Article Text

Abstract
Objective The systemic spread of colorectal cancer (CRC) is dominated by the portal system and exhibits diverse patterns of metastasis without systematical genomic investigation. Here, we evaluated the genomic evolution of CRC with multiorgan metastases using multiregion sequencing.
Design Whole-exome sequencing was performed on multiple regions (n=74) of matched primary tumour, adjacent non-cancerous mucosa, liver metastasis and lung metastasis from six patients with CRC. Phylogenetic reconstruction and evolutionary analyses were used to investigate the metastatic seeding pattern and clonal origin. Recurrent driver gene mutations were analysed across patients and validated in two independent cohorts. Metastatic assays were performed to examine the effect of the novel driver gene on the malignant behaviour of CRC cells.
Results Based on the migration patterns and clonal origins, three models were revealed (sequential, branch-off and diaspora), which not only supported the anatomic assumption that CRC cells spread to lung after clonally expanding in the liver, but also illustrated the direct seeding of extrahepatic metastases from primary tumours independently. Unlike other cancer types, polyphyletic seeding occurs in CRC, which may result in late metastases with intermetastatic driver gene heterogeneity. In cases with rapid dissemination, we found recurrent trunk loss-of-function mutations in ZFP36L2, which is enriched in metastatic CRC and associated with poor overall survival. CRISPR/Cas9-mediated knockout of ZFP36L2 enhances the metastatic potential of CRC cells.
Conclusion Our results provide genomic evidence for metastatic evolution and indicate that biopsy/sequencing of metastases may be considered for patients with CRC with multiorgan or late postoperative metastasis.
- colorectal cancer
- colorectal metastases
- gene mutation
Data availability statement
Data are available on reasonable request. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, HX (xuheng81916@scu.edu.cn). Most data supporting the findings of this study are available within the article and onlin supplemental files. The raw sequencing data from this study have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, under accession numbers HRA000046 (BioProject: PRJCA001380) that can be accessed at http://bigd.big.ac.cn/gsa-human. All other data are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Anatomically, colorectal cancer (CRC) metastasis is assumed to follow a sequential manner, where liver is the first and most common site, followed by extrahepatic organs, such as lung.
Although extrahepatic metastasis is frequently observed concurrent with liver metastasis in clinic, it may also arise as oligometastasis in several patients with CRC.
Targeted treatment of metastatic CRC is challenging, and the drug response predominantly relies on the mutation status of driver genes such as KRAS and BRAF.
What are the new findings?
Three metastatic models (ie, sequential, branch-off and diaspora) were revealed based on the distinct migration patterns and clonal origins.
The anatomic assumption that CRC cells may spread to lung after clonally expanding in liver was genomically evidenced. Our data also support the direct seeding of extrahepatic metastases from primary tumours.
Extrahepatic metastases may originate from advanced tumour clones with additional driver gene mutations acquired, leading to late metastases with intermetastatic driver gene heterogeneity.
Enriched trunk mutations of ZFP36L2 were identified in metastatic CRC with rapid disseminations and poor treatment outcomes.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The sequential model enforces the importance of timely resection of liver metastases to prevent further metastatic progression of liver-derived tumour clones.
Given the intermetastatic driver gene heterogeneity, biopsy and sequencing of metastases may be considered for therapeutic decision making in patients with CRC with multiorgan or late postoperative metastasis.
Comparative evaluation of driver genes, including ZFP36L2, may improve the risk stratification and refine the personalised therapy recommendations, leading to optimal oncological outcomes.
Introduction
Distant metastasis is a lethal consequence of cancer progression in patients with colorectal cancer (CRC), accounting for the majority of cancer-related deaths.1 The presence of clinically overt metastases in distant organs not only indicates end-stage disease but also leaves patients few therapeutic options.2 Since main venous drainage of the large intestine flows into liver via the portal system, metastatic pattern of CRC is considered to follow a stepwise manner, where the regional lymph nodes (RLN) are the first-station, followed by liver and subsequent extrahepatic organs (eg, lung).3 However, such an anatomic assumption was neither explored by genomic investigation nor fully supported by the diverse clinically observed metastatic patterns, where extrahepatic metastasis arises regardless of lymph node and/or liver metastasis.4 5 Moreover, recent genomic studies revealed that lymph nodes can be seeded by multiple tumour clones and liver metastases may originate from independent subclones in the primary tumour directly, reinforcing the uncertainty of canonical metastatic models.6–8 These findings support the notion that metastasis is determined not only by extrinsic factors (eg, physical accessibility), but also by intrinsic traits of cancer cells.
Cancer cells acquire metastatic potential as a result of random mutations, genetic drift and nonrandom selection.9 As such, primary and metastatic cancer cells are expected to be genetically heterogeneous, which constitutes the rational basis for phylogenetic analysis.10 To address the genomic evolution of metastasis, clones with metastatic potential are distinguished from subclones in primary tumours based on genomic features of patient-matched metastatic samples.6 11 However, although cancer metastasis is known as a systemic disease with multi-organ involvement, genomic analyses of distant metastasis thus far predominately focus on single pairing of primary tumour/metastasised organ (eg, primary tumour paired with liver metastasis).8 12–15 Thus, direct genomic evidence for systemic metastatic evolution in CRC is still lacking.
