Article Text

Abstract
Objectives Coeliac disease (CD) is a complex autoimmune disorder that develops in genetically susceptible individuals. Dietary gluten triggers an immune response for which the only available treatment so far is a strict, lifelong gluten free diet. Human leucocyte antigen (HLA) genes and several non-HLA regions have been associated with the genetic susceptibility to CD, but their role in the pathogenesis of the disease is still essentially unknown, making it complicated to develop much needed non-dietary treatments. Here, we describe the functional involvement of a CD-associated single-nucleotide polymorphism (SNP) located in the 5’UTR of XPO1 in the inflammatory environment characteristic of the coeliac intestinal epithelium.
Design The function of the CD-associated SNP was investigated using an intestinal cell line heterozygous for the SNP, N6-methyladenosine (m6A)-related knock-out and HLA-DQ2 mice, and human samples from patients with CD.
Results Individuals harbouring the risk allele had higher m6A methylation in the 5’UTR of XPO1 RNA, rendering greater XPO1 protein amounts that led to downstream nuclear factor kappa B (NFkB) activity and subsequent inflammation. Furthermore, gluten exposure increased overall m6A methylation in humans as well as in in vitro and in vivo models.
Conclusion We identify a novel m6A-XPO1-NFkB pathway that is activated in CD patients. The findings will prompt the development of new therapeutic approaches directed at m6A proteins and XPO1, a target under evaluation for the treatment of intestinal disorders.
- celiac disease
- methylation
- gluten
- intestinal gene regulation
- inflammation
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplemental information. Online available data analyzed in this study are: miCLIP (GSE128699) miCLIP (GSE128699): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128699 and RNAseq (GSE84745): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84745. Data is held in GEO repository and it is freely available for reuse.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
rs3087898 single-nucleotide polymorphism, in the 5’UTR of the XPO1 gene, is associated with coeliac disease (CD) susceptibility.
XPO1 protein is involved in the regulation of the nuclear factor kappa B (NFkB) pathway.
NFkB pathway, and its downstream cytokines (ie, IL-8), are upregulated in the coeliac intestinal mucosa.
Alterations in N6-methyladenosine (m6A) RNA methylation have been associated with different intestinal conditions.
What are the new findings?
The XPO1 mRNA harbouring the CD-associated allele is preferentially methylated and has enhanced translation efficiency.
XPO1 translation is m6A-dependent and it is mediated by YTHDF1 protein.
Gliadin induces the activation of m6A machinery in intestinal epithelial cells leading to increased XPO1 levels.
Allele-specific increase of XPO1 activates NFkB-mediated intestinal inflammation.
Coeliac patients show allele-specific variation of XPO1 protein levels, a gluten-dependent increase of m6A, XPO1, METTL3 and YTHDF1, together with downstream inflammatory effects.
How might it impact on clinical practice in the foreseeable future?
Our discoveries will prompt the development of novel therapeutic approaches for CD treatment focused on m6A machinery proteins and XPO1 targeting. Molecules targeting these proteins are currently being evaluated or used for the treatment of other intestinal diseases.
Introduction
Coeliac disease (CD) is a chronic inflammatory and autoimmune condition that affects primarily the small intestine and develops in genetically susceptible individuals upon gluten ingestion. In patients with CD, gluten triggers a proinflammatory response that leads to the activation of the innate and adaptive immune systems.1 2 This includes presence of proinflammatory gluten specific T cells producers of interferon-γ, cytotoxic transformation of intraepithelial lymphocytes (IEL), epithelial cell stress and the release of innate proinflammatory cytokines.3 4
To date, the only effective treatment for CD patients is a lifelong, strict gluten-free diet (GFD). However, there are limitations related to dietary compliance, which may lead to complications, highlighting the unmet need for adjuvant therapies. Approximately 30% of the genetic risk for developing CD is conferred by the human leucocyte antigen (HLA) genes and fine mapping studies have revealed additional non-HLA regions that altogether account for an additional 6.5% of heritability.5 About 90% of CD patients carry HLA-DQ2 and DQ8 alleles, however, these variants are also common in the general population. Thus, functional characterisation of associated variants is required to better understand CD pathogenesis. A substantial proportion of the CD-associated single-nucleotide polymorphisms (SNPs) map to non-coding regions, making it challenging to determine their molecular functions.6
N6-methyladenosine (m6A) represents the most abundant internal chemical modification of mRNAs and non-coding RNAs. m6A modifications are involved in multiple aspects of RNA metabolism and play crucial roles in many cellular processes, thus, they are emerging as a new layer of gene expression regulation.7–10 Nowadays some m6A writers or methylases, as N6-methyltransferase protein complex (METTL3/METTL14), and m6A erasers or demethylases (Fat mass and obesity-associated protein, FTO or AlkB homolog 5 RNA demethylase, ALKBH5) are known to control the m6A levels in RNAs by adding or removing m6A methylation marks. Additionally, the so-called reader proteins as the YT521-B homology domain-containing RNA Binding Proteins (RBPs)11–13 recognise methylated RNAs and regulate their processing. Moreover, m6A dynamicity allows for a rapid response on different cellular stress signals implicated in several pathways related to autoimmunity and immune tolerance.14 15 However, the effect of SNPs associated with immune and inflammatory diseases on the variability of the so called epitranscriptome has not been systematically analysed. A recent study shows that m6A-Quantitative Trait Loci (QTL) are enriched in complex traits as autoimmune diseases and highlights the importance of performing disease-specific studies.16 Moreover, perturbation of RNA methylation in T cells has been recently associated with development of colitis.17 Thus, a better understanding of how CD-associated SNPs can alter m6A marks and consequently regulate the molecular mechanisms driving the inflammatory response caused by gluten ingestion could provide a completely new layer of information regarding the pathogenesis of this disease, opening the door to the development of new essential therapeutic strategies.
In this study, we sought to functionally characterise a CD-associated non-coding SNP, rs3087898, in exportin 1 (XPO1) gene. XPO1 is a nuclear export protein that has been described to target many proteins and RNAs involved in diverse cellular processes.18 Among others, XPO1 has been related to the regulation of nuclear factor kappa B (NFkB) pathway,19 which is, in turn, upregulated in CD patients.20 21 In addition, upregulation and mutations in XPO1 have been implicated in different malignancies, evolving as an interesting therapeutic target for diverse types of cancer.22 23 We demonstrate that, a CD-associated non-coding and differentially m6A methylated SNP in the 5’UTR (untranslated region) of the XPO1 mRNA alters protein translation, contributing to the inflammatory environment in intestinal epithelial cells (IEC) characteristic of CD. The study highlights a previously unknown link between gluten exposure and alterations in the m6A-related machinery.
