Article Text

Abstract
Objective Intrahepatic cholangiocarcinoma (ICC)—a rare liver malignancy with limited therapeutic options—is characterised by aggressive progression, desmoplasia and vascular abnormalities. The aim of this study was to determine the role of placental growth factor (PlGF) in ICC progression.
Design We evaluated the expression of PlGF in specimens from ICC patients and assessed the therapeutic effect of genetic or pharmacologic inhibition of PlGF in orthotopically grafted ICC mouse models. We evaluated the impact of PlGF stimulation or blockade in ICC cells and cancer-associated fibroblasts (CAFs) using in vitro 3-D coculture systems.
Results PlGF levels were elevated in human ICC stromal cells and circulating blood plasma and were associated with disease progression. Single-cell RNA sequencing showed that the major impact of PlGF blockade in mice was enrichment of quiescent CAFs, characterised by high gene transcription levels related to the Akt pathway, glycolysis and hypoxia signalling. PlGF blockade suppressed Akt phosphorylation and myofibroblast activation in ICC-derived CAFs. PlGF blockade also reduced desmoplasia and tissue stiffness, which resulted in reopening of collapsed tumour vessels and improved blood perfusion, while reducing ICC cell invasion. Moreover, PlGF blockade enhanced the efficacy of standard chemotherapy in mice-bearing ICC.
Conclusion
PlGF blockade leads to a reduction in intratumorous hypoxia and metastatic dissemination, enhanced chemotherapy sensitivity and increased survival in mice-bearing aggressive ICC.
- cholangiocarcinoma
- hepatic fibrosis
Data availability statement
Data are available on reasonable request. Data are available on reasonable request from the corresponding author DGD. The authors used deidentified participant data at Fundeni Clinical Institute, Bucharest, Romania, University Hospital Cologne, Germany, and Massachusetts General Hospital, Boston, USA.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Intrahepatic cholangiocarcinoma (ICC) is a rare liver malignancy with limited therapeutic options at advanced stages (chemotherapy).
ICC is characterised by aggressive progression, desmoplasia and vascular abnormalities.
What are the new findings?
Placental growth factor (PlGF) is highly expressed in ICCs compared with normal liver tissue, primarily in tumour stromal cells, and is associated with poor outcomes.
PlGF blockade enriched for more quiescent, less activated cancer-associated fibroblasts, characterised by high gene transcription levels related to the Akt pathway, hypoxia and glycolysis.
PlGF blockade reduced desmoplasia, decreased tissue stiffness and reopened collapsed tumour vessels in vivo, resulting in improved blood perfusion, a reduction in intratumoral hypoxia and metastatic dissemination, enhanced chemotherapy sensitivity and increased survival in mice-bearing ICC.
How might it impact on clinical practice in the foreseeable future?
These findings demonstrate a new mechanism by which PlGF mediates ICC desmoplasia and progression.
Blockade of PlGF is an immediately translatable strategy to reprogramme the desmoplastic and hypoxic ICC microenvironment and inhibit its progression and resistance to standard chemotherapy.
Introduction
Intrahepatic cholangiocarcinoma (ICC) is a cancer type encompassing diverse epithelial tumours arising in the liver.1 ICC has an aggressive phenotype and limited therapeutic options, which makes its prognosis dismal.2 Moreover, ICC incidence is increasing.3 Overall survival rate remains extremely poor: 5-year survival rates are 15% for patients with early-stage ICC, 6% for those with regional lymph node involvement and 2% for those with metastatic disease. Meanwhile, various genetic alterations in ICC have been identified, leading to multiple clinical trials of molecularly targeted agents for advanced disease.4 5 However, these approaches benefit only the fraction of patients with appropriate genetic alterations. Systemic chemotherapy using gemcitabine and cisplatin (Gem/Cis) remains the standard of care for all patients with advanced ICC, but the benefits are limited.3 6 New treatment options are urgently needed.
A defining feature of ICC is the abundant tumour stroma (desmoplasia), which has complex functional interactions with cancer and normal stromal cells.7 The desmoplastic stroma consists of cancer-associated fibroblasts (CAFs), which closely surround the neoplastic bile ducts and can provide ICC cells with proliferative and prosurvival/antiapoptotic signals. These CAFs may be activated hepatic stellate cells (HSCs) or portal fibroblasts with a myofibroblast-like phenotype.8–10 Activated CAFs produce extracellular matrix (ECM) components and communicate with endothelial, perivascular, immune and cancer cells to induce angiogenesis and immunosuppression, thus promoting tumour progression.10 In addition, the characteristic vascular abnormalities and tissue hypoxia and desmoplasia also impede drug delivery into the tumours, thereby contributing to resistance to conventional chemotherapy.11
Placental growth factor (PlGF), which is a member of the vascular endothelial growth factor (VEGF) family, binds to VEGFR1 but not VEGFR2, as well as to neuropilin-1 and -neuropilin2 (Nrp1 and Nrp2). PlGF binding to VEGFR1 and Nrp1 on endothelial cells can lead to a crosstalk between Nrp1, VEGFR1 and VEGFR2, which consequently enhances VEGF-driven responses.12 However, PlGF can also transmit prosurvival and promigration signals to cancer cells directly through Nrp1 in gastrointestinal cancer.13 Conversely, tumour cells can produce PlGF to activate stromal cells and promote angiogenesis, inflammatory and stromal cell recruitment and metastatic progression.14–17
PlGF expression, unlike that of VEGF, is undetectable in healthy conditions but is highly upregulated in pathological conditions, including hypoxia.18 Therefore, PlGF inhibition is expected to selectively inhibit pathological angiogenesis, without affecting the growth or maintenance of physiological vessels, which is likely to cause fewer side effects.14 19 In support of this, blockade of PlGF in mouse models has resulted in antitumour effects mediated by changes on myeloid cells.15 Similarly, Nrp1 expression level is significantly higher in cholangiocarcinoma tissues compared with adjacent normal biliary tissues, as detected by quantitative real-time-PCR analyses and immunohistochemistry (IHC).20 Nrp1 inhibition also reduced the proliferation and migration of cholangiocarcinoma cells and suppressed tumour growth and lung metastasis in experimental models.20 Previously, we reported that the PlGF/Nrp1 axis in cancer cells is a driver of paediatric medulloblastoma.21 This work has led to a phase I trial in children with brain tumours (NCT02748135).
Here, we examined the impact of PlGF inhibition on the desmoplastic stroma and ICC cell viability and invasion. We also evaluated the impact of genetic or pharmacologic PlGF inhibition on tumour growth and metastatic progression in mice bearing orthotopic ICC in p53-null/Kras-mutant and Idh/Kras-mutant driven tumours, and on tumour response to chemotherapy.
Materials and methods
Mouse models of ICC and treatments
We used two models of orthotopically implanted ICC, using murine ICC cell lines derived from spontaneously arising tumours.22 23
Primary culture and isolation of CAFs from murine tumours
CAFs were isolated from fresh ICC tissues by the outgrowth method, as previously described.24 25
Single-cell RNA sequencing: droplet encapsulation and library preparation
Single-cell RNAseq (scRNAseq) was performed using a modified version of the DropSeq protocol.26
Measurement of Young’s modulus (stiffness)
The Young’s modulus (stiffness) of tumours was determined by an unconfined compression test.27
For additional details, see online supplemental methods and table S1.