Because metastasis affects the effectiveness of clinical treatment, elucidating the paths and origins of metastasis may optimise treatment decisions for patients with CRC. For instance, timely lesion resection will be immensely important if extrahepatic metastases originate from liver metastases, while treatment of metastatic CRC will be more complicated if multiorgan metastases derive from different tumour clones.16 A recent mathematical model demonstrated the homogeneity of driver mutations among metastases, suggesting that a single biopsy adequately represents the driver mutations for therapeutic decision making.17 However, since liver acts as an anatomic barrier in venous drainage of the large intestine, genomic evolution of systemic metastases in CRC may not be comparable to tumours that spread in systemic circulation directly (eg, lung cancer). To date, the phylogenetic evolutions from the primary site to, and between liver and extrahepatic organs in CRC have barely been characterised.10 It is thus unclear whether the treatment strategies are needed to be modified according the diverse models of systemic metastases in CRC.
Here, we analysed the genomic evolution of distant metastases based on multiregion sequencing of CRC with multiorgan metastases to reconstruct phylogenetic trees, and identified three distinct metastatic models, including branch-off model (polyphyletic seeding from primary tumour to liver/lung), sequential model (monophyletic seeding from liver to lung), and diaspora model (monophyletic seeding from primary tumour to liver/lung). Moreover, we also identified a potential novel driver gene, recurrent mutations of which were validated in independent cohorts.
Materials and methods
Patients
Medical records of 14 216 patients with CRC in West China Hospital (WCH) from 2009 to 2018 were systemically reviewed. We selected six cases with available formalin-fixed paraffin-embedded surgical samples from matched normal colon/rectum tissues, primary tumour (PRM) and multiorgan metastases (figure 1A). Meanwhile, 146 patients with CRC with matched PRM and liver/lung metastasis, and 210 consecutive patients with CRC with available PRM samples only were included for validation (online supplemental figure S1,S2).
Supplemental material

Tracking tumour evolution of distant metastases in colorectal cancers. (A) Study design schematic. samples from primary tumour (PRM), regional lymph node metastasis (RLN), liver metastasis (LIM), lung metastasis (LUM), thoracic lymph node metastasis (TLN) and normal tissue from six patients with colorectal cancer were selected based on pathological evaluation and sent for whole-exome sequencing (WES). Phylogenetic trees were reconstructed to infer the metastatic seeding pattern of each case. D1–D3 stand for different driver mutations acquired during metastatic evolution. Multiregion genomic data from each patient were jointly analysed. The candidate novel features were validated in two independent cohorts. (B) Mutation counts shared by at least n tumour samples in each patient. (C) The total number of somatic mutations is visualised as stacked bars classified by private mutations, shared mutations (recur in at least two tumour samples) and trunk mutations (shared by all tumour samples) in each individual (top). The trunk and shared mutations were clustered to illuminate the phylogenetic relationship of each mutation among tumour samples (bottom). (D) Fractions of driver gene mutations are higher in trunk mutations compared with non-trunk mutations. (*p<0.05, two-sided paired t-test) (E) (left) Most LIM are more closely related to the PRM than to LUM. The plot shows the d(LIM to PRM)/d(LIM to LUM) – 1. (The distance of each LIM to its closest PRM, divided by its distance to its closest LUM, minus one) (right) Analogous plot for LUM, showing d (LUM to PRM)/d(LUM to LIM) – 1.
Cell lines
Humans CRC cell lines HCT15, HCT116, and SW480 were purchased from ATCC, and were cultured with recommended medium (online supplemental methods) at 37℃ with 5% CO2.
Supplemental material
Data analysis
Details were described in online supplemental methods.
Results
Overview of samples
To comprehensively analyse the lineage between primary tumour and distant metastases, we collected surgical samples of six CRC cases who underwent multiple operations, with clinical information shown in online supplemental table S1. Overall, a total of 74 patient-matched samples passed pathological review and quality control for subsequent analysis (online supplemental methods, figure S2 and data S1). Briefly, all patients had one primary lesion except two for patient C01. Both liver metastasis (LIM) and lung metastasis (LUM) were collected for each patient, with two synchronous LIM in patient C03 and two metachronous LUM in patient C01. Additionally, we also obtained metastatic samples from RLN of patient C01 and C02, and thoracic lymph node (TLN) of patient C01. In most cases, different regions of each tumour were sampled, with the total number of 18 PRM, 22 LIM, 20 LUM, 5 RLN and 3 TLN.
