Article Text

Abstract
Objective The intestinal microbiome affects the prevalence and pathophysiology of a variety of diseases ranging from inflammation to cancer. A reduced taxonomic or functional diversity of the microbiome was often observed in association with poorer health outcomes or disease in general. Conversely, factors or manifest diseases that determine the long-term stability or instability of the microbiome are largely unknown. We aimed to identify disease-relevant phenotypes associated with faecal microbiota (in-)stability.
Design A total of 2564 paired faecal samples from 1282 participants of the population-based Study of Health in Pomerania (SHIP) were collected at a 5-year (median) interval and microbiota profiles determined by 16S rRNA gene sequencing. The changes in faecal microbiota over time were associated with highly standardised and comprehensive phenotypic data to determine factors related to microbiota (in-)stability.
Results The overall microbiome landscape remained remarkably stable over time. The greatest microbiome instability was associated with factors contributing to metabolic syndrome such as fatty liver disease and diabetes mellitus. These, in turn, were associated with an increase in facultative pathogens such as Enterobacteriaceae or Escherichia/Shigella. Greatest stability of the microbiome was determined by higher initial alpha diversity, female sex, high household income and preserved exocrine pancreatic function. Participants who newly developed fatty liver disease or diabetes during the 5-year follow-up already displayed significant microbiota changes at study entry when the diseases were absent.
Conclusion This study identifies distinct components of metabolic liver disease to be associated with instability of the intestinal microbiome, increased abundance of facultative pathogens and thus greater susceptibility toward dysbiosis-associated diseases.
- colonic microflora
- fatty liver
- diabetes mellitus
- pancreas
- E. coli
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The gut microbiome represents a complex ecosystem exerting vital functions and its disturbance can contribute to the pathogenesis of many diseases—particularly when microbiome diversity is reduced.
Little, however, is known about which host phenotypes and diseases affect the long-term stability of the gut microbiome or render it instable.
What are the new findings?
At population level, the overall faecal microbiome community structure is remarkably stable over time.
Individuals suffering from metabolic liver disease, diabetes mellitus or related phenotypes show the vastest long-term microbiome perturbations.
Factors associated with the greatest long-term stability of the faecal microbiota composition are high initial microbial (alpha) diversity, female sex, high household income and preserved exocrine pancreatic function.
Microbiota instability in general, and specifically fatty liver disease and diabetes mellitus are linked to a long-term increase in opportunistic pathogens like Enterobacteriaceae or Escherichia/Shigella.
A specific gut microbiota signature is associated with an increased susceptibility toward incident metabolic liver disease.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Individuals suffering from metabolic liver disease or metabolic syndrome may exhibit greater susceptibility towards dysbiosis-associated disorders.
Whether attempts to maintain or restore the gut microbiome community structure can positively affect the onset and course of metabolic liver disease needs to be investigated in interventional trials.
Introduction
The human gut constitutes a habitat for the complex ecosystem which encompasses trillions of micro-organisms, together known as the intestinal microbiome. Bacterial cells represent the largest fraction of this biotope even outnumbering their human host by cell number.1 Jointly the gut microbiota carry out important metabolic functions,2 which are critical for the maintenance of human health.3 At the same time dysbiosis, a state of structural and functional microbiota disruption, can contribute to a variety of disorders including metabolic liver disease.4 5 The gut microbiota structure is not static over time but subject to continuous adaptive changes. The high interpersonal gut microbiota variation at a very young age decreases substantially until adulthood when the gut microbiome becomes more stable.6 But even afterwards, marked alterations as a response to environmental stimuli, for example, changes in diet, are possible but can still be largely reversible if the trigger is discontinued.7 Other environmental changes such as permanent shifts in geographic location, however, may cause enduring gut microbiota alterations.8 Ultimately, over time each individual faces numerous life events which will cumulatively shape the individual gut microbiota profiles. Previous investigations which tried to determine the specific changes occurring in the gut microbiota during adulthood mostly relied on cross-sectional data.6 9 This approach can be problematic as people of different ages have often been subjected to very different environmental conditions in the early years of their lives, which is a period of particular sensibility for the initial gut microbiota constitution.10 Moreover, cross-sectional analyses cannot determine whether and how the presence of an overt disease subsequently affects the microbiota composition and stability. Intraindividual comparisons using paired long-term follow-up data are therefore necessary to determine the contribution of different phenotypic factors to the gut microbiome changes over time. We therefore investigated intestinal microbiota profiles by 16S rRNA gene sequencing in 2564 paired faecal samples from 1282 participants of the Study of Health in Pomerania (SHIP), which were collected at two time points with a 5-year interval (SHIP-2 and SHIP-3, respectively) in this longitudinal cohort study (figure 1).

Scheme of study design. Paired microbiota profiles from Study of Health in Pomerania (SHIP)-2 and SHIP-3 participants (n=1282 at each time point) and corresponding SHIP-2 phenotype data were included into the analysis.
Methods
Study participants
SHIP is a longitudinal cohort study located in Northeast Germany.11 It was designed to determine the incidence and prevalence of subclinical disorders, clinical disease and their risk factors.12 13 The population-based sample was drawn in 1996 from a population of 213 057 using a two-stage stratified cluster approach including only individuals aged 20–79 years. The net sample included 6265 subjects of which 4308 participated in the study. Follow-up investigations are scheduled in 5-year intervals and have already been performed from 2002 to 2006 (SHIP-1, n=3300), 2008–2012 (SHIP-2, n=2333) and 2014–2016 (SHIP-3, n=1718). Faecal samples for microbiota determination were collected at SHIP-2 and SHIP-3. Paired faecal microbiota data at both study time points were available for 1366 individuals. Of these, 84 datasets were removed due to intake of antibiotics at the time of sample collection, a history of gastrointestinal cancer, liver cirrhosis or missing data on a history of liver cirrhosis, leaving 1282 datasets for analysis. All participants provided written informed consent and the study was approved by the local ethics committee.