Supplemental material
Supplemental material
Results
PlGF expression is increased with ICC progression in patients
We examined tissues from an archival cohort of patients diagnosed with different disease stages of ICC (N=43). We assigned grading scores based on staining intensity and area (negative, low, moderately high and high) using IHC28 29 (online supplemental table S2). The vast majority of cases showed moderately high/high expression for both PlGF and Nrp1; only 14% of cases were negative for PlGF expression (online supplemental figure S1A). Moreover, PlGF expression scoring was significantly associated with disease stage (p=0.042) (online supplemental figure S1B) but not with overall survival (not shown).
Supplemental material
Supplemental material
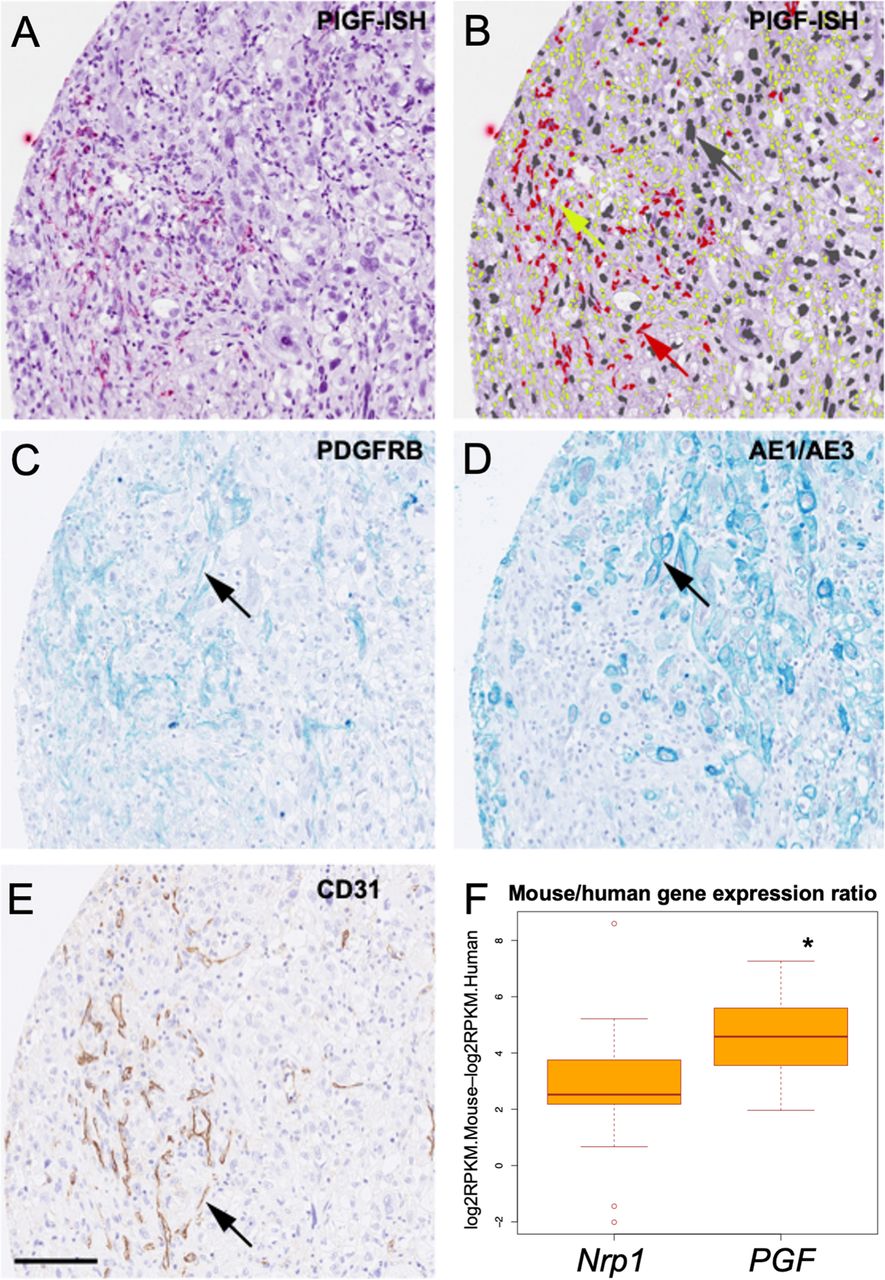
Since PlGF is a secreted protein, we next used RNA in situ hybridisation to identify the cells expressing PlGF in tissue samples from a clinically annotated cohort of ICC patients (N=52) (online supplemental figure S1C). Nrp1 expression was assessed by IHC. PlGF expression was predominantly found in CAFs and endothelial cells, and rarely in cancer cells; in contrast, Nrp1 was expressed by both ICC cells and CAFs (figure 1A–E and online supplemental figure S1D,E). Expression of PlGF or Nrp1 was rarely detected in surrounding liver tissues.

Expression of PlGF in human intrahepatic cholangiocarcinoma (ICC). (A) Representative in situ hybridisation (PlGF-ISH, red dots) staining of PlGF in ICC human tissue. (B) Same image of (A) showing the overlay of classifying cells according to cell type (red, PlGF-positive CAF; yellow, PlGF-negative CAF; grey, PlGF-negative ICC cell). (C) Representative serial section showing PDGFRβ staining (arrow highlights positive cell) to identify fibroblasts. (D) Representative serial section showing AE/AE3 staining (arrow highlights positive cell) to identify cancer cells. (E) Representative serial section showing CD31 staining (DAB, arrow highlights positive cell) of endothelial cells (scale bar 250 µm). (F) Differential levels of the Nrp1 and PGF gene expression calculated by subtracting logRPKM for human (ICC cells) from mouse (stromal cells) in a collection of 22 patient-derived xenograft (PDX) ICC tissues (online supplemental table S3; note the significantly higher expression levels in murine vs human PGF, *p<0.05). CAFs, cancer-associated fibroblasts; PlGF, placental growth factor;
Supplemental material
In addition, we analysed RNA-sequencing data from 28 patient-derived xenograft samples after separating them into mouse and human reads (online supplemental table S3). Median expression of PGF was significantly higher (16-fold) in the mouse stroma compared with the human ICC cells (p<0.01) (figure 1F). Median expression of Nrp1 was fourfold higher in the mouse-derived stromal cells compared with human ICC cells, but below the threshold of p=0.01. Of note, log2RPKM density plots for all genes in mouse and human were largely overlapping. Finally, we examined the levels of blood circulating PlGF in ICC patients enrolled in clinical studies. We found elevated levels of PlGF in patients with advanced disease (online supplemental figure S1F).
These data were in line with analyses of The Cancer Genome Atlas (TCGA) database,30 which showed that the PGF gene was expressed at higher levels in human ICC tissues compared with normal liver tissue (online supplemental figure S1G). Furthermore, mining data from published scRNAseq database31 showed that PGF was expressed at higher levels in fibroblasts and endothelial cells in human ICC tissues compared with other cell types (online supplemental figure S1H).
Thus, PlGF expression is predominant in tumour stroma and is increased in advanced disease stages, while Nrp1 is broadly expressed in ICC tissues.