Supplemental material
Supplemental material
Next, we performed whole-exome sequencing and identified a total of 1899 somatic mutations, and 1588 of which were validated with a targeted sequencing approach, with a median depth of sequencing of 384× (ranging from 53× to 1121.5×) (online supplemental table 1). No CRC-related germline mutation, somatic hypermutation or microsatellite instability (MSI) case was identified. Shared somatic mutation counts gradually decreased with the increasing number of tumour samples in each case, and eventually maintained at the level of ~70 mutations (figure 1B). Notably, no single mutation was shared by more than 18 tumour samples (n=21) in patient C01 probably due to the distinct genetic origin of synchronous primary tumours. Thus, the mutations shared by 18 tumour samples were considered as clonal in this case and those from the independent tumour lesion were excluded in most subsequent analysis. Overall, an average of 76 (ranging from 69 to 82) clonal mutations were identified across these patients and classified as trunk mutations (figure 1C and online supplemental table S3).
Based on The Cancer Genome Atlas (TCGA) consensus list of putative driver genes,18 mutations were further classified into driver (n=48) and passenger (n=1408) mutations. Trunk mutations exhibited significantly higher fractions of driver genes compared with non-trunk mutations (6.0% vs 1.9%; p=0.02) (figure 1D), supporting the positive selection pressures during cancer progression. Pairwise genetic distances, defined as the total number of non-shared mutations across the entire exome for two samples,6 19 were then calculated to illuminate the evolutionary relationships (online supplemental figure S3). For 12 out of 22 (54.5%) LIM, their distance to PRM was shorter than that to LUM, and 6 out of 20 (30%) LUM had a shorter genetic distance to PRM than to any LIM (figure 1E), indicating that both LIM and LUM probably originated from PRM directly in some cases. On the other hand, several LUM demonstrated a shorter genetic distance to LIM than to any PRM, attributing these LIM to LUM. Together, this result supports diverse models of systemic metastasis in CRC, which merits further exploration.
Phylogenetic reconstruction of metastatic spread in CRC
To analyse the genomic evolution, we reconstructed the phylogeny of metastases in each case by using Treeomics.20 A hepatic origin of LUM was demonstrated in two out of six patients, which supported the anatomic assumption and explained why most patients with LUM also experienced LIM.4 Briefly, patient C01 had synchronous tumours in sigmoid colon and rectum (figure 2A). No shared driver mutation was present between these two lesions, revealing distinct genetic origins even in the same individual. The phylogenic analysis attributed subsequent regional and distant metastases to the rectal cancer, where cancer cells spread from PRM to liver and lung in a sequential manner, supporting a model of sequential metastasis (hereafter referred to as ‘sequential model’). Similarly, the tumour migration pattern of patient C02 resembled that of sequential model (figure 2B). Although LIM samples were obtained later than LUM and had a longer exposure to chemotherapy, a liver derived LUM was still demonstrated. In these two cases, RLN descend from subclones in PRM over time rather than give rise to each other, supporting the wider evolutionary bottleneck of RLN than distant metastases.7 21 Additionally, although it was assumed that cancer cells migrated from lymph node may spread to lung through venous circulation via subclavian vein,6 neither LIM nor LUM in these two patients was seeded through RLN.