Materials and methods
Human samples
Small bowel biopsy samples were obtained from Biocruces Bizkaia Health Research Institute, from Hospital de Galdakao-Usansolo and from the Celiac Disease Center (Columbia University) according to the criteria in force at the time of recruitment.
Cell lines
Human intestinal cell line HCT116 (Sigma-Aldrich, #91091005), murine intestinal cell line C26 (kindly provided by Dr Beatriz Arteta) and human Jurkat T cell line (Sigma-Aldrich, #88042803) were used.
Animals
Specific pathogen-free Ythdf1 KO mice were provided by CH. C57BL/6 JRj was purchased from Janvier Labs (France) and used as wild-type mice. Tissues from mice transgenic for the HLA-DQ2 gene (human haplotype DR3-DQ2) with mouse CD4+ cells exclusively, and originally generated at the University of Melbourne24 were received from EFV (McMaster University).
Overall design
Pepsin Trypsin digested Gliadin (PTG) stimulations were performed in human samples, human cell lines and mouse models for further m6A methylation, RNA and protein expression analyses. Detailed methods for all studies and mouse treatments are provided in online supplemental materials and methods.
Supplemental material
Patient and public involvement
The patients and the public were engaged during the recruitment of volunteers via informative flyers to CD patients and at outreach activities open to the general public. The clinical and basic investigators have regular involvement with the local and national CD patient societies. The findings of the study will be disseminated in events organised by the societies and in National and International CD meetings.
Results
The CD-associated XPO1 allele is preferentially methylated and has enhanced translation efficiency
Mostly due to their localisation in non-coding regions, the molecular functions of many immune and inflammatory disease-associated SNPs remain unexplained.6 We observed that the CD-associated SNP rs3087898 (GRCh38:chr2:61538039) is located in the 5’UTR of XPO1, close to three m6A consensus motifs (GGACT). An examination of the MeT-DB V2.0 m6A database25 revealed m6A peaks in the 5’UTR of XPO1 in both mouse and human mRNAs (see online supplemental figure 1A). Moreover, Vienna Package online tool26 predicts a change in the secondary structure of the 5’UTR according to the SNP genotype (see figure 1A and online supplemental figure 1B). As different cell types are involved in the activation of the inflammatory response in CD, we first quantified m6A levels in the 5’UTR of XPO1 in the main cell types implicated in CD pathogenesis (T cells and IEC). We confirmed the methylation of this region by m6A RNA Immunoprecipitation (meRIP), followed by qPCR in the human IEC line HCT116 while the Jurkat T cell line showed very low methylation levels, pointing to a specific function of these m6A marks in enterocytes (see online supplemental figure 1C). We further confirmed m6A methylation in the XPO1 5’UTR in human duodenal whole biopsies and specifically in the epithelial fraction (see online supplemental figure 1D). Using online available m6A individual-nucleotide-resolution cross-linking and immunoprecipitation from HCT116 cells27 we observed that the three m6A consensus motifs in the 5’UTR are methylated (see figure 1B) and confirmed these results using an RT-qPCR-based method (see online supplemental figure 1E).28 In addition, taking advantage of the heterozygosity of the HCT116 cell line for the CD-associated SNP, we observed that the mRNA transcript carrying the CD-risk allele (XPO1*T) is more methylated than its alternative, protective form (XPO1*C) (see figure 1C). As m6A methylation is involved in diverse RNA metabolic processes, we wanted to elucidate the effects of this differential methylation on the XPO1 mRNA. We did not observe any differences in mRNA cellular localisation (see online supplemental figure 1F) or stability (see online supplemental figure 1G) when comparing the protective (XPO1*C) and risk (XPO1*T) mRNA forms. However, the analysis of translation efficiency using a luciferase reporter preceded by the 5’UTR of XPO1 showed higher luciferase activity in the presence of the T allele (see figure 1D), suggesting an implication of this SNP in the translation process. These results were confirmed by cloning both 5’UTR forms upstream of the XPO1 coding sequence, where we observed higher XPO1 protein production from the risk allele form (see figure 1E).
Supplemental material
Supplemental material

The CD-associated XPO1 allele is preferentially methylated and has enhanced translation efficiency. (A) Zoomed secondary structure of each of the human 5’UTR XPO1 forms as predicted by Vienna package. rs307898 SNP (green arrows) and putative m6A methylated adenines (red arrows) are highlighted in yellow. (B) Quantitative representation of miCLIP reads (orange) and mutations (blue) in the 5’UTR of XPO1. miCLIP data from HCT116 cells (heterozygous for rs3087898) was downloaded from GEO repository (GSE128699). Grey squares represent GGACT motifs, red dots represent methylated As and the green triangle and bar correspond to the associated SNP. (C) Allele-specific m6A levels in the 5’UTR of XPO1 in HCT116 cells as assessed by meRIP-qPCR. RNAs were immunoprecipitated with an anti-m6A antibody (normal IgG as control) and enrichment of m6A was determined by qPCR, n=4. (**p<0.01 according to a two-way ANOVA test. Enrichment relative to control IgG #p<0.05, ####p<0.0001 according to a two-way ANOVA test). (D) A dual-luciferase reporter system was used to assess the translation efficiency of each allele of the 5’UTR of XPO1. HCT116 cells were cotransfected with a Renilla luciferase plasmid and a plasmid with the Firefly luciferase CDS preceded by the 5’ UTR of XPO1. Bioluminiscence was measured 48 hours post-transfection, n=6. (****p<0.0001 according to a two-tailed Student’s t-test). (E) Both forms of the 5’UTR were cloned upstream the XPO1 cDNA and constructs were transfected into HCT116 cells. XPO1 and GAPDH (load control) were detected by Western blotting 48 hours post-transfection. Left, representative immunoblot; right, quantitative summary data, n=4. (*p<0.05 according to a two-tailed Student’s t-test). All values are mean±SEM. 5’ UTR, 5′ untranslated region; ANOVA, analysis of variance; m6A, N6-methyladenosine; miCLIP, m6A individual-nucleotide-resolution cross-linking and immunoprecipitation.