PlGF is secreted by ICC cells and HSCs and is increased in hypoxic conditions
We next examined PlGF secretion in vitro and the impact of hypoxia, a common feature of ICC. To this end, we examined a panel of available murine ICC cells (425 and SS49), human ICC cells (HUCCT1), and HSCs (one of the CAF precursors in ICC), cultured in normoxic and hypoxic conditions. We measured PlGF protein concentration in culture supernatant using ELISA. All ICC cells and the HSCs secreted detectable levels of PlGF, and the concentration was significantly increased in hypoxic conditions (1%O2 for 48 hours) and reduced by short-hairpin (sh)-RNA constructs for PlGF (online supplemental figure S1I). We also found that high expression levels of Nrp1—but not of Nrp2 or VEGFR1—were detectable in most murine and human ICC cell lines; these levels were comparable to those seen in human umbilical vein endothelial cells (online supplemental figure S1J).
Thus, PlGF expression is detectable and increased under hypoxic conditions in HSCs and cancer cells, and Nrp1 is expressed across human and mouse ICC lines.
PlGF blockade enriches for a subset of quiescent CAFs characterised by a hypoxia-like phenotype, increased cell glycolysis and Akt-related genes
To examine the role of PlGF/Nrp1 in ICC cells and CAFs, we first generated orthotopic tumours by intrahepatic injection of 425 (p53/Kras-mutant) murine ICC cells. CAFs were isolated from primary cultured tumour tissues using outgrowth and clonal isolation methods. We obtained CAF clones after limited dilution, which were expanded in individual wells of 96-well plates. Then, we validated CAF identity using the mesenchymal cell markers fibroblast activation protein (FAP), platelet-derived growth factor receptor (PDGFR)β and α−smooth muscle actin (SMA)32 to distinguish them from other stromal or cancer cells (online supplemental figure S2). In separate experiments, we dissociated the primary ICC tissue and subjected the heterotypic tissue cultures to an antimouse PlGF antibody (5D11D4) or IgG (control). This short term in vitro culture method yielded a high degree of cell viability, allowing scRNAseq analysis. scRNAseq analysis was performed using cells collected after 24 hours of anti-PlGF antibody treatment of the whole tumour lysate, in the absence of hypoxia, to remove the confounding factors related to hypoxia-induced changes. In addition, we performed scRNAseq analyses of 425 ICC cells and CAFs cultured in vitro and used their transcription profile as a reference for the whole tissue culture scRNAseq analyses.
In the whole tissue culture, transcriptional profiling using scRNAseq analysis identified distinct ICC cell and CAF populations (figure 2A). ICC cell clustering was driven primarily by cell cycle related genes and pathways. Since we found no significant difference in proportions after anti-PlGF treatment, we grouped all ICC populations for subsequent analysis. The cell population most impacted by PlGF blockade was a CAF subset (labelled as CAF_2). This population was strongly enriched after treatment with anti-PlGF antibody, with treated cells representing about 80% of the cluster (figure 2B). This is a far greater proportion than what would be expected if cell cluster partitioning in the scRNAseq analysis was not dependent on treatment (p<0.0001; Fisher’s exact test).

