the effectivity of a first-line gemcitabine/cisplatin treatment in 50 patients with advanced gBRCA1/2, PALB2-mutated PDAC (2-year OS 30.6% and 3-year OS 17.8%).86 Various retrospective studies confirmed the superior tumour response to platinum derivates in DDR-deficient PDAC (summarised in table 3).84 85 87 FOLFIRINOX is the only platinum-containing treatment for patients with advanced PDAC established in a positive phase III trial.8 However, only about 25% of patients are eligible for FOLFIRINOX, due to its high level of adverse effects.88 Therefore, appropriate patient selection, including the Eastern Cooperative Oncology Group performance status and the genetic make-up of the tumour, is similarly critical. A retrospective analysis of 32 curatively resected patients with a germline BRCA1, BRCA2 or PALB2 mutation revealed a significant survival benefit when treated with platinum, compared with 62 HRP patients (mOS: HRD 47.7 months vs HRP 23.1 months, p=0.032). Interestingly, the subgroup of HRD patients benefited to a relative extent from perioperative platinum-based chemotherapy (mOS: HRD not reached vs HRP 23.1 months).89 If platinum was omitted the survival rates were similar between both cohorts. Various other studies reported similar trends (summarised in table 4).
Retrospective studies on platinum-based chemotherapy in patients with advanced homologous recombination-deficient PDAC
Retrospective studies on platinum-based chemotherapy in patients with resected homologous recombination-deficient PDAC
In view of the data shown above, several conclusions can be drawn: (i) in patients with unselected advanced PDAC the only approved platinum-based regimen in a first-line setting is FOLFIRINOX (level of evidence 1b); (ii) platinum does not seem to be the main critical effector in HR-proficient PDAC in either an adjuvant (level of evidence 1a)90 or palliative setting (level of evidence 1a)91; (iii) platinum derivates such as oxaliplatin seem to determine the treatment response to a combination regimen such as FOLFIRINOX in advanced HRD PDAC.3 92 93
PARP inhibitors
PARPs are activated by DNA breaks and catalyse the transfer of ADP-riboses to form long-branched chains to target proteins involved in processes such as transcription and DNA damage repair. The resulting scaffold of negatively charged polymers further recruits DDR effectors (figure 1). PARP1 was originally described in SSB repair through base excision but is now well-accepted as also participating in DSB repair, stalled replication fork sensing and in the recruitment of DNA repair proteins at DNA damage sites94 (figure 1). As PARP1/2 are crucial enzymes during HR-mediated DSB repair in most cancer cells, targeting these enzymes in HRD tumours seems to be an elegant method and underpins the principle of synthetic lethality.94 However, side effects in highly proliferative healthy cells can occur as well. PARP inhibitors are nicotinamide mimetics that directly inhibit PARP1 activity and further act by trapping the PARP protein at SSBs, forcing a lethal accumulation of DNA damage and cell death consecutively. While the efficacy of PARP1 inhibitors is established in BRCA-associated cancer types,37 their specific role in PDAC has only recently been demonstrated. However, monotherapy with PARP1 inhibitors displayed only modest activity in HRD PDAC, mostly BRCA1/2-mutated95–97 (table 5), and studies without genetic stratification did not show a meaningful benefit.98 Besides the missing stratification, factors such as previous therapies are meaningful, for example, as shown in a phase I trial in pretreated BRCA1/2-mutated patients with PDAC, in which only patients who were not refractory to platinum responded (PR or CR) to rucaparib.95 However, in proving the effectiveness of gemcitabine/cisplatin (as shown in ‘Platinum analogues’ section) in gBRCA/PALB2-mutated PDAC, a phase II trial failed to demonstrate an additional benefit of veliparib in 27 patients (mOS G/C+V 15.5 months vs G/C 16.4 months, p=0.6) (table 5).86 In line with this, a currently not fully published phase I/II trial in patients with advanced PDAC showed a significantly higher objective response rate (ORR) of 50% in HRD patients (n=16) compared with 17% HRP patients (n=41) for the combination of folinic acid, fluouracil, oxaliplatin and veliparib (table 5)99 The most promising evidence of PARP inhibition in advanced PDAC was published recently from the phase III POLO trial.100 Patients with germline BRCA1/BRCA2-mutated advanced PDAC without progress to platinum-based first-line chemotherapy (≥16 weeks) received either olaparib as maintenance treatment or placebo. Olaparib nearly doubled the median PFS compared with placebo (7.4 vs 3.8 months; p=0.004), while OS remained similar in both arms (18.9 vs 18.1 months).100 The interim analysis of a single-arm phase II trial with rucaparib as maintenance therapy after platinum-based induction in the case of any pathogenic BRCA1, BRCA2 or PALB2 mutation also demonstrated an ORR of 37% with minimal toxicity in 19 patients.101 In summary, PARP inhibitors represent the first targeted therapy to show efficacy in a subpopulation of patients with advanced PDAC in a phase III clinical trial.100
Clinical trials with PARP inhibitors on patients with advanced PDAC
Toxicity and therapeutic pitfalls