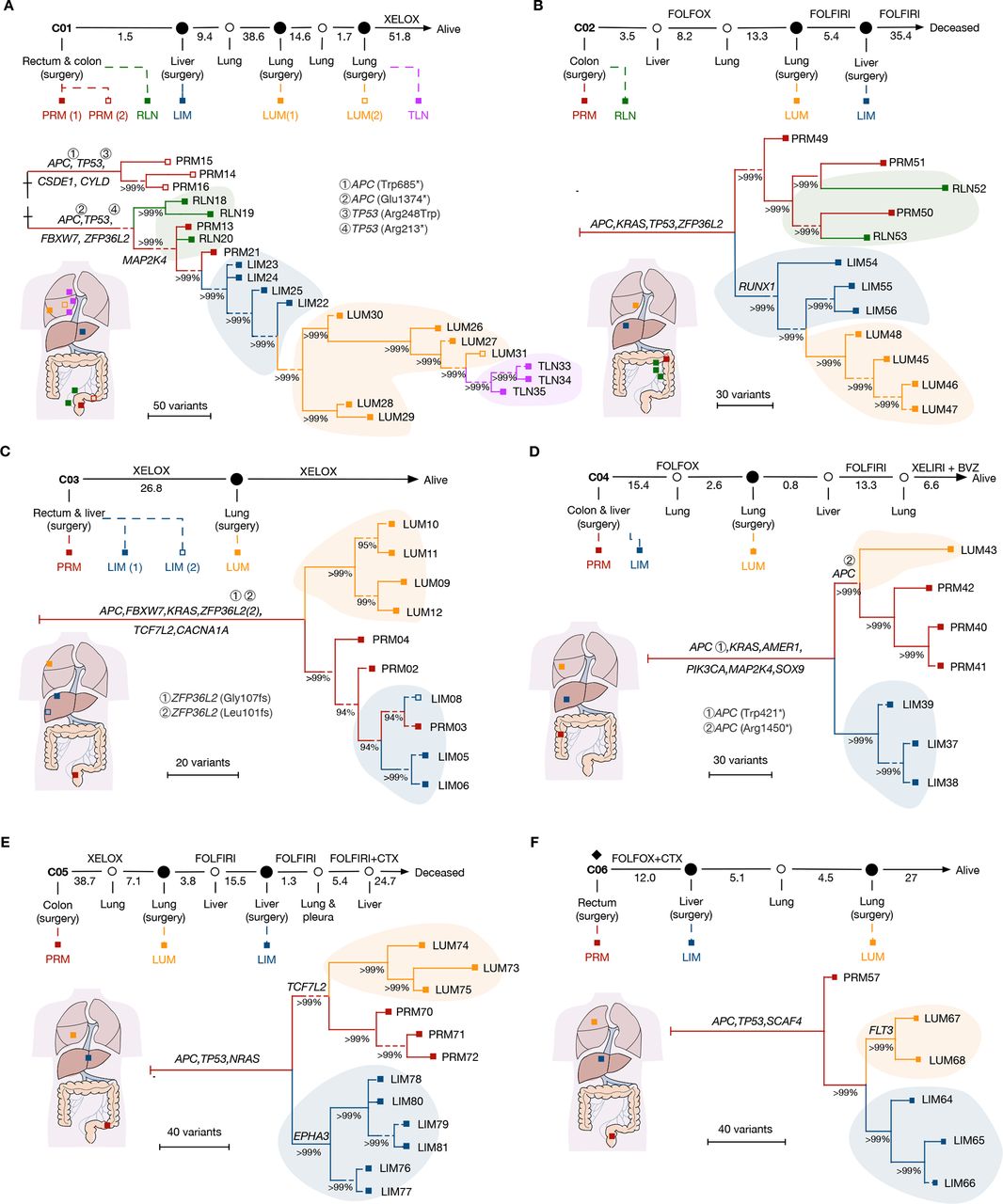
Phylogenetic reconstruction of metastatic spread in CRC. (A–F) The clinical course is shown from the time of surgical resection of primary tumours to death or last follow-up. A new metastasis clinically observed is annotated as hollow circles. Rhombus indicates the use of neoadjuvant therapy. A hollow square indicates synchronous lesions. Time intervals between each event are shown in months. Systemic treatments are indicated in the intervals. A schematic diagram was provided for each case to demonstrate the origin site of each sample. All trees were reconstructed with phylogenomic methods and scaled to demonstrate the metastatic pattern. Seeding events that gave rise to primary tumour (PRM), regional lymph node metastasis (RLN), liver metastasis (LIM), lung metastasis (LUM), thoracic lymph node metastasis (TLN) are shaded in red, green, blue, orange and purple, respectively. Putative driver gene mutations are annotated on the trunk or branches of the trees. The length of the dashed line is not taken into account in the scale bar. BVZ, bevacizumab; CRC, colorectal cancer; CTX. cetuximab; FOLFIRI, 5-fluorouracil, leucovorin, irinotecan; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; XELIRI, capecitabine plus irinotecan; XELOX, capecitabine plus oxaliplatin.
The other four cases were classified as a different pattern, exhibiting independent origin of LIM and LUM from PRM, supporting a model of branch-off metastasis (hereafter referred to as ‘branch-off model’). Patient C03 underwent resection of two metastases in the right liver lobe (figure 2C). Phylogenetic reconstruction clustered two liver metastatic lesions to the same clade with several regions of the primary tumour, indicating recent divergence. Notably, an early branching of metastatic clone to lung was observed, implicating LUM occurred ahead of LIM. Patient C04 and C05 exhibited a similar pattern, but with early occurrence of LIM. Meanwhile, the CRC evolved further and independently gave rise to LUM (figure 2D,E). Although only one PRM sample was obtained from patient C06, distinct lineage between LIM and LUM was also illustrated (figure 2F).
To assess the robustness of our inference, we used other methods (eg, Lineage Inference for Cancer Heterogeneity and Evolution (LICHeE)22; and Minimum Event Distance for Intra-tumour Copy-number Comparisons (MEDICC)23) to infer phylogenetic trees. Although slightly different trajectories were constructed, the basic tree structures of phylogeny for all patients were consistent (online supplemental figure S4, S5). Collectively, at least two migration patterns in CRC were identified: a sequential model that liver metastases were seeded from primary tumour initially and spread to extrahepatic organs (eg, lung) after clonally expand in liver; and a branch-off model with distant metastases in different organs originated from primary tumours independently.