Single cell RNA sequencing analysis of gene expression changes after PlGF blockade. (A) tSNE plot (each dot represents a cell) demonstrating different populations of CAFs and ICC cells based on transcriptional profiles in whole tumour lysate from orthotopic 425 ICC model cultured in vitro with anti-PlGF antibody. The CAF_2 (arrow) cluster was strongly enriched after PlGF blockade. (B) Expression of the PGF gene (encoding for PlGF) across CAF and ICC cell clusters. (C) Proportion plot demonstrating representation of treated and control cells in each cluster. (C–E) The cluster of cells affected by PlGF blockade showed a gene expression pattern associated with increased expression in genes related to hypoxia (hyp) (C) glycolysis (glyc) (D) and Akt pathways (path) (E). *P<0.05; **p<0.001; ***p<0.0001. CAF, cancer-associated fibroblast; ICC, intrahepatic cholangiocarcinoma; PIGF, placental growth factor; tSNE, t-distributed stochastic neighbor embedding.
Initial examination of this CAF_2 population indicated low levels of proliferation/G1 arrest and low expression of collagen type I, PDGFR-β and transforming growth factor (TGF)-β relative to the other CAF populations (online supplemental figure S3). A pathway analysis demonstrated this CAF_2 population exhibits high glycolysis and a hypoxia-like phenotype (online supplemental table S4), despite being cultured in normoxic conditions (figure 2C,D and online supplemental figure S4). Aside from hypoxia and glycolysis related pathways, the CAF_2 population exhibited significant alterations in the AKT pathway-related genes when compared with the other CAF populations (figure 2E).
Supplemental material
Thus, the scRNAseq analysis identified the CAFs as the major cell population affected by PlGF blockade. The primary transcriptomic disruption induced by PlGF blockade involved pathways related to cell proliferation and hypoxia. These effects are potentially mediated by the AKT pathway in this more quiescent CAF subset.
PlGF promotes a myofibroblast-like phenotype in CAFs via Akt/NF-κB signalling
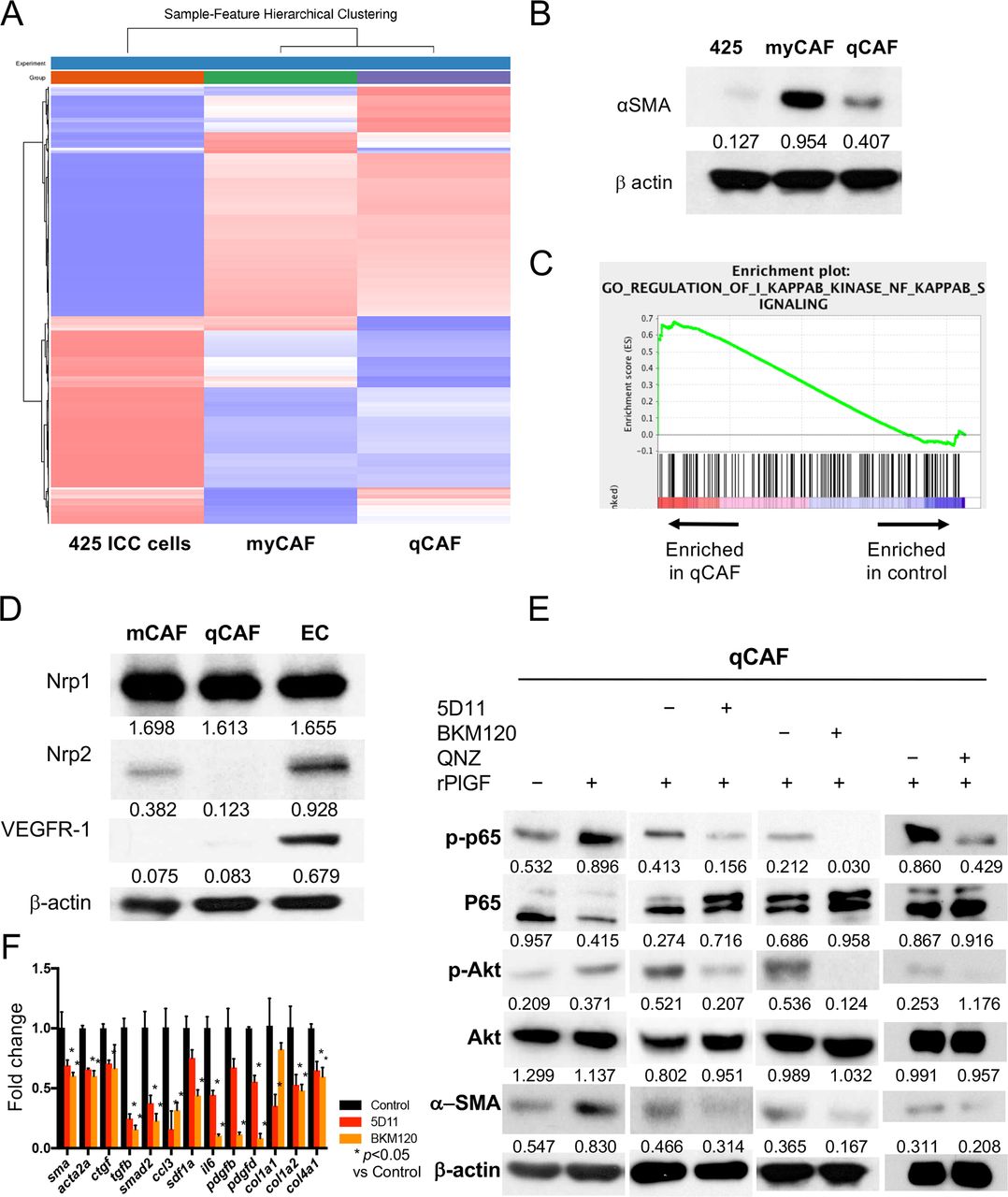
We next dissected the role of the PlGF/Akt axis using the CAFs isolated from murine ICC tissue. The CAF subsets expressed fibroblast markers (FAP, PDGFR-β) but exhibited a differential activation phenotype and are herein referred to as myofibroblast-like (my)CAFs (highly activated, α-SMAhi) or quiescent (q)CAFs (figure 3A,B and online supplemental figure S2A,C). Western blotting and qPCR analyses showed that qCAFs expressed lower gene and protein levels of profibrotic markers (sma, acta2 and ctgf) and fibrosis-induced growth factors (tgfb, smad2, ccl3, sdf1a) (online supplemental figure S2B). By bulk RNA sequencing analysis, we found that qCAFs showed a transcriptional enrichment in factors related to the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway (figure 3C and online supplemental figure S2D,E). The NF-κB pathway is known as one of the downstream targets of the Akt pathway. Moreover, using Western blotting and IHC, we found that Nrp1 was expressed in qCAFs (figure 3D and online supplemental figure S2F).
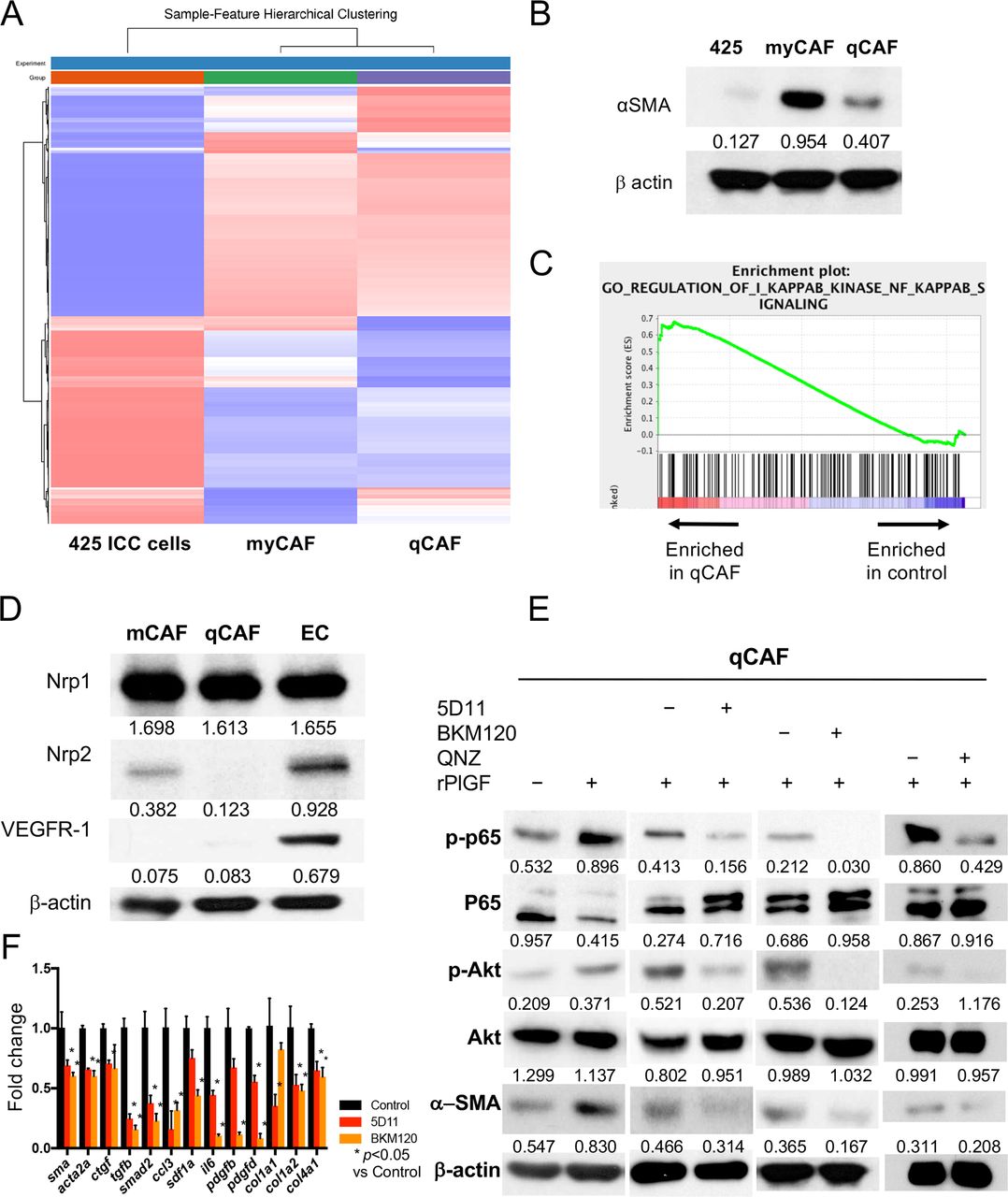
PlGF promotes a myofibroblast-like phenotype in CAFs via increased NF-kB/Akt activation. (A, B) RNAseq analysis of two CAF clones (A) comparing high α-SMA-expressing myCAF versus low α-SMA-expressing qCAF versus 425 cells (B) (see online supplemental dataset 1). (C) GSEA shows enrichment in NF-kB pathway-related genes in qCAF cells versus control (425 ICC cells). (D) High protein expression of Nrp1 but not Nrp2 or VEGFR1 in CAFs. (E) Recombinant PlGF further activates NF-kB/Akt axis and increases α-SMA expression in qCAFs, while PlGF blockade using 5D11D4 (5D11) treatment downregulates NF-kB/Akt pathway and α-SMA expression. Moreover, inhibition of PI3K using the compoundBKM120 and NF-kB with the compound QNZ also inhibited activation of NF-kB and α-SMA expression. (F) Both PlGF blockade and PI3K inhibition reduced the expression of profibrotic genes in qCAFs (analysed in triplicate). CAF, cancer-associated fibroblast; PIGF, placental growth factor.