PARP inhibitors cause side effects as seen with high-dose veliparib, causing unacceptably high haematological toxicity and a sporadic association with acute myeloid leukaemia in the long term.102 Similarly, veliparib plus gemcitabine/cisplatin and olaparib plus cisplatin/irinotecan/mitomycin C were associated with a marked increase of mainly haematological Common Terminology Criteria for Adverse Events (CTCAE) ≥grade 3 toxicities.103 Adverse events CTCAE ≥grade 3 also emerged in 40% of patients on olaparib monotherapy, mainly involving haematological and GI side effects, as well as fatigue/asthenia.95 96 100 However, health-related quality of life in the POLO trial revealed no clinically meaningful difference between the olaparib and placebo groups.104 Thus, the toxicity of PARP inhibitors has to be taken into account, particularly when combined with chemotherapy and when used at high doses. Although this has not been ultimately solved, there might be potential solutions for optimising these therapies, such as (i) more elaborated application regimens, (ii) novel drug formulations and/or (iii) the synergistic action of several drugs each used at an ineffective dose range on their own. So-called ‘nanomedicine’ strategies could help to overcome drug delivery challenges by reducing systemic toxicities on the one hand and by overcoming diffusion-limiting PDAC stroma on the other. As suggested by the approved nab-PTX novel concepts with superior activity could arise, including surface-optimised nanocarriers.9 105
Another pitfall is the lack of predictive knowledge about a given HRDness-causing mutation, that is, not every mutation of a given DDR gene assigns sensitivity to PARP inhibitors.106 At the moment, most of the data collected on BRCA1/2 mutations and on non-BRCA mutations are purely preclinical70 107 (figure 2). In ovarian cancer, it has already been shown that both somatic genetic alterations and modifications in epigenetic markers may lead to functional restoration and revert HRD, but whether the same can happen in PDAC tumours is elusive.108 109 We need to develop better in silico prediction algorithms that take into account the general role of a gene in a signalling network and its precise mutation. Finally, the additive value of PARP inhibitors together with or after a platinum-based regimen is questionable and response predictors in this clinical setting are lacking.37 95–97 100
Some open questions still remain and a multimodal approach including preclinical models, advanced in silico algorithms and explorative basket trials also including non-BRCA1/2 mutations is required. Here, it might be reasonable to restrict these to BRCA-associated cancers, as in non-BRCA-associated cancers at least BRCA1/2 mutations frequently appear to work in a biologically neutral manner.37
Inducing homologous recombination deficiency and future directions
New treatment modalities currently aim at inducing or maintaining an HRD state independent of the mutational make-up of a given tumour. This could increase the efficacy of DDR-interfering agents and increase the patient cohort. Such strategies might also bypass drug resistance, since PARP inhibitor resistance can occur, for example, by HR restoration (figure 3). Specifically, hypoxia might impair HR, for instance, by silencing the BRCA1 promoter or downregulation of RAD51/52.110 Anti-angiogenic agents (AA) counteract hypoxia-induced angiogenesis and, in turn, lower blood perfusion and thereby oxygen tension. This hypothesis is in contrast with transiently AA-induced repair termed tumour ‘vessel normalisation’. Nevertheless, concrete evidence supporting the fact that vessel normalisation is decisive for AA functioning is lacking, whereas induced hypoxia appears to be the major driver.111 Indeed, trials testing bevacizumab and cediranib combined with olaparib did not show unexpected toxicities, but showed promising antitumour response at least in ovarian cancer.94 Phase II basket trials including PDAC are currently recruiting (NCT02498613; table 6).
Ongoing clinical trials of therapies affecting DNA damage repair pathways that include patients with pancreatic cancer
The phosphoinositide 3-kinase (PI3K)-protein kinase B-mammalian target of rapamycin pathway seems relevant in maintaining HR,94 thereby blocking PI3K sensitising to PARP inhibitors via BRCA1/2 downregulation in triple-negative breast cancer cells.112 However, robust data are lacking and clinical evidence only supports tolerability.113 MEK inhibition causes repression of both HR and NHEJ repair activity in PDAC cells. A clinical trial exploring these preclinical data is currently enrolling (NCT03162627; table 6). Furthermore, inhibition of the nuclear serine/threonine kinase WEE1 can induce HRDness in PDAC. The DNA damage checkpoint WEE1 impairs unscheduled replication origin firing and thus prevents nucleotide pool depletion and replication stress, ultimately resulting in DSBs.114 Preclinically, the WEE1 inhibitor AZD1775 sensitises to radiotherapy, an observation currently examined in a clinical trial (NCT02194829; table 6). Another approach might be molecules mimicking BRCA2 mutations that disrupt the RAD51-BRCA2 complex.115 Finally, clinical grade ATM inhibitors (AZD0156, KU60019, AZD1390), which directly prevent downstream ATM phosphorylation, can sensitise tumour cells to DDR interfering strategies. Preliminary data from the AToM study, a phase I clinical trial in advanced cancers, showed promising partial responses on a combination with olaparib.116
The limited success of treatments based on single DDR inhibiting agents is mostly due to compensatory pathways, drug toxicity and a lack of reliable response predictors. Moreover, the appropriate stratification of clinical trials for the different DDR alterations may help to identify the best treatment, as it has already done in prostate cancer (eg, Triton2 study, NCT02952534). Exploiting vulnerabilities by (i) seeking a synthetic lethal interaction within a given PDAC genotype and/or by (ii) a synergistic interaction in the low-dose range between applied drugs could boost both efficacy and tolerability.71 Thus, the multiple targeting of distinct DDR pathways could be a promising concept, provided that precise dose-escalation studies are warranted together with extensive preclinical testing. The combinatorial use of ATR inhibitors with conventional chemotherapy or PARP inhibitors in patients with ATM-deficient PDAC seems interesting, as it could assign HRDness to a given cancer.107 This is currently being assessed in various recruiting trials; for more details see table 6.