XPO1 translation is m6A-dependent and it is mediated by YTHDF1 in vitro and in vivo
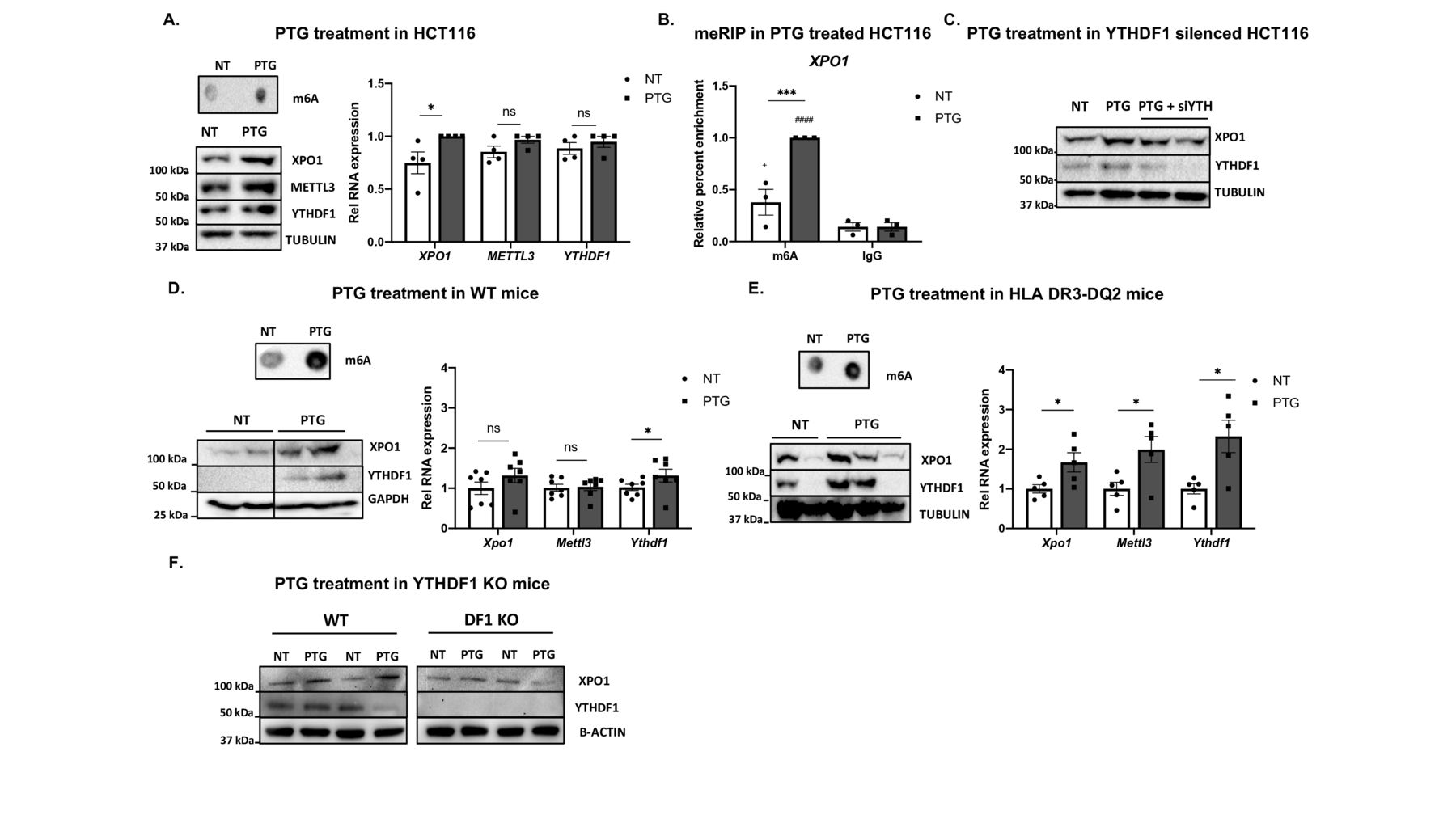
To assess whether the elevated m6A levels present in the XPO1*T form are responsible for the enhanced XPO1 translation efficiency, we analysed the effect of m6A methylation on XPO1 protein amounts. For this purpose, we overexpressed METTL3 writer protein, increasing overall m6A methylation (see online supplemental figure 2A, B), but we did not observe any significant differences in XPO1 mRNA or protein levels (see figure 2A). Since XPO1 translation is allele-specific and the risk allele shows higher methylation, based on a recent m6A-QTL study confirming translation upregulation effect by YTHDF1 in human cells,16 we hypothesised that the m6A reader protein YTHDF1, could differentially bind each of the alleles influencing translation.8

XPO1 translation is m6A-dependent and it is mediated by YTHDF1 in vitro and in vivo. (A) HCT116 cells were transfected with a METTL3 expression plasmid or an empty vector (EV) and effects were analysed 48 hours post-transfection. Overall RNA methylation was measured by dot blot using an anti-m6A antibody. XPO1 mRNA levels were quantified by RT-qPCR. (ns, non-significant according to a two-tailed Student’s t-test). XPO1 and METTL3 protein levels were measured by immunoblot with GAPDH as a loading control, n=3. Top right, representative dot blot; bottom right, representative immunoblot. (B) HCT116 cells were transduced with lentiviral particles of either an empty pLKO vector or constructs with shRNAs targeting METTL3 or YTHDF1, and cells were selected for puromycin resistance. Overall RNA methylation was measured by dot blot using an anti-m6A antibody. XPO1 mRNA levels were quantified by RT-qPCR. (*p<0.05; ****p<0.0001, according to a two-tailed Student’s t-test). XPO1, METTL3 and YTHDF1 protein levels were measured by immunoblot and actin was used as a loading control, n=4. Top right, representative dot blot; bottom right, representative immunoblot. (C) METTL3 was immunoprecipitated (IP) with an anti-METTL3 antibody, and allele-specific XPO1 levels were quantified by RT-qPCR, n=3. (ns, non-significant according to a two-way ANOVA test. Enrichment relative to control IgG (#p<0.05 according to a two-way ANOVA test). Right, representative immunoblot of the IP experiment with HSP90 as negative control for the IP. (D) YTHDF1 was immunoprecipitated (IP) with an anti-YTHDF1 antibody, and allele-specific XPO1 levels were quantified by RT-qPCR, n=4. (*p<0.05 according to a two-way ANOVA test. Enrichment relative to control IgG ##p<0.01, ####p<0.0001 according to a two-way ANOVA test). Right, representative immunoblot of the IP experiment with HSP90 as a negative control for the IP. (E) The duodenum was harvested from WT and Ythdf1 KO mice, and XPO1 and YTHDF1 protein levels were quantified by immunoblot, with tubulin as a loading control. Left, representative immunoblot; right, quantitative summary data (+/-: control mice; DF1 KO: Ythdf1 knockout mice), n=7. (*p<0.05, according to a two-tailed Mann-Whitney U test). All values are mean±SEM. ANOVA, analysis of variance; m6A, N6-methyladenosine; WT, wild-type.
To obtain a stable reduction of the m6A machinery, METTL3 or YTHDF1 were knocked down in the HCT116 cells (see online supplemental figure 2C, D). We observed an overall decrease of m6A levels on METTL3 knock down, (see figure 2B and online supplemental figure 2E), including reduced m6A levels in the 5’UTR of XPO1 (see online supplemental figure 2F), which resulted in lower XPO1 mRNA and protein amounts (see figure 2B and online supplemental figure 2G). XPO1 mRNA and protein levels were both decreased when YTHDF1 was knocked down (see figure 2B and online supplemental figure 2G), although m6A levels did not vary (see figure 2B, online supplemental figure 2E and 2F). These results show that methylation on the 5’UTR is necessary to maintain XPO1 transcript levels and adequate translation and suggest that its translation is mediated through recognition of the methylated 5’ UTR mRNA by YTHDF1.