16S rRNA gene sequencing
Sequencing was performed as described before in detail.14 15 In brief, faecal samples were collected by the study participants at home, stored in a tube containing stabilising DNA buffer and then transported to our laboratory by the participants or courier. After DNA from faecal samples was isolated (PSP Spin Stool DNA Kit; Stratec Biomedical AG, Birkenfeld, Germany), it was stored at −20°C until analysis by 16S rRNA gene sequencing of the V1–V2 region on a MiSeq platform (Illumina, San Diego, California, USA).
Assignment of taxonomic annotation and predicted metagenomics
More details on taxonomy assignment and predicted metagenomics are given in the online supplemental methods. In brief, the open-source software package DADA216 (V.1.10) was used for amplicon data processing which enables a single-nucleotide resolution of amplicons. The functional genomic potential of the faecal microbiota were predicted with PICRUSt2.17 All samples were normalised to 10 000 16S rRNA gene read counts for analysis.
Supplemental material
Metabolomic measurements
In a subsample of 911 SHIP-2 individuals plasma metabolites were determined using the AbsoluteIDQ p180 Kit (BIOCRATES LifeSciences AG, Innsbruck, Austria) on an AB SCIEX 5500 QTrap mass spectrometer (AB SCIEX, Darmstadt, Germany) with electrospray ionisation combined with a HPLC system (Agilent 1260 Infinity Binary LC, Santa Clara, California, USA). Following quality control and data processing, 177 metabolites were available for statistical analyses.
Phenotype data
Determination of laboratory parameters (alanine aminotransferase (ALT), creatinine, high-density lipoprotein (HDL), low-density lipoprotein (LDL), N-terminal prohormone of brain natriuretic peptide (NT-proBNP), thyroid-stimulating hormone (TSH)) was performed on a Dimension VISTA platform (Siemens Healthcare Diagnostics, Eschborn, Germany). Glycated haemoglobin (HbA1c) concentrations were determined using a high-performance liquid chromatography (Bio-Rad Diamat, Munich, Germany). Estimation of glomerular filtration rate (eGFR) was done using the CKD-EPI equation.18 NT-proBNP measurements were treated as missing in case of eGFR ≤30 mL/min due to possible accumulation. The trait diabetes mellitus was assigned to all individuals with a positive history of diabetes mellitus in combination with a history of current treatment (diet, medication or insulin), a random blood glucose ≥11.1 mmol/L or an HbA1c ≥6.5%. Alcohol consumption was estimated in grams alcohol per day, by counting the number of alcoholic beverages that had been consumed on average per day over the last 30 days and calculation of its average alcoholic content. Smoking was quantified as on average smoked cigarettes per day. Fatty liver disease (FLD) was determined based on ultrasound findings in case of hyperechoic liver tissue (in the absence of a history of liver cirrhosis) using a high-resolution device (Vivid i, GE Healthcare, Chicago, Illinois, USA) in B mode in combination with serum ALT levels belonging to the upper 25% of the investigated population. The trait arterial hypertension was assigned in case of antihypertensive medication or a systolic blood pressure ≥140 mm Hg or a diastolic blood pressure ≥90 mm Hg in the third of three consecutive measurements. Carotid intima–media thickness was measured bilaterally (Vivid i) and thereafter the average of both sides calculated. Manifest atherosclerotic disease (as opposed to carotid intima–media thickness) was assigned if the participants had a history of myocardial infarction and/or stroke. Hypothyroidism was assumed when TSH was ≥4 mU/L or in case of thyroid hormone replacement therapy. A food frequency score was used to estimate the balance of diet, with higher food frequency scores suggesting a more balanced diet as described elsewhere.19 20 For calculation of this score, ordinal consumption data from 15 food categories (meat, sausage, fish, boiled potatoes, pasta, rice, raw vegetables, boiled vegetables, fruits, whole grain/black/crisp bread, wheat oats/cornflakes, eggs, cake/cookies, sweets and savoury snacks) was evaluated. The net household income was evaluated in classes (‘1’: <€500, ‘2’: €500–€900, ‘3’: €900–€1300, ‘4’: €1300–€1800, ‘5’: €1800–€2300, ‘6’: €2300–€2800, ‘7’: €2800−€3300, ‘8’: €3300–€3800, ‘9’: >€3800). Exocrine pancreatic function was determined using a pancreatic elastase ELISA (BIOSERV Diagnostics, Germany) based on faecal samples as described before.14
Data analysis
All statistical analyses were performed using 'R' (v.3.6.3).21 Alpha diversity scores were determined with the 'diversity' function (vegan).22 The Bray-Curtis (BC) dissimilarity, Euclidean and Jaccard distance were calculated using the function 'vegdist' (vegan). Principal coordinate analysis (PCoA) was carried out by the 'cmdscale' function (vegan). The emergence of negative eigenvalues during PCoA was avoided by square root transformation of the BC dissimilarity prior to the ordination. To test for significant differences of the PCo1–PCo5 scores between SHIP-2 and SHIP-3, paired two-sided Wilcoxon-signed rank tests were performed. For permutational analysis of variance, the function 'adonis' (vegan; 1000 permutations) was used. Comparison of the relative abundance data between SHIP-2 and SHIP-3 of all taxa that were present in at least 10% of all SHIP-2 samples and with a mean abundance of at least 0.1% was done using the paired two-sided Wilcoxon-signed rank tests. Comparison of the taxon presence–absence profiles between SHIP-2 and SHIP-3 of all taxa that were present in at least 10% but less than 80% of all samples was performed using McNemar’s test ('mcnemar.exact' function, exact2×2). Differences in genomic pathways between SHIP-2 and SHIP-3 were evaluated using the paired two-sided Wilcoxon-signed rank tests. Only pathways with a presence of at least 25% of samples at SHIP-2 were considered for this analysis. Alpha