Supplemental material
Next, we investigated whether PlGF stimulation mediates CAF activation. We found that exposure to recombinant (r)PlGF activated Akt and p65, and induced α−SMA expression in murine CAFs and human HSCs (figure 3E and online supplemental figure S5A,B). Consistent with the scRNAseq cell results, anti-PlGF treatment downregulated Akt/NF-κB pathway activation in both CAFs and HSCs—despite maintenance of high total Akt levels—and inhibited α-SMA and TGF-β expression in a dose-dependent manner (figure 3E and online supplemental figure S5A,B). Moreover, direct inhibition of PI3K/Akt by the inhibitor BKM120 or of NF-kB pathway using the compound QNZ showed decreased α−SMA expression in CAFs and HSCs (figure 3E and online supplemental figure S5A). Furthermore, both PlGF and PI3K inhibition downregulated myofibroblast-related genes in CAFs (figure 3F). Finally, examination of a publicly available dataset showed a significant direct correlation between PGF and ACTA2 genes in human ICC (online supplemental figure S5C).
These results indicate that PlGF activates Akt/NF-kB pathway and promotes the myofibroblast-like (α-SMAhi) phenotype in CAFs.
CAFs promote ICC cell invasion via PlGF expression
To further study the paracrine interaction between CAFs and ICC cells mediated by PlGF, we compared the 3-D invasion capabilities using real-time analyses of heterotypic spheroid co-cultures. The co-culture of 425 ICC cells with qCAFs showed larger spheroid formation than when ICC cells were cultured alone (figure 4A,B). To evaluate ICC cell invasion in real time, we used a time-lapse 3-D culture system with an Olympus IX81 Confocal microscope equipped with the Fluoview software (lens:10X air; slice thickness: 1–5 µm). In brief, spheroids were grown on top of a 1.5% agarose gel under serum-free conditions. When these spheroids were transferred to Matrigel-coated wells, they showed increased invasion into the surrounding matrix. By measuring the invasion area, we found that co-culture significantly enhanced cell invasion area when compared with ICC cells alone; this enhancement was almost completely inhibited by repression of PlGF in the ICC cells or PlGF blockade with antibodies (figure 4C,D and online supplemental movie S1).
Supplementary video
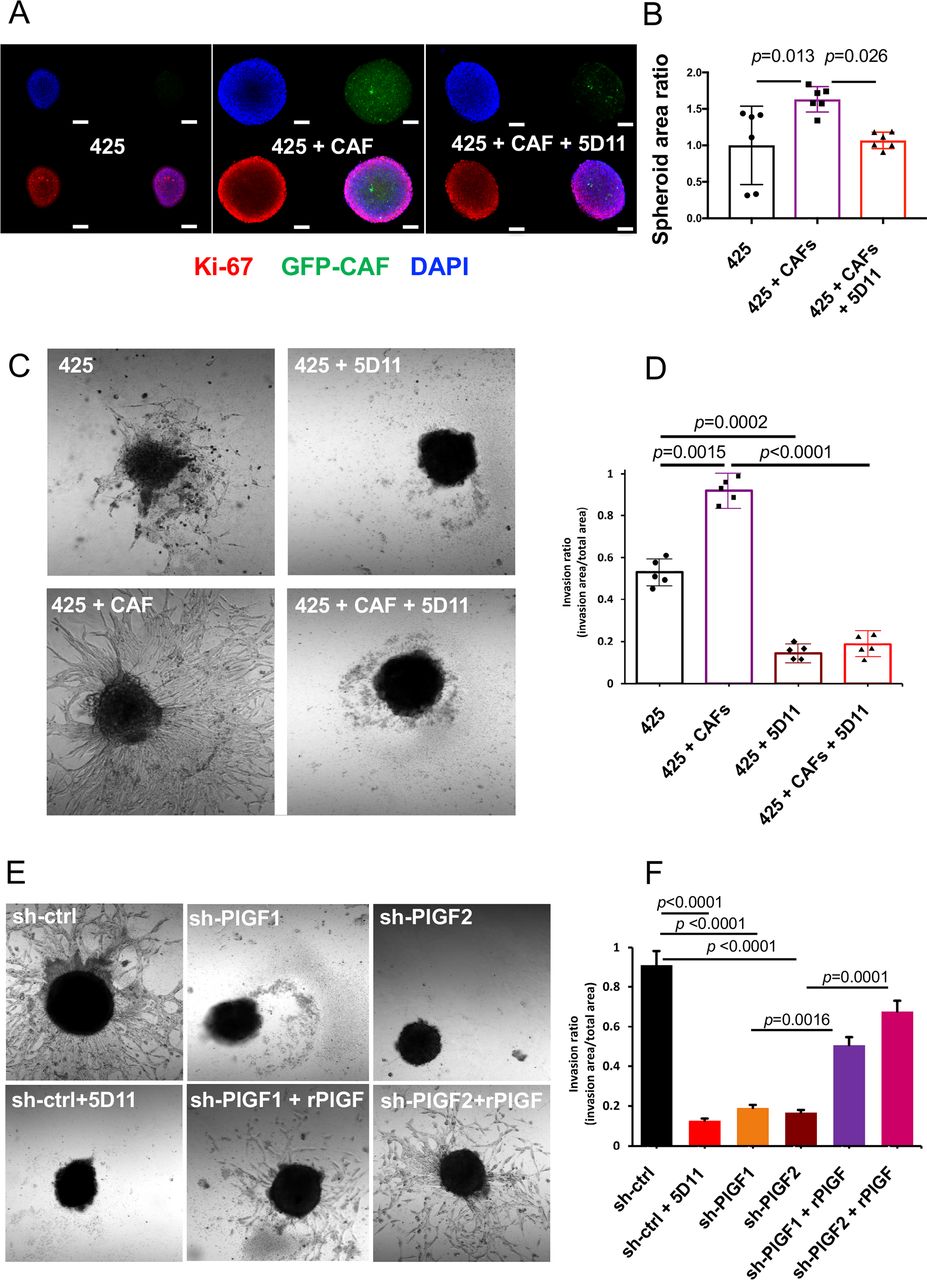
Impact of PlGF inhibition on ICC cell invasion. (A–D) Representative images of in vitro 3-D coculture of 425 ICC cells with GFP-expressing qCAFs versus ICC cells alone (A); CAFs increased the spheroid formation compared with 425 ICC cells alone, and this effect was suppressed by PlGF blockade with 5D11D4 (5D11) antibody (B); Scale bar, 160 µm. In addition, 3-D co-culture of ICC cells with CAFs (C) further increased ICC cell invasion, and PlGF blockade with 5D11 antibody inhibited this effect (D). (See online supplemental movie S1). (E, F) Analysis of ICC cells alone in 3-D (spheroid) invasion assays (E) showed that PlGF knockdown or anti-PlGF treatment using 5D11D4 (5D11) inhibited 425 ICC cell invasion in Matrigel (F); this effect was rescued by addition of recombinant (r)PlGF exposure in PlGF knockdown cells (see online supplemental movie S2). All experiments were performed in triplicate; p values from Tukey’s test. CAF, cancer-associated fibroblast; ICC, intrahepatic cholangiocarcinoma; PlGF, placental growth factor.