Clonal origin and spread of metastatic CRC
Next, we reconstructed genomic architecture of each sample and tracked the clonal dynamic to determine the clonal origin of metastasis. By combining sequencing coverage with inferred copy numbers and estimated tumour purities, the cellular prevalence was estimated after filtering out neutral tail mutations.24 25 Mutations with similar cellular prevalence across all samples in each patient were inferred as a tumour clone (online supplemental figure S6). For these six patients, a median of 5 clones (range from 4 to 6) was identified. The possible hierarchies of the tumour clones in each patient were estimated using a genetic algorithm (ie, MACHINA) and mapped to the spread path.26 The routes of clonal evolution were visualised with a tree representing the metastasis direction, from primary tumour to liver and lung (figure 3A–F and online supplemental figure S7).

Clonal origin and spread of metastatic CRC. (A–F) Tumour clones were identified as clusters of mutations with similar cellular prevalence in each patient. Each tumour clone was represented by its estimated clonal frequency (left). Evolution trees of tumour clone for each case representing the metastasis direction from the primary tumour to liver and lung were illustrated (middle). The migration history was inferred for each case using machina (right). The driver genes of each clone were annotated. The most recent common ancestor for liver metastasis and lung metastasis were shaped as a square. Subclones were shaped as a hollow circle/square. CRC, colorectal cancer; LIM, liver metastasis; LUM, lung metastasis; PRM, primary tumour; TLN, thoracic lymph node.

Characterisation of the metastasizing clones in CRC. (A) Illustration of the monophyletic and polyphyletic seeding. D1–D4, driver gene mutations (B) the relative time of metastases after surgery for primary tumours were compared between monophyletic and polyphyletic seeding cases. metastases seeded by advanced subclones with additional driver gene mutations were coloured as red. Liver metastases and lung metastases are shaped as circle and inverted triangle respectively. (C) Metastases seeded by advanced subclones were observed later than those seeded by early clones. (*p<0.05, two-sided t-test). (D) Six mutational subtypes in primary tumours, liver metastases and lung metastases is shown. (*p<0.05, Wilcoxon rank-sum test). (E) The relative contributions of COSMIC mutational signatures in mutations occurred in primary site, liver and lung of each patient (left) and pie charts indicating signature distributions (right) were demonstrated between monophyletic and polyphyletic seeding cases. (F) The mutation characteristics of signature 17 is presented (left). The relative weights of signature 17 were significantly elevated in mutations occurred in liver/lung (n=12) compared with those in primary site (n=6). (*p<0.05, Wilcoxon rank-sum test). (G) Correlations between relative weight of signature 17 and the relative time of metastases after surgery. Shading indicates the 95% CI of the linear regression. Pearson correlation coefficient and p value are reported. CRC, colorectal cancer; LIM, liver metastasis; LUM, lung metastasis; PRM, primary tumour.
In the reconstructed evolution tree, clone containing the full complement of alterations common to all subsequent subclones in LIM and LUM was annotated as the most recent common ancestor (MRCA).27 The MRCA of patient C01 and C02 in PRM (ie, clone 3 in C01–C02) was disseminated to liver and transformed into dominant clone in LUM (figure 3A,B). For patient C03, metastatic competence is acquired within the MRCA (clone 2), which drives dissemination to multiple sites of liver and lung (figure 3C), supporting the recent divergence of metastases (figure 2C). Meanwhile, for patient C04-C06, LUM emerged from more advanced subclones in PRM, (ie, clone 3 in C04; clone 4 in C05 and clone 5 in C06), while LIM originated from the MRCA directly except for patient C05, whose LIM also emerged from another advanced subclones in PRM (ie, clone 2 in C05) (figure 3D–F). Based on the clone trees, migration history was inferred for each case, which supported the migration pattern reconstructed by Treeomics.
Characterisation of metastasizing clones in CRC
As driver mutations (eg, KRAS and BRAF) increasingly inform treatment decisions in metastatic CRC,28 heterogeneity of these alterations among metastases should be of clinical interest. Based on results above, we observed intermetastatic driver gene heterogeneity in three cases (C04-C06) (figure 3D–F), supporting the polyphyletic seeding of distinct organs.10 Whereas all metastases in patient C01–C03 were attributed to monophyletic seeding events with no additional driver gene observed (figures 3A–C and 4A). Notably, the rapid progression with multiple clonal drivers in patient C03, which is the only monophyletic seeding case with branch-off model, supports a model of diaspora metastasis involving multiple sites as previously reported in other tumours (hereafter referred to as ‘diaspora model’).12 29 In another hand, LUM in cases with polyphyletic seeding were all evolved from advanced subclones with additional driver mutations in PRM (secondary APC mutation of clone 3 in C04, TCF7L2 mutation of clone 4 in C05, FLT3 mutation of clone 5 in C06), which were present at clonal frequencies in LUM as determined by target sequencing. With the time it takes to anticipate additional driver mutations, the probability of intermetastatic driver gene heterogeneity negatively correlates with metastatic potential of tumour clones.17 To estimate and compare the metastatic potential of cases with monophyletic and polyphyletic seeding, we reviewed the time elapsed from primary tumour resection to the diagnosis of metastases (figure 4B and online supplemental data S1). Metastases seeded by advanced clones with additional driver mutations were predominantly observed later than those seeded by early clones (p=0.045) (figure 4C). These observations provide a possible connection between the metastatic potential of tumour clones and their consequent metastatic model.