diversity scores of SHIP-2 and SHIP-3 samples were compared using the paired two-sided Wilcoxon-signed rank test. Assessment of association between phenotypic factors at SHIP-2 and faecal microbiota changes toward SHIP-3 (microbial (in-)stability) expressed as log-transformed BC dissimilarity, Euclidean or Jaccard distance was performed as follows: metric variables were cleaned from outliers which were more than 3 SD away from the mean. Metric variables without zeros (eg, HbA1c) were log-transformed, whereas square root transformation was performed in case of present zeros (cigarettes per day or alcohol consumption). Afterwards, Pearson, Spearman or point biserial correlations were calculated for metric (age, alcohol, body mass index (BMI), carotid intima–media thickness, HbA1c, LDL/HDL ratio, eGFR, pancreatic elastase, number of household members, NT-proBNP, species richness (N0), smoking, TSH), ordinal (food frequency score, net household income) or nominal data (arterial hypertension, diabetes mellitus, dyslipidaemia, FLD, female sex, hypothyroidism, manifest atherosclerotic disease, proton pump inhibitor usage), respectively ('cor.test' function, stats). Afterwards, the significance of those factors identified to correlate with microbiota (in-)stability was confirmed by performing a linear regression model including possible confounding factors as stated in the online supplemental table S4. The associations of incident diseases at SHIP-3 with SHIP-2 and SHIP-3 microbiota profiles were determined by performing permutational analysis of variance (vegan function 'adonis'; 1000 permutations). For linear regression analysis, microbiota data were cleared from zeros (treated as missing), log-transformed and standardised. Metabolome data were also log-transformed and standardised before linear regression analysis. Included covariates for regression models are listed in the online supplemental tables S4–S11 of the respective analyses. All p-values derived from comparisons of PCo scores, phenotype–microbiota associations, taxon comparisons of relative abundance, presence–absence, genomic pathway or alpha diversity data were corrected for multiple testing by the method of Benjamini-Hochberg ('p.adjust' function, stats) and thereafter called q-values. P-values or q-values <0.05 were considered significant. All p-values and q-values were rounded to three significant digits.
Supplemental material
Data availability
All microbiome and phenotype data were obtained from the SHIP data management unit and can be applied for online through a data access application form (https://www.fvcm.med.uni-greifswald.de/dd_service/data_use_intro.php).
Patient and public involvement statement
SHIP is a regionally confined epidemiological study that regularly informs participants about current investigations and recent findings by newsletter, YouTube videos (eg, https://www.youtube.com/watch?v=t7C8dGNJ_rk) and its website (https://www2.medizin.uni-greifswald.de/cm/fv/ship/). Participants serve on the advisory board of the SHIP study and SHIP organises public hearings for question and answer sessions and for input from the public regarding its research focus.
Results
Overall faecal microbiota community profile
The overall faecal microbiota community profile was remarkably stable over time with Bacteroides, Prevotella and Faecalibacterium remaining the three most abundant taxa at follow-up (figure 2A). To further investigate the temporal stability of the faecal microbiota, an analysis of beta diversity, which provides information how microbiota communities or samples differ from each other, was carried out using BC dissimilarity. Subsequent PCoA based on BC including all paired 2564 baseline and follow-up samples confirmed a largely stable faecal microbiota composition between SHIP-2 and SHIP-3 (figure 2B). However, detailed analysis of the PCo scores revealed significant structural microbiota changes (figure 2C) indicating specific microbiota shifts in some individuals.

Stability of the faecal microbiome from Study of Health in Pomerania (SHIP)-2 to SHIP-3. (A) Stacked bar plots show the average faecal microbiota composition at SHIP-2 and SHIP-3. (B) Principal coordinate analysis (PCoA) of 2564 faecal microbiota samples obtained from 1282 individuals at SHIP-2 (orange dots) and SHIP-3 (blue dots). (C) Boxplots display the PCo1–PCo5 scores comparing SHIP-2 (orange boxes) to SHIP-3 (blue boxes) samples. Numbers in brackets below denote the percentage of variation explained by the respective PCo. The overall faecal microbiome landscape was quite stable from SHIP-2 to SHIP-3 with small but significant shifts along PCo1, PCo3 and PCo5. (D) Shown are the percentage changes in abundance of the 18 taxa with a significant increase (red) or decrease (blue) in abundance over time. All taxa are ordered according their mean abundance at SHIP-2 from top (most abundant) to bottom (least abundant). (E) Analysis of the taxon presence distribution from SHIP-2 to SHIP-3. Shown are the ORs and their respective 95% CIs at SHIP-3 compared with SHIP-2 as baseline of all 16 taxa with significant changes in their presence profiles. An OR over 1 indicates an increased likelihood for the respective taxon to be present in a SHIP-3 sample if it was not present in the same subject at SHIP-2, whereas an OR below 1 implies the loss of the respective taxon in SHIP-3 samples compared with SHIP-2. All taxa are ordered according to the percentage of samples with presence of the respective taxon at SHIP-2 from top (high presence) to bottom (low presence). (F) Comparison of alpha diversity between SHIP-2 and SHIP-3. Species richness (N0) was slightly reduced at follow-up, whereas no change was observed in Simpson diversity number (N2) or Shannon diversity index (H). Whiskers are drawn up to 1.5 times the IQR (outliers not shown). *Indicates significant difference. c, class; f, family; o, order; p, phylum.