PDOs may in future also provide guidance for selecting the most appropriate combination(s). A prerequisite for such an approach, particularly in a disease like PDAC with extraordinary high intertumoural heterogeneity, is the sequencing of the tumour or the tumour DNA in liquid biopsies to determine the presence of a particular DDR mutation that can be appropriately targeted.
Homologous recombination deficiency and immunotherapy
Checkpoint inhibitors have revolutionised the treatment of some cancers, with the highest efficacy in mismatch repair-deficient tumours carrying a significant mutational burden. As mentioned above, MMR proteins correct the erroneous incorporation of bases during DNA replication. In PDAC, MMR deficiency is very rarely found in around 1% of cases and is associated with a better prognosis.117 MMR deficiency is predictive of an improved response to immunotherapy across multiple types of cancer including PDACs.118 PARP inhibitors can increase immune response and induce PD-L1 expression.117 Nonetheless, whether this establishes a synergistic axis between PARP and immune checkpoint inhibition in HRD PDAC remains unclear. In patients with PDAC, low ATM expression inversely correlates with PD-L1 expression, and the preclinical inhibition of ATM increased interferon signalling and sensitised to immune checkpoint inhibition.119 A phase Ib/II trial is currently recruiting to estimate the efficacy and safety of PARP inhibition with either ipilimumab (anticytotoxic T-lymphocyte-associated protein 4) or nivolumab (antiprogrammed cell death protein 1) (table 6; NCT03404960). Supporting data come from phase II trials combining olaparib with durvalumab (anti-PD-L1), showing tolerable toxicity and promising disease control rates in patients with breast and prostate cancer.120 ,121 In general, immunotherapy in PDAC appears to be restricted to cases with specific mutations leading to neoantigen expression. As in an unselected population with metastatic PDAC, ORR was far below expectations.122
Conclusions
Impaired DNA damage repair is a relevant characteristic of PDAC, frequently with an inherited origin. Loss-of-function mutations in genes involved in DNA damage repair justify therapeutic targeting with a platinum agent in the polychemotherapy and/or PARP inhibitors as a reasonable option, particularly for later lines of treatment. At least for BRCA-associated cancer types (pancreatic, prostate, breast or ovarian cancer), BRCA1/2 mutations remain clinically relevant, independent of their pathogenic or somatic origin. Maintenance with PARP inhibitors after induction chemotherapy is a promising approach that is likely to be incorporated in clinical practice for patients with a BRCA1/2 germline mutation. With the ultimate goal of hitting PDACs ‘hard and early’ and avoiding the emergence of resistant clones, more studies are urgently needed to demonstrate the efficacy of combinatorial approaches to DDR inhibition and to identify the best combinations for the respective targets in the DDR machinery. Furthermore, potential ‘HRDness inducers’ creating artificial vulnerabilities, in combination with DNA-damaging drugs such as PARP inhibitors and/or alternative DDR inhibitors, could provide a significant benefit for a larger group of patients with PDAC and open up a new era in the field of PDAC treatment.
Acknowledgments
We are deeply grateful to Kuhn Elektro-Technik GmbH for supporting our research to fight pancreatic cancer. The authors thank Sabine Geller from the Media and Design department of the University of Ulm for her support in illustration editing.
References
Footnotes
TS and AK contributed equally.
Correction notice This article has been corrected since it published Online First. The joint author statement has been added.
Contributors All authors wrote and revised the manuscript.
Funding The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: main funding was provided by the German Cancer Aid grant to AK (111879). Additional funding came from the Deutsche Forschungsgemeinschaft (DFG, KL 2544/1–1, 1–2, 5-1, 7-1), the BIU fund (Böhringer Ingelheim), the INDIMED-Verbund PancChip and the Else-Kröner-Fresenius Memorial funding to AK. AK also received funding from the DFG within the Heisenberg programme (KL2544/6-1) and from the Baden-Württemberg Foundation via ExPo Chip. ANR-DFG collaborative research project (ANR-18-CE92-0031, DFG KL 2544/5–1) to AK. AK, LW, TS were funded by the DFG HEIST RTG GRK 2254/1. LP was funded by Bausteinprogramm of Ulm University Hospital. LW was further supported by the German Cancer Aid Priority Program Translational Oncology (70112504).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.