Supplementary video
We next assessed the direct effect of PlGF inhibition on ICC cell viability and invasiveness. PlGF-knockdown induced a significant but small reduction of proliferation in vitro (p<0.05) in murine ICC cells (425 and SS49), while PlGF blockade had no impact on the size of established spheroids (online supplemental figure S5D,E). However, evaluation of ICC cell invasion in real-time showed that PlGF knockdown or blockade with the antimouse PlGF antibody 5D11D4 significantly suppressed the invasive capabilities of 425 ICC cells, including in the presence of CAF-conditioned media. The invasive ability was rescued by addition of rPlGF to PlGF-knockdown ICC cells (figure 4E–F and online supplemental figure S5F, movie S2).
Using Western blotting, we found that both PlGF knockdown and 5D11D4 treatment decreased the level of phosphorylated Akt in 425 and SS49 murine ICC cells; this effect was prevented by adding rPlGF to the PlGF-knockdown cells (online supplemental figure S5G,H). In addition, knockdown of Nrp1 expression in human ICC cells resulted in a decrease in Akt phosphorylation (online supplemental figure S5I). Finally, stimulation of ICC cells with rPlGF activated Akt in Nrp1 intact cells but not in Nrp1-knockdown cells (online supplemental figure S5J).
Taken together, these results show that CAFs stimulate the invasive 3-D growth of ICC cells, which can be inhibited by PlGF blockade.
Genetic or pharmacological PlGF inhibition in ICC delays tumour progression in mice
We next investigated the in vivo role of PlGF in tumour progression using orthotopic ICC models generated by intrahepatic injection of cells derived from tumours spontaneously arising in p53 KO/Kras G12D mice (425 cells) and Idh2 R172K/Kras G12D mice (SS49 cells). These ICC models exhibited excessive ECM accumulation and abundance of α−SMA-positive myofibroblast-like CAFs surrounding vascular structures associated with neoplastic ducts. Moreover, they displayed a hypoxic tumour microenvironment and similar clinical symptoms to human ICC, such as abundant bloody ascites and frequent metastases to distant organs.
We first evaluated the effect of PlGF expression knockdown in cancer cells using shRNA constructs for PlGF (sh-PlGF) vs a scrambled sh-RNA (sh-control) (online supplemental figure 1G and figure 5C), with or without anti-mouse PlGF antibody (5D11D4) vs IgG control. Measurement of plasma PlGF concentration confirmed the near complete blockade of circulating PlGF in the mice treated with 25 mg/kg 5D11D4 (online supplemental figure S6A). We found that genetic ablation of PlGF expression in ICC cells significantly delayed the initial growth of 425 ICC tumours in SCID mice, and that complete pharmacologic blockade with 5D11D4 significantly delayed the growth of established 425 ICCs (figure 5A,B). Moreover, anti-PlGF treatment of established sh-PlGF tumours further inhibited ICC growth compared with all other groups (figure 5A,B).
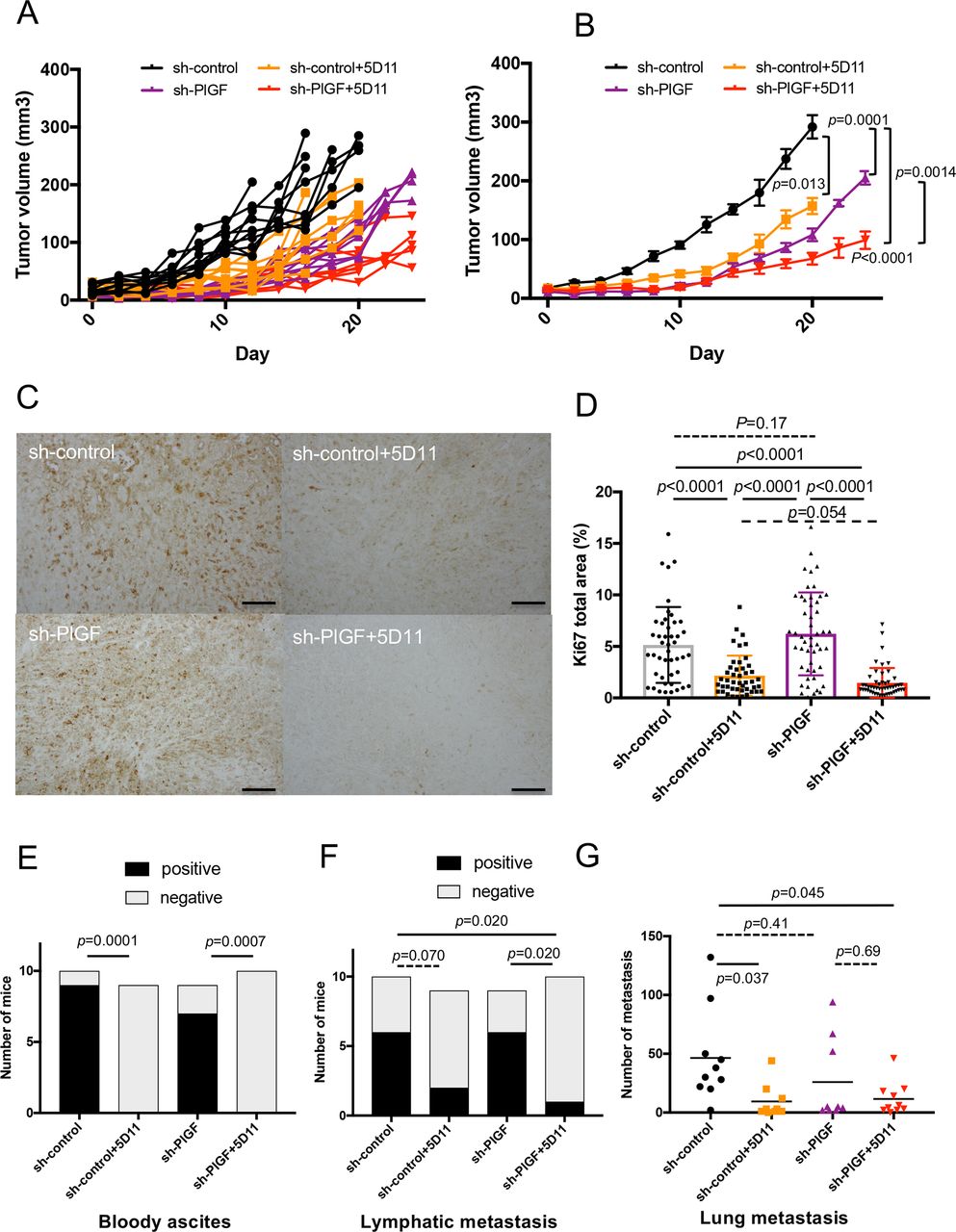
Impact of genetic inhibition of PlGF in ICC cells on ICC growth and/or pharmacologic blockade of PlGF on established tumour progression. (A, B) PlGF knockdown in ICC cells delayed tumour onset, while complete pharmacological blockade significantly delayed established ICC growth in the orthotopic 425 model (individual growth curves in (A) and average tumour size with SD in surviving mice in (B)). (C, D) Representative IHC for Ki-67 as a marker of cell proliferation in tumour sections from the four treatment groups (C); PlGF blockade—but not PlGF knockdown in ICC cells—significantly reduced in vivo ICC cell proliferation in size-matched orthotopic 425 tumours (D). (E) PlGF blockade significantly reduced the occurrence of bloody ascites. (F, G) Combined genetic and pharmacologic inhibition of PlGF reduced metastasis to the lymph nodes (F) and lungs (G). P values from Mann-Whitney U test (A, B), Tukey’s test (D, G) and Fisher’s test (E, F). In vivo studies were performed in duplicate; n=10 mice per group. Scale bars, 100 µm in (C). ICC, intrahepatic cholangiocarcinoma; IHC, immunohistochemistry; PlGF, placental growth factor.