To clarify whether METTL3 and YTHDF1 proteins interact with the XPO1 5’UTR in an allele-specific manner, we performed RIP assays and quantified the amount of immunoprecipitated allele-specific XPO1 mRNA (see online supplemental figure 2H, 1). While METTL3 did not show preferential binding to any of the alleles (see figure 2C), YTHDF1 was preferentially bound to the risk allele (XPO1*T) (see figure 2D), in accordance with the higher translation efficiency observed in the presence of this allele. The murine intestinal cell line C26 also showed methylation at the 5’UTR of Xpo1 (see online supplemental figure 2J), and quantification of XPO1 levels in IEC isolated from the duodenum of WT and Ythdf1 KO mice confirmed the involvement of YTHDF1 in the regulation of XPO1 protein amounts in vivo. In fact, Ythdf1 KO mice showed significantly lower XPO1 protein levels in the epithelial cells isolated from the duodenum than their WT littermates (see figure 2E and online supplemental figure 2K, L).

Gliadin induces the activation of the m6A machinery in intestinal epithelial cells leading to increased XPO1 in vitro and in vivo. HCT116 cells were treated with a low dose of 30 µg/mL pepsin-trypsin digested gliadin (PTG) for 48 hours and stimulated with 350 µg/mL PTG for 24 hours as shown in SF3.A. (A) Overall RNA methylation was measured by dot blot using an anti-m6A antibody. Top left, a representative dot blot of four independent experiments is shown. Bottom left, XPO1, METTL3 and YTHDF1 protein levels were measured by immunoblot in untreated (NT) and PTG-treated cells, with tubulin as a loading control. A representative immunoblot of 4 independent experiments is shown. Right, relative expression measured by RT-qPCR for XPO1, YTHDF1 and METTL3 in HCT116 cells, n=4. (*p<0.05, **p<0.01, ****p<0.0001, ns: non-significant according to a two-tailed Student’s t-test). (B) m6A levels in the 5’UTR of XPO1 were assessed by meRIP-qPCR. RNA from untreated (NT) and PTG-treated cells was immunoprecipitated using an anti-m6A antibody and normal IgG as a control. Enrichment in m6A was determined by qPCR, n=3. (***p<0.001 according to a two-way ANOVA test). Enrichment relative to the control IgG +p<0.1; ####p<0.0001 according to a two-way ANOVA test). (C) Two independent siRNAs targeting YTHDF1 were transfected 16 hours prior to PTG stimulation. XPO1 and YTHDF1 protein levels were measured by immunoblot in untreated (NT), PTG-treated (PTG) and PTG-treated and YTHDF1-silenced (PTG +siYTH) cells, with tubulin as a loading control. Representative immunoblot of three independent experiments. (D) C57BL/6 mice (WT) and (E) HLA-DQ2 mice. Mice were gavaged with PTG and CT (PTG) during 3 weeks, once a week. Control mice received only CT (NT). Top left, overall RNA methylation was measured by dot blot using an anti-m6A antibody. Bottom left, representative immunoblot for XPO1 and YTHDF1 protein levels in untreated (NT) and PTG-treated mice (PTG), GAPDH and tubulin were used as a loading control. Right, relative expression measured by RT-qPCR for Xpo1, Ythdf1 and Mettl3, n=5–7. (*p<0.05, ns, non-significant according to a one-tailed Mann-Whitney U test). (F) Epithelial cells derived from WT or Ythdf1 KO mice were stimulated with 250 µg/mL for 4 hours. Representative immunoblot of XPO1 and YTHDF1 protein levels in untreated (NT) and PTG-treated cells from WT or Ythdf1 KO (DF1 KO) mice, with actin as a loading control. All values are mean±SEM. ANOVA, analysis of variance; KO, knockout; m6A, N6-methyladenosine; WT, wild type.
Gliadin induces the activation of the m6A machinery in IEC leading to increased XPO1 in vitro and in vivo
m6A marks in 5’UTRs of mRNAs have been described to be key factors in the activation of translation on stress stimuli.29 In order to determine whether gliadin stimulation (the environmental trigger of CD) intensifies m6A marks and leads to an increase in XPO1 levels, we stimulated intestinal cells with PTG. HCT116 cells were cultured for 48 hours with a low-dose PTG concentration (30 µg/mL) and subsequently challenged with a higher dose of PTG (350 µg/mL) for 24 hours (see online supplemental figure 3A). We observed an increase in overall m6A levels, together with higher METTL3, YTHDF1 and XPO1 expression, as well as higher protein levels (see figure 3A and (online supplemental figure 3B). As expected, we also observed that the levels of m6A in the 5’UTR of XPO1 were higher in the presence of PTG (see figure 3B), suggesting that the gliadin-induced increase in XPO1 protein levels is mediated by an m6A-dependent mechanism. Further supporting this idea, when PTG stimulations were performed in YTHDF1-silenced cells, XPO1 levels did not vary (see figure 3C and online supplemental figure 3C). Taken together, our results show that gliadin activates the m6A machinery, increasing methylation levels in the 5’UTR of XPO1 and leading to a YTHDF1-dependent increase of XPO1 protein amounts.
To analyse whether gliadin is also able to enhance the m6A machinery and boost XPO1 translation in vivo, we treated C57BL/6 mice, previously naïve of gluten exposure, with PTG and CT (see online supplemental figure 3A). In accordance with the in vitro model, we observed that tissues from mice treated with PTG had increased m6A methylation together with Ythdf1 and Xpo1 induction at the mRNA and protein levels (see figure 3D and online supplemental figure 3D), confirming the previously observed gliadin induction of m6A and XPO1 in vivo. Interestingly, when small intestinal samples from HLA-DQ2 mice were studied, a stronger response to gliadin could be observed at RNA levels in mice that had been treated with PTG+CT versus controls kept in gluten free diet (see figure 3E and online supplemental figure 3E). In addition, YTHDF1 involvement in gliadin-mediated XPO1 induction was further confirmed as epithelial cells from Ythdf1 KO mice barely showed XPO1 increase when compared with WT (see figure 3F and online supplemental figure 3F).