Determination of changes in taxon abundance and predicted metagenomics from baseline to follow-up
A total of 18 taxa were identified with significant abundance changes at follow-up compared with baseline (figure 2D and online supplemental table S1). A comparison between baseline and follow-up samples based on presence–absence data revealed 16 taxa with significantly altered presence profiles (figure 2E and online supplemental table S2). No major changes were found in alpha diversity, which describe the taxon variation within a sample, although median N0 was slightly lower at follow-up (q=0.012) compared with baseline (figure 2F). Corresponding to the changes in several taxa, alterations in 164 predicted genomic microbial pathways were found (online supplemental table S3, figures S1 and S2). These pathways belonged to the MetaCyc23 superclasses biosynthesis (n=100), degradation/utilisation/assimilation (n=43), generation of precursor metabolites and energy (n=16) and others (n=5) (figure 3). Several of the metabolic pathways for short-chain fatty acid (SCFA) production were either increased or decreased in abundance, similarly to the taxonomic changes in SCFA producers, yet the combined predicted microbial genetic potential for SCFA production, however, did not change. Analysis of taxon abundances revealed several Gram-negative bacteria such as Enterobacteriaceae, Escherichia/Shigella and Citrobacter to be increased at follow-up (figure 2D). Correspondingly, the genetic microbial potential for the production of lipopolysaccharides (LPS) was increased at SHIP-3 compared with SHIP-2.
Supplemental material
Supplemental material
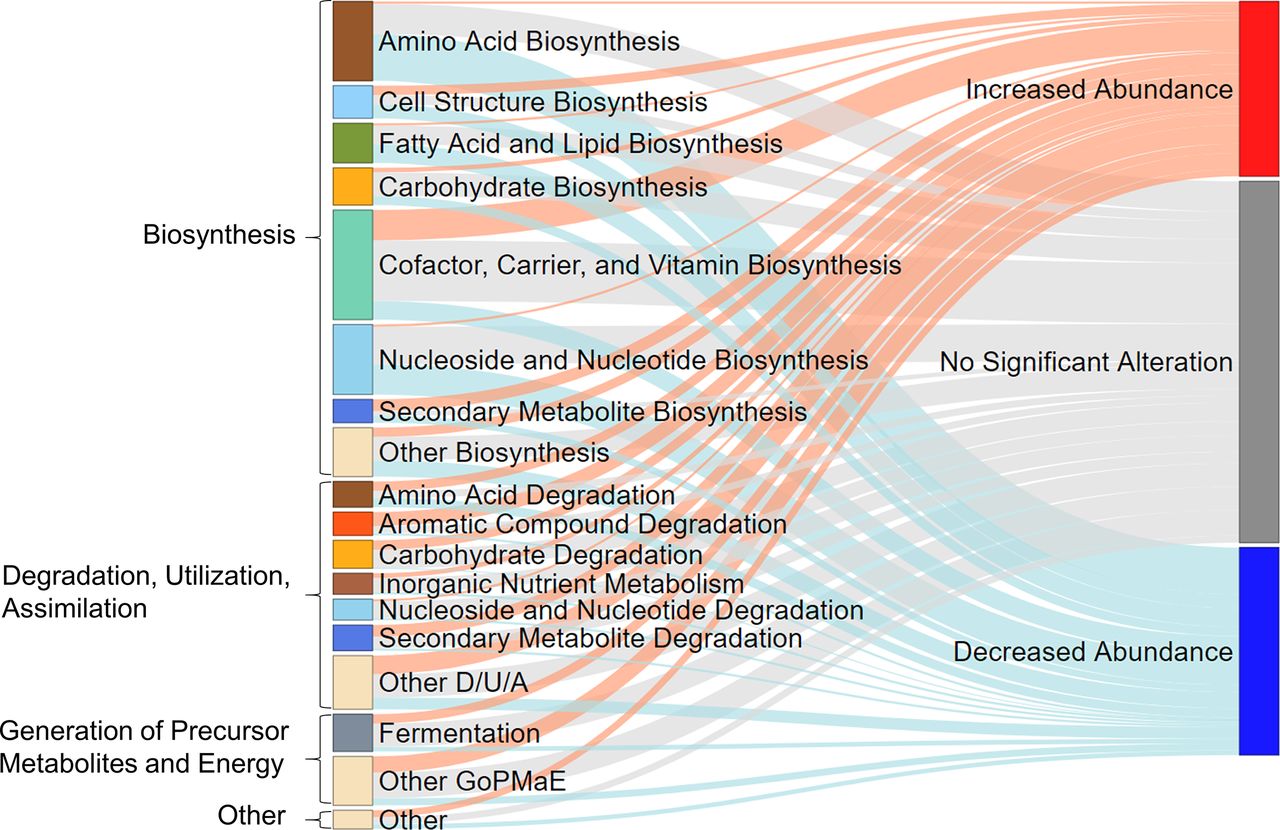
Sankey plot displays the proportions of predicted metagenomic pathways with significantly (q<0.05) increased or decreased abundance or no significant alteration. D/U/A, degradation, utilisation, assimilation; GoPMaE, generation of precursor metabolites and energy.