To investigate the mechanism of benefit of PlGF inhibition, we conducted a time-matched study in which all mice were sacrificed on day 11 after the start of treatment. We found that anti-PlGF treatment significantly decreased the cell proliferation index in orthotopic ICC tumour tissues, assessed by Ki67 staining (figure 5C,D). Furthermore, PlGF inhibition prevented the formation of bloody ascites and significantly reduced regional (lymph node) and distant (lung) metastasis (figure 5E–G). These results were confirmed in the SS49 model of ICC, in which PlGF blockade significantly delayed tumour growth (online supplemental figure S6B).
Finally, to interrogate the specific roles of stroma-derived versus tumour-derived PlGF, we orthotopically implanted 425 ICC cells in PlGF-proficient vs PlGF-deficient C57Bl/6 mice. Tumour formation was reduced after implantation of ICC cells with decreased PlGF expression and when stromal cells were deficient in PlGF, and completely prevented when both tumour and stromal cell PlGF expression was inhibited (online supplemental figure S6C).
Thus, PlGF expression plays a critical role in the initial ICC growth, and stroma-derived PlGF is mediating tumour formation and progression in two genomically distinct murine ICC models.
PlGF blockade reduces tumour desmoplasia and tissue stiffness, and results in increased blood perfusion in ICC
Next, we studied the functional impact of PlGF inhibition on relevant components of the tumour microenvironment. Analysis of ICC tissues showed a high vascular density, but many of the tumour vessels were collapsed (figure 6A), similar to human ICC (figure 1E and online supplemental figure 6D). Here, vessel collapse was associated with desmoplasia (ie, high density of myofibroblast-like (α-SMA+) CAFs and collagen I expression (figure 6B–E, online supplemental figures S6E and S7), and increased tissue stiffness (figure 6F). PlGF blockade—but not PlGF knockdown in ICC cells—decreased the expression of collagen I, as well as tissue stiffness, resulting in the opening of blood vessels (figure 6A–D and online supplemental figure S6E). A significant reduction in α-SMA required both PlGF knockdown in ICC cells and antibody blockade (figure 6E). Opening of collapsed blood vessels resulted in improved blood perfusion and significantly reduced intratumorous hypoxia, reflected by reduced hif1 transcription after treatment (figure 6G–H). There were no significant differences in tumour-associated macrophage numbers or polarisation (online supplemental figure S8).

PlGF inhibition reduces ICC desmoplasia and improves tumour blood perfusion by opening collapsed vessels. (A) PlGF inhibition induced opening of collapsed blood vessels (representative IHC of vessels in inset). (B) Representative immunofluorescence (IF) for collagen I and CK19 (upper panel) and α-SMA and CK19 (lower panel) in tumours after PlGF inhibition. (C–E) Quantification of IF data showing that PlGF blockade and PlGF knockdown in ICC cells reduced CK19-positive cell area (C) and collagen I expression (D); α-SMA+ ‘myofibroblast-like’ CAF density was reduced in the PlGF knockdown groups, most substantially after PlGF blockade (E). (F) PlGF blockade significantly decreased tumour stiffness. (G) Representative OCT of patent blood vessels in ICC with or without PlGF blockade. (H) Anti-PlGF treatment significantly reduced intratumour hypoxia, reflected by reduced hif1 transcription. P values from Tukey’s test (A, C–E) and Mann-Whitney U teat (F, H). Scale bars, 500 µm in (C). ANOVA, analysis of variance; ICC, intrahepatic cholangiocarcinoma; IHC, immunohistochemistry; OCT, optical coherence tomography; PlGF, placental growth factor.
These results indicate that tumour stromal cells are a key factor in PlGF-mediated desmoplasia by deposition of collagen I in murine ICC.
Anti-PlGF treatment enhances the efficacy of standard chemotherapy against ICC in mice
Having revealed the impact of PlGF blockade on vascular function and desmoplasia, we examined the impact of PlGF blockade on ICC response to a standard chemotherapy regimen. We compared the efficacy of anti-PlGF therapy when combined gemcitabine and cisplatin (Gem/Cis) versus each therapy alone or control IgG in the orthotopic 425 ICC model. We found that combination therapy (Gem/Cis plus anti-PlGF antibody) for 3 weeks (ie, until mice in control group were sacrificed) significantly enhanced tumour growth delay (figure 7A,B). This benefit was associated with significant decreases in bloody ascites and pleural effusions (online supplemental figure S9A,B). Moreover, when treatment was given continually, combination therapy significantly increased overall survival compared with each treatment alone, with an HR of 0.04 vs control (p<0.0001) and an HR=0.31 vs Gem/Cis (p=0.018) (figure 7C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PlGF blockade enhances the anti-tumour effects of conventional chemotherapy and increases survival. (A, B) The combination of anti-PlGF antibody 5D11D4 (5D11) with conventional gemcitabine plus cisplatin (GC) chemotherapy for 3 weeks significantly delayed tumour growth compared with GC or 5D11 alone (individual growth curves in A) and average tumour size with SD in surviving mice in B). (B) Kaplan-Meier survival distributions after continuous treatment with GC, 5D11 or their combination vs control in mice with established ICC. In vivo studies were performed in duplicate; n=10 mice per group. (D) Addition of 5D11 did not further reduce the viability of 425 cells after gemcitabine (Gem) treatment in vitro. P values from Mann-Whitney U test (B) and log-rank test (D). (E) Proposed mechanism of action of PlGF blockade in ICC: PlGF is produced primarily by CAFs and affects both cancer and stromal cells in an autocrine and paracrine manner in ICC. PlGF inhibition reduces ICC cell invasion and tumour desmoplasia and stiffness, which enhances blood perfusion and the efficacy of chemotherapy. CAF, cancer-associated fibroblast; ICC, intrahepatic cholangiocarcinoma; PlGF, placental growth factor.
We next examined whether PlGF blockade impacted ICC cell sensitivity to chemotherapy. In vitro evaluation of cytotoxicity using an MTT assay showed that exposure to Gem induced a dose-dependent cell killing in 425 cells. However, the cytotoxicity of Gem was not affected by concomitant PlGF or NF-kB blockade in ICC cells (figure 7D and online supplemental figure S9C).
Collectively, these results show that PlGF blockade can substantially enhance the efficacy of chemotherapy in ICC in vivo via tumour stroma-mediated mechanisms.
Discussion
ICCs are characterised by aggressive (invasive) growth and an abundant stroma (desmoplasia), mediated by several molecular pathways and a hypoxic tumour microenvironment.33 PlGF can promote the formation of an abnormal vasculature and increased hypoxia via elevated matrix deposition and increased tissue stiffness—all of which are determinants for tumour aggressiveness and resistance to treatment.14 18 34 35 We report here that PlGF and Nrp1 are broadly expressed in human ICC, particularly in the stroma, and that PlGF expression levels are elevated by hypoxia and in advanced disease. We found that PlGF is highly expressed by CAFs in ICC, promoting desmoplasia by the induction of CAF activation and invasive tumour growth through CAF/ICC cell interactions (figure 7E).