Next, we conducted mutational spectrum analysis, and identified significant enrichment of T>G transversions in metastatic samples compared with primary tumours (6.3% vs 2.9%, p=0.041) (figure 4D). To explain such mutational shift and elucidate the dynamics of mutational signatures over time, we quantified the relevant contributions of COSMIC mutational signatures in mutations stratified by sample site (figure 4A). To exclude the possibility of overfitting, we only fitted signature 1, 5, 6, 10 and 17 as recommended for CRC/pancancer.30 31 Interestingly, the relative weight of signature 17 was significant higher in liver and lung compared with those in primary site (p=0.014) (figure 4E,F), accounting for the enrichment of T>G transversions in metastatic samples. Moreover, the relative weight of signature 17 positively correlated with time elapsed between primary surgery and metastasis diagnosis (r=0.524, p=0.026; figure 4G), which is consistent with previous reports.32 As it has been proposed that signature 17 is elevated in brain metastasis of CRC,31 our data provide a dynamic vision of mutational shift from liver to extrahepatic organs and implicate signature 17 as a potential feature of metastatic evolution.
Candidate driver gene in metastatic CRC
Somatic mutations may contribute to metastatic phenotypes, while metastatic competence can arise from heterogeneous cancer cell populations without need for acquisition of additional mutations.33 Although metastatic advantage provided by additional driver mutations is limited,34 such competence can still benefit from metastatic phenotypes conferred by initial mutations in the trunk of tumour evolution.35 Therefore, we analysed the landscape of driver mutations in all tumour samples. Overall, recurrent trunk mutations were observed not only in well-known driver genes (eg, APC and TP53), but also a novel candidate (ie, ZFP36L2) (figure 5A). Notably, ZFP36L2 mutations were enriched in the trunk clones of cases with monophyletic seeding, highlighting the potential role of ZFP36L2 in tumourigenesis and metastasis. In total, four ZFP36L2 somatic trunk mutations were identified in three out of six patients, with one patient (C03) carrying biallelic mutations (figure 5B and online supplemental figure S8A,B). The only point mutation (ie, C206Y) carried by patient C01 located in the conserved domain and was predicted to impair the interaction between ZFP36L2 and zinc ion (figure 5B), which is crucial for protein-RNA binding. The other three frameshift mutations induced loss of ZFP36L2 expression in primary and metastatic tumour samples (figure 5C). Moreover, downregulation of ZFP36L2 was also observed in LIM/LUM of patient C06 with no ZFP36L2 coding mutation, possibly due to epigenetic silencing.
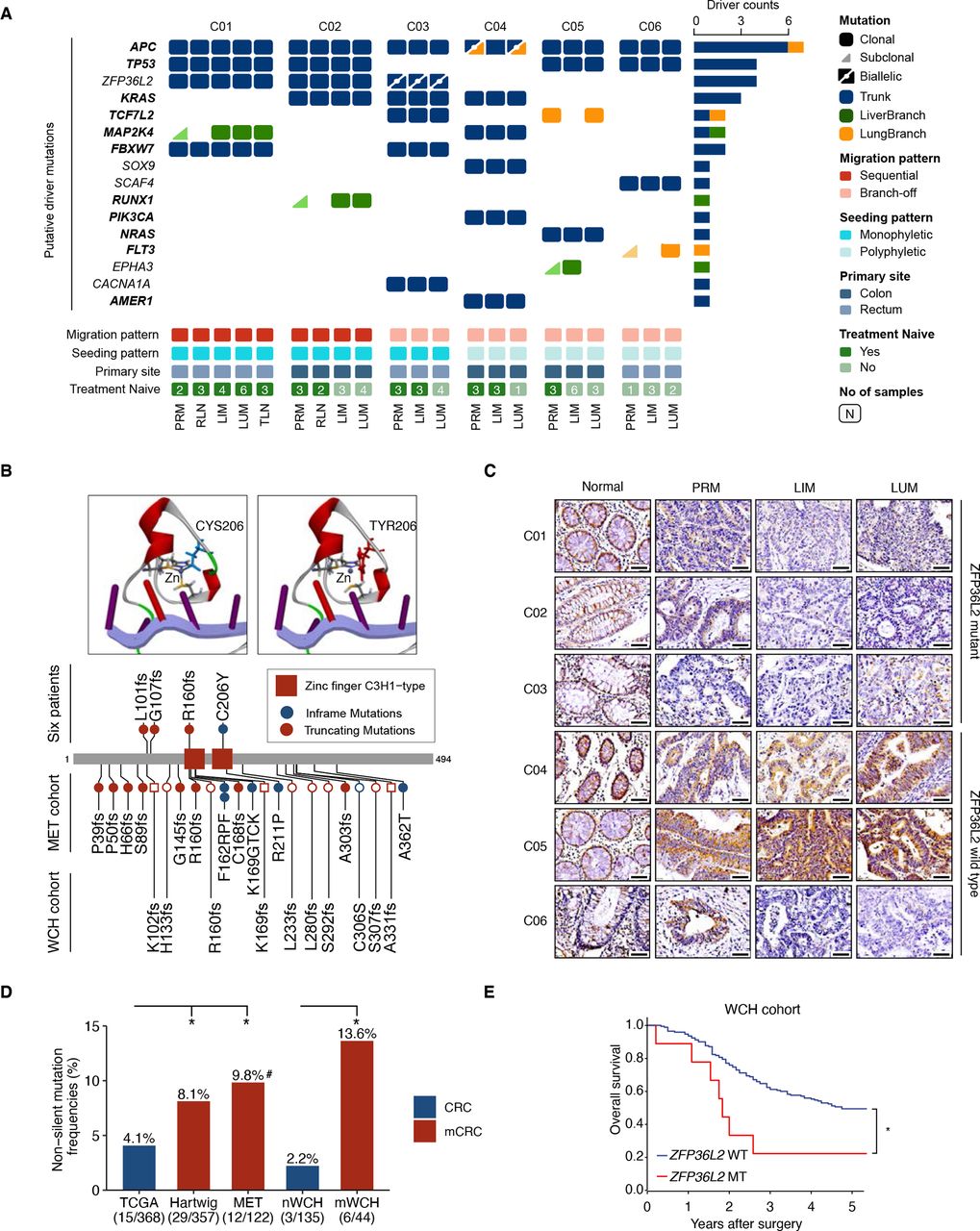