Determination of phenotypic factors associated with stability or instability of the faecal microbiome
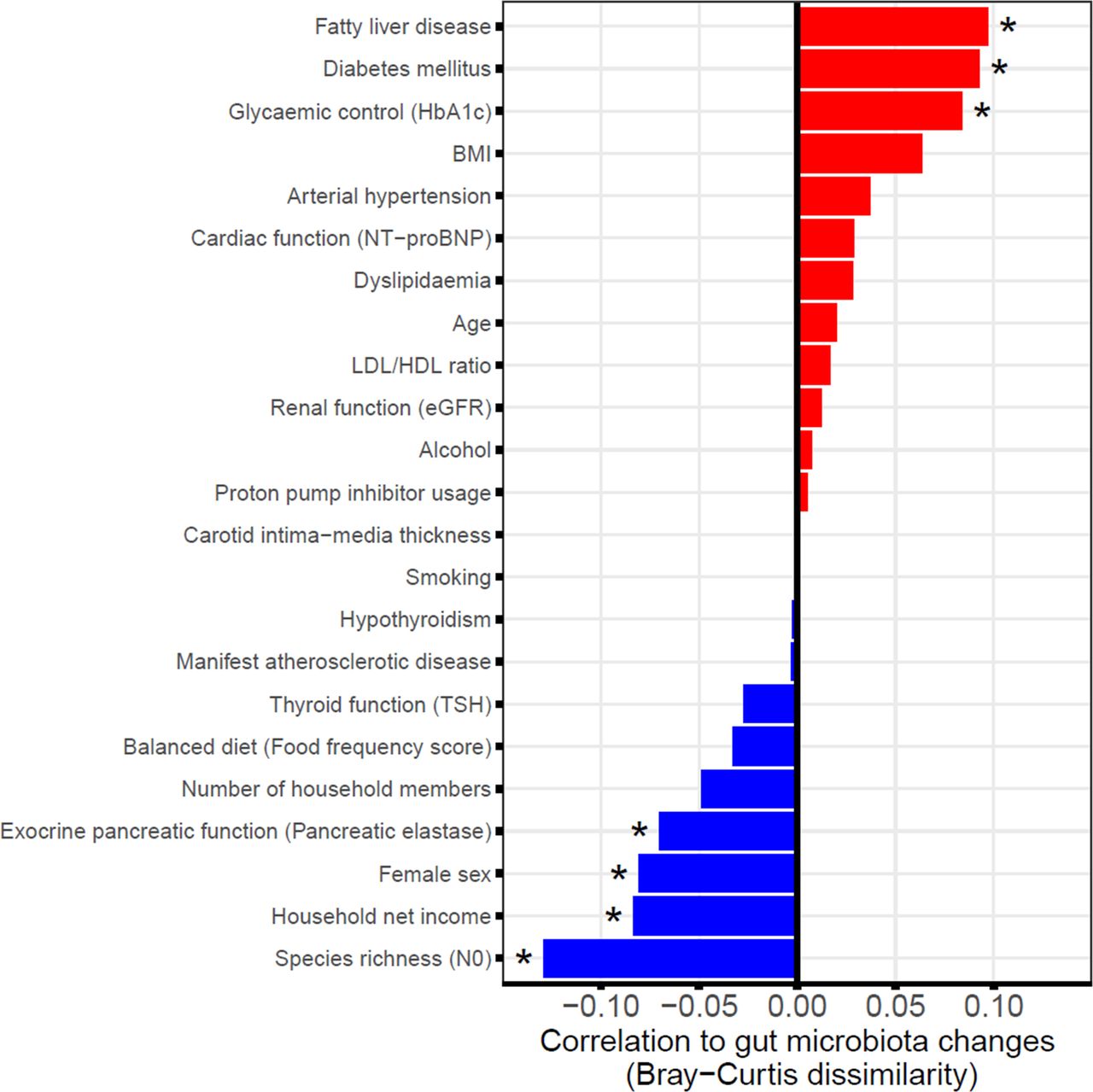
Several mostly cross-sectional studies have been conducted to identify the factors that contribute to the gut microbiota variation showing associations with age, sex, exocrine pancreatic function, obesity, diabetes mellitus, FLD, diet or medication among other factors.7 14 24–27 We therefore investigated whether these and other phenotypic factors (table 1) were also associated either positively or negatively to longitudinal faecal microbiota changes from baseline to follow-up (expressed as beta diversity). As a result, numerous host characteristics were found to be associated with either increased or decreased faecal microbiota stability (figure 4). A positive correlation with changes in the faecal microbiome (BC) over time was shown for FLD (q=0.006), diabetes mellitus (q=0.007) and the level of glycaemic control (HbA1c, q=0.014). An increase in BMI exhibited a smaller positive but non-significant (q=0.067) association. Several other phenotypic traits were predictive of faecal microbiota stability (negative association to BC), among them high initial microbial diversity (N0 at SHIP-2, q<0.001), female sex (q=0.015), high net household income (q=0.014) and preserved exocrine pancreatic function (q=0.039). Additional regression analyses with adjustment for possible confounding factors (online supplemental table S4A), including the oral antidiabetic medications metformin28 and dipeptidyl peptidase-4 (DPP-4) inhibitors29 in case of the traits diabetes mellitus and glycaemic control confirmed the significance of the aforementioned associations. Using Jaccard distance (online supplemental figure S3A and table S4B) as measurement of beta diversity, which only includes presence/absence information of the bacteria, additionally revealed PPI usage to be positively associated with larger gut microbiota changes whereas a balanced diet was linked to gut microbiota stability over time. Using the Euclidean distance instead did not reveal further additional findings (online supplemental figure S3B and table S4C).