Allele-specific increase of XPO1 effectuates intestinal inflammation in vitro and in vivo
XPO1 inhibitors have been widely tested as anticancer drugs due to their ability to downregulate NFkB activity.30 It is also known that gliadin induces NFkB activation and further cytokine release in human immune20 and IEC.31 However, the cell-type specificity and the mechanisms underlying gliadin-induced inflammation are not totally understood. Thus, we wanted to determine whether the activation of the m6A machinery and subsequent XPO1 increase in response to PTG is able to activate the NFkB and downstream inflammatory pathways in human intestinal cells. As described above, the higher m6A levels present in the risk allele form (XPO1*T) lead to greater XPO1 protein amounts (see figure 1E). In accordance with our hypothesis, we observed that cells overexpressing the XPO1*T form showed significantly higher induction and activation of the NFkB subunit p50 (see figure 4A,B and online supplemental figure 4A).
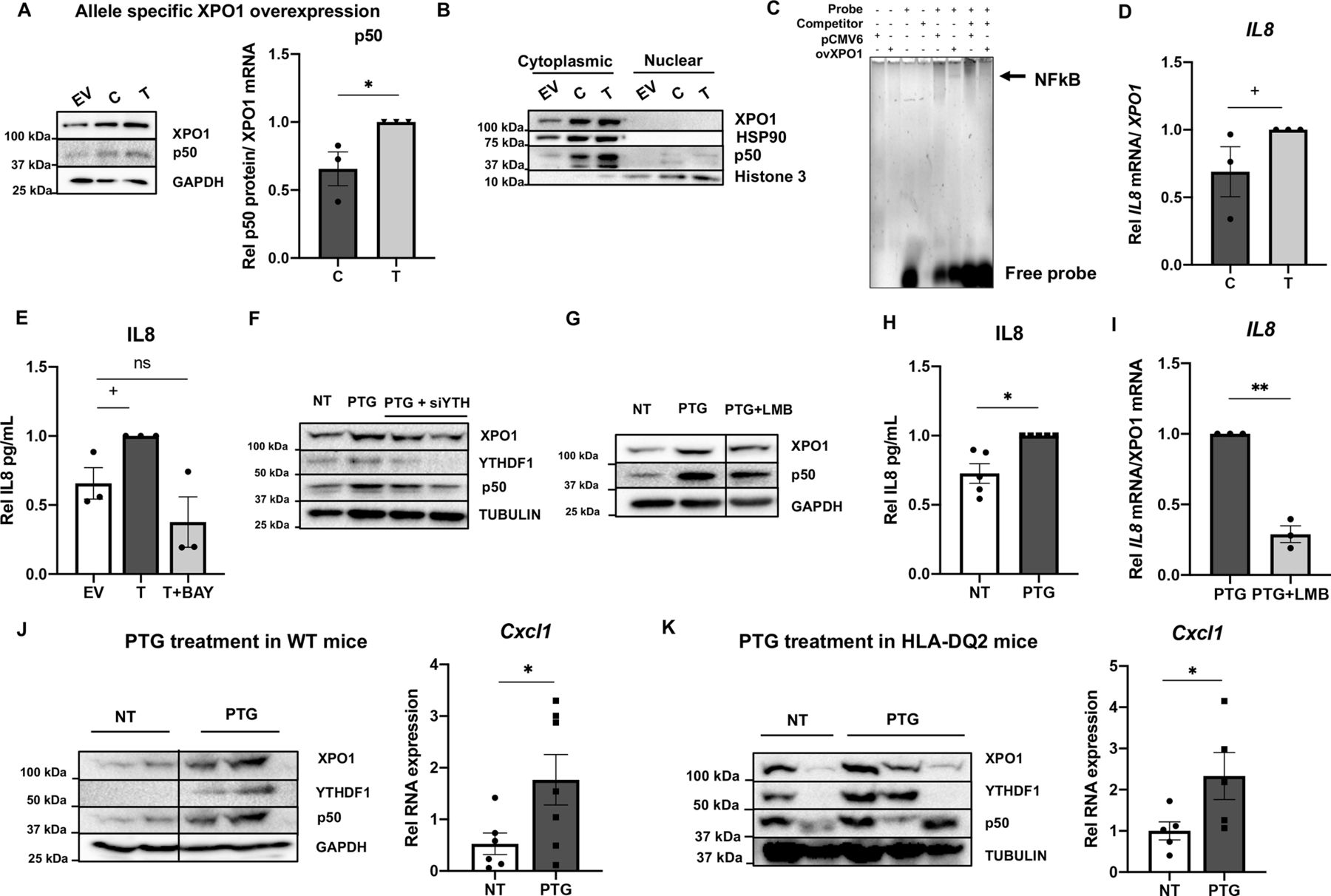
Allele-specific increase of XPO1 effectuates intestinal inflammation in vitro and in vivo. Expression plasmids with each form of the XPO1 5’UTR upstream the cDNA or the empty vector (EV) were transfected into HCT116 cells. (A) XPO1 and p50 protein levels were quantified by immunoblot, with GAPDH as a loading control. Left, representative immunoblot of 3 independent experiments; right, quantitative summary data of p50 relative to XPO1. (*p<0.05. according to a two-tailed Student’s t-test). (B) The cytoplasmic and nuclear fractions of cells overexpressing each 5’UTR form were separated and XPO1 and p50 protein levels were quantified by immunoblot, HSP90 was used as a cytoplasmic loading control and H3 as a nuclear loading control. (C) Nuclear extracts from HCT116 cells transfected with an EV or XPO1 expression plasmid (ovXPO1) were analysed by gel shift assay for their ability to bind to an oligonucleotide with the IL8 promoter NFkB consensus sequence, with 10X unlabeled probe as competitor. A representative gel of 2 independent experiments is shown. (D) IL8 expression was quantified by RT-qPCR in cells overexpressing both forms of XPO1; n=3. (+p<0.1, according to a one-tailed Student′s t-test). (E) Secreted IL8 was measured by ELISA in the medium of cells transfected with an EV or overexpressing XPO1*T in the absence (T) or presence of the NFkB inhibitor Bay 11–7082 (T+BAY), n=3. (+p<0.1, ns: non-significant according to a one-way ANOVA test). (F) XPO1, YTHDF1 and p50 protein levels were measured by immunoblot in untreated (NT), PTG-treated (PTG) and PTG-treated +YTHDF1-silenced (PTG +siYTH) cells, with tubulin as a loading control. A representative immunoblot of three independent experiments is shown. (G) Xpo1 and p50 protein levels were quantified by immunoblot in untreated (NT), PTG-treated (PTG) and cells treated with PTG + the XPO1 inhibitor leptomycin B (PTG +LMB), with GAPDH as a loading control. A representative immunoblot of 3 independent experiments is shown. (H, I) IL8 expression and secreted IL8 levels were quantified by RT-qPCR and ELISA, in untreated (NT), PTG-treated and PTG+LMB-treated cells, n=3–5 (*p<0.01; **p<0.01 according to a two-tailed Student′s t-test). (J) Left, representative immunoblot of p50 protein levels in untreated (NT) and PTG-treated WT mice, with GAPDH as a loading control. Right, expression of mouse IL8 functional homolog cytokine Cxcl1 quantified by RT-qPCR in untreated (NT) and PTG-treated WT mice, n=7. (*P<0.05, according to a one-tailed Mann-Whitney U test). (K) Left, representative immunoblot of p50 protein levels in untreated (NT) and PTG-treated humanised HLA-DQ2 mice, with tubulin as a loading control. Right, expression of mouse IL8 functional homolog cytokine Cxcl1 quantified by RT-qPCR in untreated (NT) and PTG-treated humanised HLA-DQ2 mice, n=2–3. (*P<0.05 according to a one-tailed Mann-Whitney U test). All values are mean±SEM. 5’UTR, 5′ untranslated region; ANOVA, analysis of variance; IL8, interleukin 8; LMB, leptomycin B; NFkB, nuclear factor kappa B; PTG, Pepsin Trypsin digested Gliadin.