Identification of ZFP36L2 as a candidate driver gene in metastatic CRC. (A) An overview of somatic putative driver mutations detected in matched primary and metastatic tumours across six patients. The mutation events of each gene across all patients are shown. Genes in the COSMIC cancer gene census are bolded. Trunk, LiverBranch and LungBranch mutations were colour-labelled as blue, green, and orange respectively. Biallelic mutations are shown as triangles with a dot-labelled split line. (B) The wide-type and C206Y mutated zinc finger domain structure of ZFP36L2. C206Y mutated ZFP36L2 lost the interaction with zinc atom (top). Detailed information of ZFP36L2 mutations in different cohorts. circle and square indicate patients in mWCH and nWCH, respectively (bottom). (C) Immunohistochemical staining of ZFP36L2 in matched primary and metastatic tumours across six patients; scale bar, 50 µm. (D) Non-silent mutation frequencies of ZFP36L2 in five cohorts of microsatellite stable CRC: TCGA, primary tumour samples from COAD/READ in TCGA; Hartwig cohort, metastatic colorectal cancer samples from Hartwig cancer cohort; MET cohort, distant metastatic samples; nWCH, primary tumour samples from patients with no distant metastasis; mWCH, primary tumour samples from patients with distant metastasis; (*p<0.05; χ2 test; #mutated in both primary tumours and liver metastasis); (E) Kaplan-Meier estimates of overall survival in the WCH cohort stratified by the ZFP36L2 mutation status (*p<0.05, log-rank test). CRC, colorectal cancer; LIM, liver metastasis; LUM, lung metastasis; PRM, primary tumour; TCGA, The Cancer Genome Atlas.
ZFP36L2 is frequently mutated in CRC (online supplemental figure S8C) but rarely estimated in metastatic CRC except a recent investigation in Hartwig cohort.36 In this case, we conducted target sequencing for ZFP36L2 in two independent validation cohorts, including 146 patients with CRC with primary tumour and matched liver/lung metastasis (MET cohort), and 210 consecutive patients with CRC with primary tumours only (WCH cohort) (online supplemental methods, online supplemental figure 1). Patients in WCH cohort were further classified into metastatic CRC (mWCH) and non-metastatic CRC (nWCH) based on the development of distant metastasis before or within 3 years after primary surgery. After excluding MSI cases (online supplemental figure S8D, E), non-silent mutations of ZFP36L2 (mostly frameshifts, figure 5B) were significantly enriched in metastatic CRC (12/122, 9.8% in patient-matched primary tumour and metastasis from MET cohort; 29/357, 8.1% in metastatic CRC from Hartwig cohort; and 6/44, 13.6% in mWCH cohort), which is higher than that in CRC cohort of TCGA (15/368, 4.1%) and nWCH (3/135, 2.2%) (figure 5D and online supplemental table S4, S5). Moreover, patients with ZFP36L2 mutations were associated with poor overall survival in WCH cohort (p=0.015) (figure 5E). These observations, in conjunction with the recurrent ZFP36L2 mutations in monophyletic seeding cases, underscore a potential causal role of ZFP36L2 loss in CRC metastasis.