Supplemental material
Distribution of phenotype variables at SHIP-2

Contribution of phenotypic factors to faecal microbiome changes. Shown are the correlations between different phenotypic factors at SHIP-2 and global changes in the faecal microbiome from SHIP-2 to SHIP-3. Large positive associations indicate greater variation or instability in the faecal microbiome if the respective factor is distinctly elevated (continuous variables) or positive (binary variables). On the other hand, large negative associations suggest stability of the faecal microbiome. *Indicates significant associations (q<0.05). BMI, body mass index; eGFR, estimated glomerular filtration rate; HbA1c, glycated haemoglobin; HDL, high-density lipoprotein; LDL, low-density lipoprotein; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; SHIP, Study of Health in Pomerania; TSH, thyroid-stimulating hormone.
Contribution of microbial instability, FLD and diabetes mellitus to levels of facultative pathogenic bacteria
Taxon abundance comparisons between baseline and follow-up exhibited an increase of several facultative pathogenic organisms (Enterobacteriaceae, Escherichia/Shigella and Citrobacter) over time (p<0.001) (online supplemental figure S4 and table S5). Using a linear regression model, not only microbial instability (BC, p=0.027) but also FLD (p=0.038) and diabetes mellitus (p=0.007) were associated with the increase in abundance of facultative pathogens toward SHIP-3. Correspondingly, microbial instability (p=0.013) and diabetes mellitus (p=0.021) were also associated with an increase in the predicted microbial potential for LPS biosynthesis toward SHIP-3.
Supplemental material
Incident metabolic and cardiovascular disease at SHIP-3 and its association to faecal microbiota and plasma metabolites
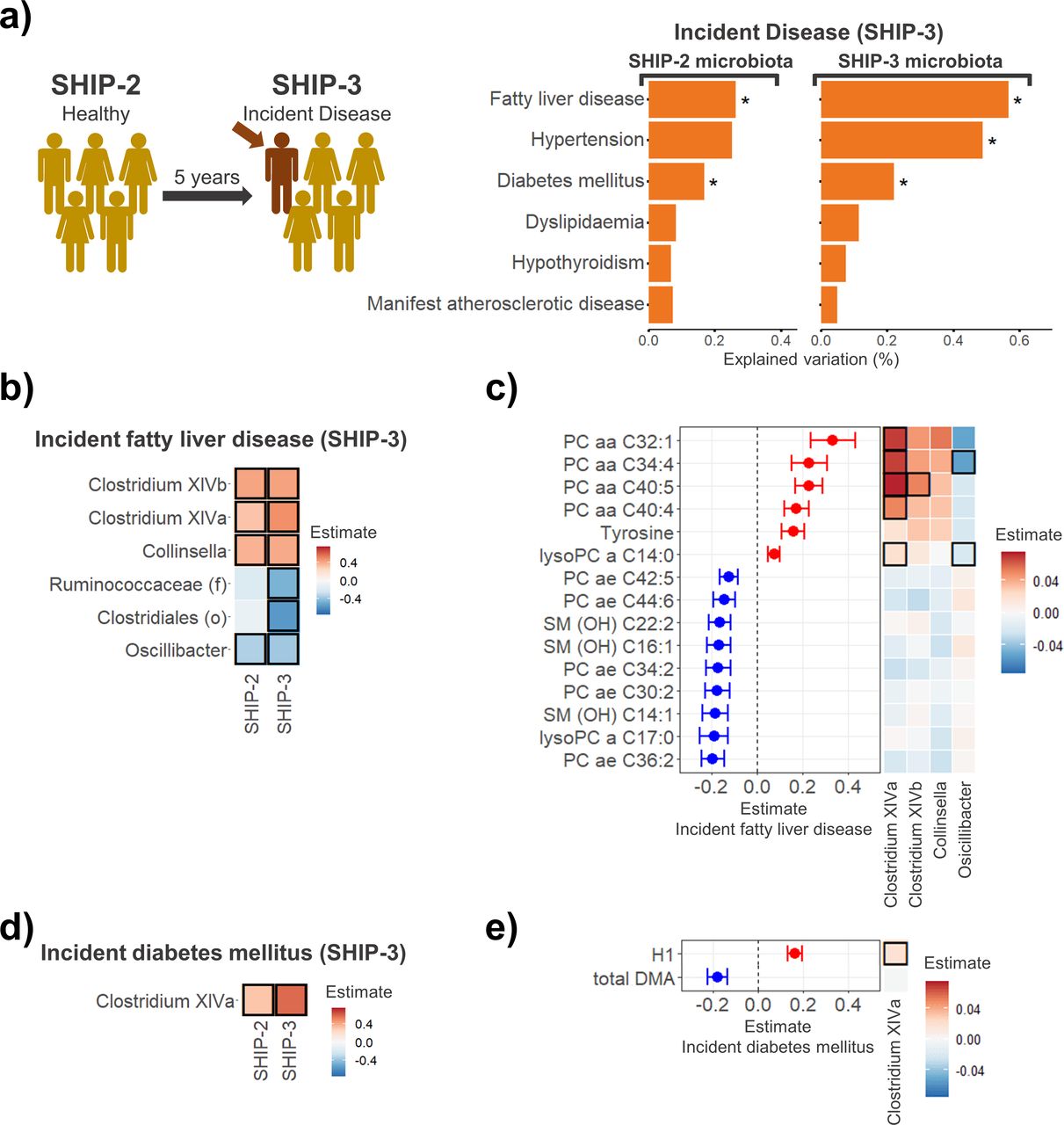
A significant proportion of participants who did not show signs of diabetes mellitus, FLD, dyslipidaemia, hypertension, manifest atherosclerosis or hypothyroidism were classified as having acquired the respective condition 5 years later in SHIP-3 (figure 5A and online supplemental table S6). We investigated whether the microbiota profiles of those individuals who had newly developed any of these diseases in SHIP-3 differed from those who remained disease free at both time points. Individuals newly classified with FLD or diabetes mellitus at SHIP-3 exhibited a significantly different gut microbiome composition compared with participants without the respective disorder (p<0.001 and p=0.008, respectively). This association could already be found at SHIP-2 before FLD or diabetes mellitus had become apparent (p=0.009 and p=0.034). Incident hypertension (p=0.003) also exhibited a significant link to the gut microbiome at SHIP-3, but not in SHIP-2. No significant link to the faecal microbiome was found for incident cases of manifest atherosclerosis, dyslipidaemia or hypothyroidism. This indicates that participants who newly developed diabetes mellitus or FLD during the 5-year follow-up already displayed significant microbiota composition changes at study entry when the diseases were still absent.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Associations between incident metabolic and cardiovascular diseases at SHIP-3 and faecal microbiota changes in the affected individuals at SHIP-2 (before the respective disease was present). (A) Individuals with incident fatty liver disease and diabetes mellitus at SHIP-3 already displayed gut microbiota changes at SHIP-2 prior to their diagnosis. (B) Associations of incident fatty liver disease with microbial taxa at SHIP-2 and SHIP-3. (C) Significant metabolite associations of incident fatty liver disease cases at SHIP-2 (left). The heatmap (right) indicates the association of the different disease-associated genera with the respective metabolites. (D) Associations of incident diabetes mellitus at SHIP-3 with microbial taxa at SHIP-2 and SHIP-3. (E) Significant metabolite associations of incident diabetes mellitus cases at SHIP-2 (left). The heatmap (right) indicates the association of the different disease-associated genera with the respective metabolites. Significant results are highlighted by a black frame. Unclassified taxa at genus level: (f): family, (o): order. DMA, dimethylarginine; H1, hexose; PC, phosphatidylcholine; SHIP, Study of Health in Pomerania; SM (OH), hydroxylated sphingomyelin.