The activation of NFkB implies an increased binding to target gene promoters in order to induce their transcription. Among other NFkB targets, IL8 has been identified as a cytokine that is induced by gluten,32 so we tested whether XPO1 is involved in NFkB-mediated IL8 increase in our model. First, Electrophoretic Mobility Shift Assay (EMSA) experiments showed that nuclear extracts of cells overexpressing XPO1 exhibit higher protein binding to the NFkB consensus sequence in the IL8 promoter (see figure 4C). Moreover, as it was the case for p50 protein activation, we observed that IL8 expression is higher in cells overexpressing XPO1, and again, induction is further increased in the presence of the XPO1*T form (see figure 4D). We confirmed that this XPO1-mediated IL8 increase is NFkB-dependent, as treatment with BAY 11–7082 (a NFkB inhibitor) counteracted the IL8 increase in XPO1 overexpressing cells (see figure 4E and online supplemental figure 4B).
In addition, cells treated with PTG, which show activated m6A machinery and increased XPO1 levels (see figure 3A), also showed increased amounts of p50 (see online supplemental figure 4C). We confirmed that the PTG induction of p50 is m6A-dependent, as PTG stimulation did not increase NFkB in YTHDF1-silenced cells (see figure 4F and online supplemental figure 4D). Moreover, when PTG-stimulated cells were treated with the XPO1 inhibitor leptomycin B, we observed a decline in the induction of p50, confirming that the inflammatory environment induced by PTG is, at least in part, dependent on XPO1 (see figure 4G and online supplemental figure 4E). Additionally, we confirmed that IL8 protein was also induced by PTG in an XPO1-function-dependent manner (see figure 4H,1 and online supplemental figure 4F). Altogether, these results suggest that gliadin-induced increase of both m6A and XPO1 activates NFkB pathway in IEC in vitro in an allele-specific manner. PTG-treated mice also showed induction of NFkB, together with an increase in the transcription of the IL8 mouse functional homolog Cxcl1 (see figure 4J,K and online supplemental figure 4G, H), demonstrating the physiological effect of gliadin stimulation in vivo. Moreover, PTG treatment in Ythdf1 KO mice-derived epithelial cells did not show an increase in Cxcl1 ex vivo (see online supplemental figure 4I).
Coeliac patients have allele-specific variation of XPO1 protein levels, a gluten-dependent increase in m6A, XPO1, METTL3 and YTHDF1, and downstream inflammatory markers
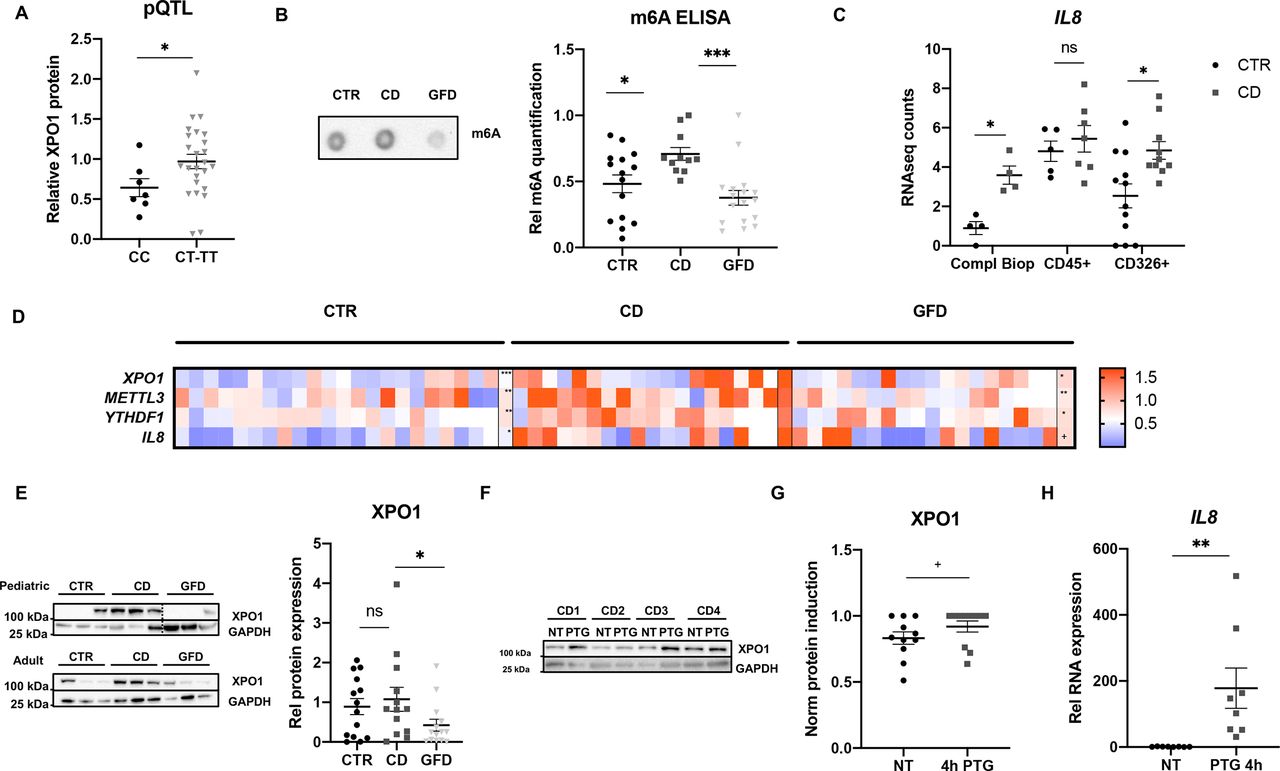
Small intestinal biopsies from coeliac patients and non-coeliac controls were used to confirm the results obtained in the cell and mouse models. Genotyping of the human samples showed that although the expression levels of XPO1 did not change significantly according to the SNP genotype (see online supplemental figure 5A), biopsies from individuals carrying the risk allele (rs3087898 genotypes CT and TT) produced higher amounts of XPO1 protein (see figure 5A). This result confirms that the genotype of the CD-associated SNP affects XPO1 protein levels (protein Quantitative Trait Loci, pQTL), which in turn influence predisposition to develop the disease [Minor Allele Frequency (MAF)controls=0.41/MAFCD=0.44].33 We quantified overall m6A levels showing that biopsies of active (newly diagnosed) CD patients have higher methylation compared with control individuals (see figure 5B). We also observed higher expression in the active CD group compared with controls in all genes (see figure 5D) as well as XPO1 protein amounts, both in paediatric and adult patients (see figure 5E). More interestingly, when gluten had been removed from the CD patients’ diet (GFD), total m6A levels (see figure 5B), expression of all genes (see figure 5D) as well as XPO1 protein amounts (see figure 5E) reverted to normal values, indicating that the induction of m6A, XPO1, METTL3, YTHDF1 and IL8 is gluten-dependent. In order to confirm the role of epithelial cells in this pathway, we quantified IL8 expression in complete biopsies and immune and epithelial cell fractions from controls and active coeliac patients. We observed that even though immune cells seem to be the main producers of IL8, the differences between active CD and control individuals come mainly from the epithelial fraction (see figure 5C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Coeliac patients have allele-specific variation of XPO1 protein levels, a gluten-dependent increase in m6A, XPO1, METTL3 and YTHDF1, and downstream inflammatory markers. (A) DNA and protein were extracted from intestinal biopsies of control (CTR) individuals, XPO1 levels were measured by ELISA and rs3087898 was genotyped using an IDT genotyping assay. XPO1 protein levels were compared between individuals with the protective genotype (CC) and individuals harbouring the risk allele (CT+TT), n=7–25. (*p<0.05 according to a two-tailed Mann Whitney test). (B) RNA was extracted from intestinal biopsies from CTR, active coeliac patients (CD) and coeliac patients on gluten free diet (GFD) and overall RNA methylation was measured. Left, representative m6A DotBlot, and right, quantitative summary of total m6A levels quantified by a commercial m6A ELISA kit, n=15–17. (*p<0.05, ***p<0.001 according to a one-way ANOVA test). (C) IL8 expression by RNAseq counts from a previous RNAseq study46 in complete biopsies, CD45+ immune cell fractions and CD326+ epithelial cell fractions from non-coeliac CTR and active coeliac disease individuals (CD), n=4–12. (*p<0.05, ns: non-significant according to a two-way ANOVA test). (D) The expression of XPO1, METTL3, YTHDF1 and IL8 was measured by RT-qPCR in RNA extracted from adult and paediatric individuals, n=6–16. Individual and mean values (last row of each group) of relative RNA expression represented in a heatmap (+p<0.1; *p<0.05, **p<0.01, ***p<0.001 according to a one-way ANOVA test). (E) XPO1 protein levels were measured by immunoblot in biopsies from paediatric and adult individuals, GAPDH was used as a loading control. Left, representative immunoblot of 5–10 samples per group; right, quantitative summary of immunoblot for XPO1 in both paediatric and adult biopsies from CTR, active CD patients and patients on GFD, n=14–16. (*p<0.05, ns: non-significant according to a one-way ANOVA test). (F, G) Biopsies from active patients were cut into two pieces and incubated with PTG for 4 hours. Representative immunoblot and XPO1 protein levels measured by ELISA in biopsies untreated (NT) and incubated with gliadin for 4 hours (PTG), GAPDH was used as a loading control, n=13. (+p< 0.1 according to a one-tailed Mann Whitney test). (H) IL8 expression was measured by RT-qPCR in untreated (NT) and PTG-incubated biopsies. (**p<0.01 according to a one-tailed Mann Whitney test). All values are mean±SEM. ANOVA, analysis of variance; IL8, interleukin 8; PTG, pepsin trypsin digested gliadin.
To further confirm the role of gluten in this scenario, biopsies from active CD patients were incubated with or without PTG for 4 hours. We detected a trend towards increased METTL3, YTHDF1 and XPO1 mRNA levels (see online supplemental figure 5B) and enhanced XPO1 protein levels (see figure 5F,G) in PTG-challenged biopsies. We could also confirm overexpression of IL8 in the stimulated biopsy samples (see figure 5H), further implying a role of gluten in this process. Interestingly, the PTG-induced increase of YTHDF1 in intestinal biopsies significantly correlated with the increase of XPO1 (see online supplemental figure 5C), confirming this m6A reader to be important for the gluten-dependent XPO1 induction in the tissue stimulation model. Altogether, these results corroborate the gliadin-induced effects observed in vitro and in vivo in human samples, and stress the importance of the activation of the m6A machinery for the consequent XPO1 increase and IL8 induction in biopsies from CD patients.
Discussion
Our work demonstrates that non-coding SNPs associated with susceptibility to immune and inflammatory diseases can alter m6A levels and functionally contribute to disease pathogenesis. This occurs via cell type-specific activation of an inflammatory response, confirming what has been recently suggested by a transcriptome-wide m6A QTL analysis.16 It was previously described that SNPs located in the 5’UTR can disrupt their secondary structure and affect translation efficiency.19 In addition, m6A marks in 5’UTRs increase in response to stress stimuli, expediting translation as a mechanism for rapid cellular response.8 Here, we show how the genotype of the CD-associated SNP rs3087898, located in the 5’UTR of the coding gene XPO1, affects mRNA methylation and subsequent translation. We found that the SNP genotype affects the binding of the RBP YTHDF1 as well as the predicted secondary structure of the 5’UTR of XPO1, what could lead to alterations in m6A levels and translational features concurring with results from a recent m6A QTL study.16 In IEC, this increased methylation confers a higher translation efficiency of the mRNA molecules that carry the risk allele (XPO1*T), an effect that is mediated by the m6A reader YTHDF1, as we have shown both in vitro and in vivo. It is important to emphasise that in contrast to enterocytes, we have observed that T cells have fewer or no methylation marks on the 5’UTR of XPO1, suggesting that this m6A-mediated mechanism is specific for IEC. Moreover, this is the first study-linking gluten, the triggering agent for CD, with alterations in m6A methylation. Interestingly, we have shown that the gluten-induced m6A-mediated increase of XPO1 activates the NFkB pathway, a hallmark of CD, opening the door to novel therapeutic approaches for CD patients.