In murine ICC models, PlGF blockade suppressed orthotopic tumour growth, in part by decreasing Akt activation and reducing the proliferation and invasion of ICC cells. Interestingly, we found that PlGF blockade had the greatest impact on subsets of CAFs exhibiting high Akt activation. Activation of the Akt/NF-κb signalling promoted a myofibroblast-like phenotype in murine CAFs with increased expression patterns associated with cell proliferation, hypoxia and glycolysis. Akt/NF-κb signalling has been associated with an activated phenotype of CAFs in breast, pancreatic, prostate and ovarian cancers.36 37 The inhibition of this pathway induced a significant reduction of chemokine and cytokine stimulation in CAFs.15 Here, we show that inhibiting PlGF or Akt and NF-kB activity reduced the myofibroblast-like phenotype in murine CAFs and HSCs, one of the precursors of CAFs in human ICC.
CAFs are the major source of matrix components in ICC.8 However, recent reports identified several subsets of CAFs based on transcriptional signatures and highlighted the benefits of targeting those that provide support to cancer cells during tumour progression.7–9 31 38–41 We provide here new mechanistic insights into the antidesmoplastic effect of PlGF blockade. Specifically, myofibroblast-like (α-SMAhi) CAFs secrete multiple profibrogenic factors, including PlGF and TGF-β, and induced morphological activation and proliferation in close interaction with cancer cells.10 42 43 The abundance of myofibroblast-like CAFs and CAF-mediated desmoplasia increases tissue stiffness, which induces collapse of tumour vessels and intratumorous hypoxia, similar to pancreatic cancer.35 The myofibroblast CAF phenotype may be regulated by several pathways, including Rho GTPase, c-Jun N-terminal kinase and Hedgehog.10 42 44 In evaluating CAF clones derived from ICC with differential activation, we found differential expression in markers such as TGF-β and α-SMA. PlGF exposure increased Akt and p65 activation, as well as myofibroblast-like expression of α-SMA and TGF-β in more quiescent CAFs. Conversely, blockade of PlGF prevented these effects.
Previous studies have investigated anti-PlGF therapy in liver cancers, including cirrhosis, hepatocellular carcinoma and cholangiocarcinoma models.16 34 45 These studies invoked mechanisms of benefit related to antiangiogenic/antivascular (vascular pruning) effects, as well as effects on inflammation (on the infiltration of tumour-associated macrophages), mediated by VEGFR1.46 Here, we are reporting a distinct mechanism of benefit for PlGF inhibition in ICC, involving decreased cancer cell invasion and myofibroblast-like activation in CAFs. These effects resulted in decreased desmoplasia and hypoxia—by opening of blood vessels as opposed to vessel pruning—as demonstrated in preclinical mouse models of ICC. This effect potentiated the tumour growth delay in two genomically distinct mouse models and increased overall survival from a treatment of Gem/Cis, the standard therapy for advanced ICC patients.
Future prospective clinical studies will be necessary to establish whether these effects of PlGF blockade will translate into a benefit in the hypovascular human ICC.33 47 However, previous studies have established that anti-VEGFR approaches have no efficacy in biliary tract cancers, including when combined with chemotherapy in randomised clinical trials of agents targeting VEGFR1.48 Moreover, as reviewed in refs.49 50, all prior attempts to improve on the efficacy of chemotherapy have failed in randomised trials over the last decade, leaving Gem/Cis chemotherapy as the only proven efficacious option currently available for biliary tract cancers, as per ABC-02 trial data.
Recent reports converge towards an interplay between tumour hypoxia, fibrosis and immune evasion as determinants of tumour progression and treatment resistance in ICC subsets.51–53 Our findings have clinical implications for other emerging therapies, such as radiotherapy or immune checkpoint blockade, which have shown promise in ICC.54 55 For example, programmed cell death 1 (PD-1) blockade has shown efficacy in microsatellite instable carcinomas, including ICC.56 PD-ligand 1 is expressed in 33%–46% of resected human ICC samples and is associated with poor prognosis.57 58 Therefore, determining whether reprogramming of the ICC microenvironment by PlGF blockade could also enhance the efficacy of radiotherapy or immunotherapy in ICC is warranted.
In summary, we demonstrate here that PlGF is a mediator of ICC progression and is a potential therapeutic target. Antibody blockade of PlGF in ICC models reprogrammed the desmoplastic microenvironment, resulting in enhanced efficacy of standard chemotherapy. These results reveal a new and actionable approach for a novel combination therapy in this disease with a dismal prognosis.
Data availability statement
Data are available on reasonable request. Data are available on reasonable request from the corresponding author DGD. The authors used deidentified participant data at Fundeni Clinical Institute, Bucharest, Romania, University Hospital Cologne, Germany, and Massachusetts General Hospital, Boston, USA.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Mark Duquette, Anna Khachatryan, Sylvie Roberge (MGH Boston) and Alexandra Florin (University of Muenster, Germany) for outstanding technical support, and Sergey Kozin and Cyril Benes (MGH Boston) for useful discussions.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SA and KI contributed equally.
Contributors SA designed and performed experiments, analysed the data and wrote the manuscript. KI, SK, JC, AM, MRN, TH, XC, SK, KK, HTN, DHS, EM, KS, HK, RRR, MI and TCES performed experiments, analysed the data and edited the manuscript. SH, IC, MJB and SS analysed the RNA sequencing data and edited the manuscript. PSP, TSH, TY, SD and IP provided human samples and analysed the data. NB provided murine reagents and analysed the data. LLM, RKJ and AXZ analysed the data and edited the manuscript. DGD designed the experiments, analysed the data, obtained funding and wrote the manuscript. All authors approved the final version of the manuscript.
Funding DD’s work was supported through NIH grants P01-CA080124, R41-CA213678 and Proton Beam/Federal Share Programme and Department of DefenseDefence grants #W81XWH-19-1-0284 and W81XWH-19-1-0482. RKJ’s work was supported through NIH grants P01-CA080124, R35-CA197743, R01-CA208205 and U01-CA224173, and by the National Foundation for Cancer Research, Harvard Ludwig Cancer Centre, Advanced Medical Research Foundation and Jane’s Trust Foundation. IP’s work was supported by the Romanian Ministry of Research and Innovation, CCCDI–UEFISCDI, project number PN-III-P1-1.2-PCCDI-2017-0797/66PCCDI, within PNCDI III. SA’s research was supported by a Postdoctoral Fellowship from Cholangiocarcinoma Research Foundation, TH received a Postdoctoral Fellowship from Astellas Foundation for Research on Metabolic Disorders, Japan, DS received a Postdoctoral Fellowship from Humboldt Foundation, KS received a Postdoctoral Fellowship from Uehara Memorial Foundation, and EM received a grant from the Philippe Foundation and the Cancéropôle PACA.
Competing interests IC is an employee of STIMIT. TY has served in a consulting or advisory role for Bristol Myers Squibb. RKJ received honorarium from Amgen and consultant fees from Chugai, Ophthotech, Merck, SPARC, SynDevRx. RKJ owns equity in Accurius, Enlight, SPARC, and SynDevRx, and serves on the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund and Tekla World Healthcare Fund. AXZ is a consultant/advisory board member for Bayer. DGD received consultant fees from Bayer, Simcere, Surface Oncology and BMS and research grants from Bayer, Exelixis and BMS. No reagents or support from these companies was used for this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.