To further explore these changes in individuals with incident FLD or diabetes mellitus, we performed linear regression analysis adjusting for different possible confounding factors (online supplemental tables S7–11). Individuals with incident FLD at SHIP-3 already presented with changes in Clostridium XIVa, Clostridium XIVb, Collinsella and Oscillibacter as well as 15 plasma metabolite levels prior to their diagnosis at SHIP-2 (figure 5B,C and online supplemental tables S7 and 8). Clostridium XIVa did not only associate with increased plasma levels of various phosphatidylcholines but also exhibited a strong association with an increase of gut microbial pathways for fatty acid biosynthesis which was independent of the individual’s diet (food frequency score) and BMI (online supplemental table S9). The gut microbiota and plasma metabolome alterations at SHIP-2 in individuals with incident diabetes mellitus at SHIP-3 were of lesser extent (figure 5D,E and online supplemental tables S10 and S11).
Discussion
In the present study, we performed a long-term intestinal microbiota follow-up of 1282 individuals to identify the phenotype factors that (de-)stabilise the microbiome. Overall, the global faecal microbiota profile of the whole population showed a largely stable composition over time. Levels of alpha diversity remained stable except for a small reduction in N0. However, levels of facultative pathogenic taxa such as Enterobacteriaceae, Escherichia/Shigella and Citrobacter were consistently increased at follow-up after 5 years. This finding was pronounced in subjects carrying a phenotype associated with less stable microbiota composition. Members of the family of Enterobacteriaceae, especially Escherichia coli, represent the most common pathogens found in either blood or bile from individuals suffering from acute cholecystitis or cholangitis,30 31 abdominal abscesses (eg, liver abscesses)32 or urinary tract infections.33 Similarly Citrobacter, another member of Enterobacteriaceae, has been identified in urinary tract infections or intra-abdominal abscesses.34 The order Enterobacterales, which comprises the aforementioned facultative pathogens, has also been linked to increased intestinal permeability in humans.35 Therefore, greater microbiota instability with increased abundance of these potential pathogens may have harmful consequences for its human hosts by increasing the risk for localised or systemic infections. Moreover, at follow-up the predicted microbial pathways for LPS biosynthesis were increased by 27%. This increase was again pronounced in subjects with less stable faecal microbiota composition, for example, diabetics. LPS triggers the production of proinflammatory cytokines and can contribute to the development of FLD and diabetes mellitus.36–38 Hence, the microbial instability may predispose to further increases in LPS secretion activity which in turn can aggravate the metabolic disorders in a vicious circle.
A further, probably disadvantageous, faecal microbiota alteration was a reduction in the amount of Bifidobacteria carriers and the predicted genetic potential for bifidobacterial lactate production which could not be attributed to other phenotypic factors. Bifidobacteria have been reported to stabilise the gut barrier by reducing its permeability for inflammatory bacterial molecules such as LPS and to alleviate LPS-associated inflammation in the liver.39–41
The individual extent of the faecal microbiota variation between SHIP-2 and SHIP-3 was variable. Disorders contributing to metabolic syndrome, metabolic liver disease and diabetes mellitus were associated with faecal microbiota instability. In our study, the association of diabetes mellitus with faecal microbiota variation could not be explained by oral antidiabetic medication such as metformin or DPP-4 inhibitors, both of which have previously been shown to be independently associated with composition changes of the gut microbiome.28 29 When analysing changes in presence/absence patterns, we also found PPI usage to be linked to gut microbiota instability over time, which supports the finding of gut microbiota changes in PPI usage from a previous cross-sectional study.42 Of note, other conditions such as atherosclerosis, cardiac function, thyroid dysfunction or renal impairment had no significant impact. Participants’ older age was not associated with faecal microbiota instability. This suggests that as long as metabolic disease is absent, even at higher age, the gut microbiome may remain largely stable and resilient to perturbations.