It is known that the rapid kinetics of m6A methylation provides cells with the ability to quickly respond to external stimuli, activating distinct pathways in different cell types.13 15 Viral infections or microbiome composition are thought to contribute to CD development, and the microbiome has been recently involved in the levels of m6A methylation present in the gut.34 However, gliadin peptides derived from gluten are the main antigens triggering CD.1 2 34 Although gliadin has been proposed as an activator of the innate immune response, little is known about the early steps that lead to breakdown of gluten tolerance. Our in vitro and in vivo experiments link gliadin exposure with an overall increase of m6A and YTHDF1, which in turn results in augmented XPO1 protein levels. This novel link between m6A modifications and gliadin exposure adds a completely new and unexplored layer to the complex scenario of CD pathogenesis. In addition, considering that viral infections and microbiome composition have been linked both to CD and to m6A methylation changes, further studies with other CD-triggering agents could help better understand the factors involved in the activation of innate immunity in CD.
XPO1 is a nuclear export receptor involved in critical signalling pathways and cellular functions and alterations of XPO1 have been associated with several malignancies. Appropriate shuttling of IkBa via XPO1 nuclear export has been linked to the activation of the NFkB pathway and downstream cytokine secretion. Moreover, it has been shown that cells treated with XPO1 inhibitors display lower NFkB activity and consequent decrease in cytokine release.35 We have shown that increased XPO1 protein levels, a consequence of the more abundant m6A marks in the XPO1*T mRNA, promote the induction of NFkB, a hallmark of CD, providing a functional explanation for the association of the SNP with CD. Moreover, we also show that the XPO1-mediated induction of NFkB is YTHDF1-dependent, supporting the implication of m6A methylation in the gluten-induced inflammatory response in IEC in CD.
CD is characterised by the destruction of the small intestinal epithelial barrier leading to IEL infiltration and an exacerbated activation of macrophages and other immune cells. A recent study using transgenic mice has highlighted the complexity of CD pathogenesis, as in the CD-like mouse model the activation and regulation of IEC as well as immune cells is needed to develop the characteristic immune response and villous atrophy that is seen in human CD patients. It has been described that different cytokines secreted by IECs play key roles in the activation and early steps of the immune response in CD.4 IL8, a neutrophil attractant chemokine, plays a key role in tissue damage and activation of the innate immune response and it has been proposed that in CD patients, IL8 is specifically secreted after gluten ingestion.36 37 In accordance with an allele-specific NFkB activation, we observed higher IL8 transcription and secretion in the presence of the XPO1 risk allele. Moreover, Cxcl1, considered functional murine homolog of IL8, was also upregulated in the intestine of mice challenged with gliadin, confirming the XPO1-mediated activation of intestinal inflammation in response to the stress stimuli. So far, most studies have been performed in CD derived immune cells and peripheral blood mononuclear cells.38–40 However, this is the first study where it is shown that CD-derived epithelial cells also express IL8, indeed, we have seen that even if they are not the main producers, epithelial cells of active CD patients show increased IL8 expression. As described for IL154, it is possible that the mechanism uncovered in the current study could be involved in an early response to gluten-mediated epithelial damage and further activation of immune cells.
Our results have clinical significance, as we determined that patients with active CD (on a gluten containing diet) have elevated m6A and XPO1, together with IL8 cytokine levels that were reversed after GFD. Additionally, we also observed that individuals harbouring the risk allele expressed higher levels of XPO1 protein, explaining the association of this SNP with disease susceptibility, as individuals homozygous for the risk allele (TT) are predicted to have higher levels of basal inflammation due to augmented amounts of XPO1 protein.
In summary, our data stress the importance of the m6A machinery in the gluten-mediated XPO1 increase that enhances NFkB expression and subsequently leads to IL8 induction in the enterocytes of CD patients. The only reliable treatment for CD so far is a strict lifelong removal of gluten from the diet, which reverses most of the disease symptoms, but negatively affects patients’ quality of life and psychological well-being. This impacts adherence to the GFD and increases the risk of complications, including gastrointestinal cancers.41 Indeed, both XPO1 and IL8 levels are increased in tumour tissues from colon and colorectal adenocarcinomas (as observed using TGCA data) while YTHDF1 has been described to regulate tumourigenicity in human colorectal adenocarcinomas.42 Interestingly, studies show that the risk to develop colon cancer in CD patients is lower in patients on GFD.43 Small molecules targeting m6A readers and erasers have already been suggested as potential therapy for different types of cancer44 and emerge as an option for protection against inflammation in CD patients. Furthermore, XPO1 inhibitors are already in use or in advanced clinical trials as anticancer therapeutic agents and have shown to have ability to decrease NFkB activity due to IkBa nuclear retention.35 45 Thus, our study provides the first experimental evidence of the involvement of a gluten-responsive m6A-XPO1-NFkB pathway in the pathogenesis of CD, putting forward novel potential therapeutic targets for this disease. The involvement of m6A, XPO1 and NFkB in diverse intestinal malignancies supports that the m6A-XPO1-NFkB axis may possibly function in response to small intestinal epithelial injury by other agents, which may have direct implications in the treatment of a variety of gastrointestinal disorders.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplemental information. Online available data analyzed in this study are: miCLIP (GSE128699) miCLIP (GSE128699): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128699 and RNAseq (GSE84745): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84745. Data is held in GEO repository and it is freely available for reuse.
Ethics statements
Patient consent for publication
Ethics approval
Experiments using these biopsy samples were approved by the corresponding ethics committee.
Acknowledgments
We thank Dr Robert Anderson and the University of Melbourne for providing and facilitating the use of the HLA-DR3-DQ2 mice model through a Materials Transfer Agreement.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it published Online First. The author's name,
Iraia Garcia-Santisteban, was amended.
Contributors AC-R designed the research. AO-G, LZ, PM, JKG, MS-D, LMM, IGS and JAR performed the experiments. IR, AH, PHG and GB recruited patients and collected human samples. AO-G, GB, LH, DS, EFV, CH, JRB and AC-R analysed the data. AO-G and AC-R wrote the paper. All authors read and approved the final version of the manuscript.
Funding This study was supported by a grant from the Spanish Ministry of Science, Universities and Innovation (PGC2018-097573-A-I00) to AC-R. JRB was funded by ISCIII Research project PI16/00258, cofinanced by the Spanish Ministry of Economy and Competitiveness and by the European Union ERDF/ESF ‘A way to make Europe’. AO-G and MS-D were funded by predoctoral fellowships from the Basque Government and the University of the Basque Country respectively. DS and LH were funded by the Spanish Ministry (MINECO) (SAF2017-83813-C3-1-R) and cofunded by the ERDF, the Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y la Nutrición (CIBEROBN) (Grant CB06/03/0001 to DS), the Government of Catalonia (2017SGR278 to DS), and the Fundació La Marató de TV3 (201627-30 to DS). CH is a Howard Hughes Medical Institute Investigator and has been funded by the National Institute of Health HG008935. We would like to thank Xuechen Yu and Justin Vargas for processing the adult CD biopsy samples obtained from Columbia University. EFV is supported by a CIHR grant 168840 and holds a Canada Research Chair.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.