On the other hand, several factors which were related to greater microbiota stability could be identified. The strongest negative association was found for alpha diversity at study entry, implying a microbiome stabilising effect. Diverse communities are less prone to the introduction of foreign species because of a greater coverage of ecological niches.43 A prominent example of how low microbial diversity can negatively affect the gut microbial integrity is Clostridium difficile, for which in vitro studies have shown an increased growth in dysbiotic microbiota with low diversity.44 In humans, this situation typically occurs after antibiotic administration, a risk factor for Clostridium difficile-associated colitis. A further protective factor of the faecal microbiome stability was preserved exocrine pancreatic function. Exocrine pancreatic function has already been shown to be the most important host factor regulating the faecal microbiota composition in a given population,14 which now includes a prominent role in determining long-term stability of the microbiome. This relationship may be explained by changes in the intestinal chyme and nutrient supply, due to maldigestion when pancreatic enzyme activity is reduced or due to impaired secretion of pancreatic antimicrobial peptides.45 The increasing faecal microbiota stability seen in individuals with higher socioeconomic status (net income) may be mediated by a reduced prevalence of disease in general, an increased utilisation of health preventive measures and/or a healthier lifestyle.
Interestingly, individuals who newly developed FLD (and to a lesser degree also diabetes mellitus) at SHIP-3 already displayed significant faecal microbiota and plasma metabolome alterations at SHIP-2 when the respective diseases were absent. Clostridium XIVa, which was increased in these individuals, also associated with an increased expression of those microbial pathways necessary for biosynthesis of saturated and unsaturated fatty acids, some of which have been described to promote hepatic lipogenesis46 47 or are correlated to increased hepatic lipogenesis.48 Therefore, fatty acids synthesised by Clostridium XIVa may increase hepatic accumulation of lipids, thus contributing to the development of FLD toward SHIP-3. Whether Clostridium XIVa could serve as a treatment target for prebiotic or probiotic therapy to reduce the incidence of FLD needs to be determined in future interventional trials.
The advantage of this investigation is the usage of longitudinal microbiome data using paired samples at two time points to overcome the limitations of cross-sectional investigations. Moreover, the extensive phenotype database of the SHIP study allowed to identify critical host factors for faecal microbiota stability. A disadvantage of the study is the use of population-based data. Only the faecal microbiota relationship with conditions or states that are frequent enough to generate sufficient statistical power can be analysed in population-based cohorts, whereas estimation of the effect of less common or rare disorders on microbiota stability and composition requires disease-specific patient cohorts.49
Here we present results from a large-scale faecal microbiota follow-up study over 5 years. With ageing, the amount of facultative pathogenic bacteria increased. However, ageing itself was not a predictor of faecal microbiota changes, whereas metabolic disorders were. Why some components of metabolic syndrome such as diabetes mellitus and FLD have a greater association than others with faecal microbiota instability and whether this is due to associated changes in gut immunology or variations in intraluminal nutrient supply needs to be investigated in human interventional trials, in which the respective disease process can actually be reversed. The association of increased instability of microbiota composition over time with metabolic syndrome is clearly suggestive of a direct causality. The observation, however, that participants who newly developed diabetes mellitus or FLD at the later time point, but 5 years earlier had no detectable manifestation of the disease, while already displaying significant changes in microbiota composition, clearly indicates that the relationship is more complex. On the other hand, a greater diversity of the intestinal microbiome, female sex and preserved exocrine pancreatic function were found to promote a greater long-term stability of the faecal microbiome and may therefore convey greater resilience against common, dysbiosis-associated disorders.
Acknowledgments
We thank Diana Krüger, Sybille Gruska, Anja Wiechert, Susanne Wiche, Doris Jordan, Ilona Urbach, Tonio Hauptmann and Ines Wulff for expert technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @mruehlemann
Contributors Author contributions to the manuscript: Planning and concept of study: MML, HV, UV, JM, AF. Acquisition of data: FF, TK, MR, CB, FUW, MS, MP, HV. Statistical analysis: FF, TK, MP, MR. Data interpretation and manuscript revision: FF, TK, GH, MML, JM, HV, UV, MN, MP, CS, AF, MR, CB, JB, MS, MD, FUW. Writing committee: FF, MML.
Funding This work was supported by the PePPP Project (ESF/14-BM-A55_0045/16), the EnErGie/P2 Project (ESF/14-BM-A55-0008/18) and the RESPONSE project (BMBF grant number 03ZZ0921E). SHIP is part of the Research Network Community Medicine of the University Medicine Greifswald which is supported by the German Federal State of Mecklenburg-West Pomerania.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the local institutional review board, University Medicine Greifswald, Germany, registration numbers BB 39/08 and BB 122/13.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data may be obtained from a third party and are not publicly available. All microbiome and phenotype data were obtained from the SHIP data management unit and can be applied for online through a data access application form (https://www.fvcm.med.uni-greifswald.de/dd_service/data_use_intro.php).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.