To determine the impact of ZFP36L2 on the metastatic potential of CRC cells, CRISPR/Cas9 engineered knockout of ZFP36L2 in three CRC cell lines (HCT15, HCT116 and SW480) were confirmed using Sanger sequencing and Western blot analysis (figure 6A and online supplemental figure S9). Metastatic potential of CRC cells was assessed through Transwell migration and matrigel invasion assays. As shown in figure 6B, loss of ZFP36L2 expression in CRC cells conferred enhanced invasion and migration ability, suggesting an inhibitory effect of ZFP36L2 on metastatic potential. Together, these data confirmed the high prevalence of ZFP36L2 deficiency in metastatic CRC and supported its impact on metastatic progression.

Loss of ZFP36L2 enhances the metastatic potential of CRC cells (A) Western blot of CRISPR/Cas9 engineered knockout of ZFP36L2 and control cells. (B) Transwell matrigel invasion and migration assay were performed in ZFP36L2-KO and parental HCT15/HCT116/SW480 cells. Representative images (left) and statistical analyses (right) of the migrated/invaded cells are shown. Scale bar, 100 µm. Data represent the mean±SD and are representative of three independent experiments. (***p<0.001, ANOVA with Tukey post hoc test). ANOVA, analysis of variance; CRC, colorectal cancer; KO, knockout.
Discussion
The direct cause of most cancer-related death is distant metastasis, pattern of which is highly variable depending on the cancer types. For cancer types with direct access to the systemic circulation (eg, lung cancer), monophyletic seeding was initially illuminated by comparative genomic analyses, indicating that additional driver mutation may not be needed for distant metastasis. This assumption is further supported by the computational estimation, claiming not enough time for the acquisition of new driver mutation and subsequent clonally expanding for polyphyletic seeding.17 As the venous drainage of large intestine is primarily into the portal vein via the mesenteric veins, liver is not only the first stop for haematogenous spread of CRC cells but also a barrier to the systemic circulation. It remains largely unknown whether the model of systemic metastasis in CRC is comparable to other cancer types.
Based on our data, the anatomic assumption that CRC cells spreads to distant organs in a colon/rectum-liver-lung order has been genetically evidenced, illustrating as sequential model. In clinical practice, a solitary liver metastasis should be removed to prevent further disease progression and improve the prognosis of patients with CRC. However, decisions regarding the resection timing and patient suitability remain controversial. The presence of sequential metastasis enforces the importance of timely resection of liver metastases, which is consistent with a recent randomised controlled trial.16 Our data also support a branch-off model where LIM and LUM originated from PRM independently. Although the lymphatic origin of distant metastasis was not supported by our data, theoretically CRC cells may seed extrahepatic metastases via lymphatic circulation by entering the subclavian vein. In addition to the portal vein system, venous drainage of rectum may also occur via middle rectal veins into the internal iliac veins, and inferior rectal veins into the internal pudendal veins, providing a bypass for the haematogenous spread of CRC cells. Moreover, formation of portosystemic shunts in the scenario of portal hypertension (eg, cirrhosis, diffuse liver metastases) may drive CRC cells into systemic circulation directly.37 Together, these anatomical features provide explanations for the direct seeding of extrahepatic organs from primary CRC.
A notable finding is that we observed intermetastatic driver gene heterogeneity between LIM and LUM in cases of branch-off model, which is in contrast to a recent genomic study showing minimal functional driver gene heterogeneity among metastases.17 A possible explanation for this distinction is that we extend our study to the scenario of systemic metastases rather than a single organ site. Liver acts as a physiological barrier capturing massive tumour cells from the portal vein,38 which may increase the time window for extrahepatic metastasis, enabling the acquisition of additional driver mutations and subsequent polyphyletic seeding. Moreover, adaptive selection of features to survival in distinct microenvironments of liver and lung may differ the spread paths.3 In fact, previous studies have determined that metastatic clones differ in their ability to colonise different organs,39–41 providing experimental evidence supporting the branch-off model. The fact that several extrahepatic metastases originated from advanced subclones coincides with the earlier finding of frequent gain of additional functional mutations in brain metastases of CRC.42 Thus, the branch-off model not only proposes an alternative explanation for the solitary extrahepatic metastasis clinically observed, but also provides genomic evidence for the organ tropism of metastatic CRC cells. More importantly, the importance of inter-metastatic driver gene heterogeneity may be determined by different responses of metastases to clinical treatments.
The driver mutations may not only participate in tumourigenesis but also confer distinct phenotypic features of tumour cells, including metastatic potential. Alterations of driver genes (eg, KRAS and PIK3CA) may provide metastatic advantages and are involved in targeted therapies for metastatic CRC.28 43 The number of clonal-mutated driver genes in the majority of CRC ranges from three to six,44 increased of which has been implicated as a causal role in rapid progression and associated with diaspora model in several cancers.12 29 45 As such, a thorough screening of potential driver genes of CRC is needed. In our study, recurrent alterations in ZFP36L2 were identified in monophyletic seeding cases and enriched in metastatic CRC. Envisaged as an RNA-binding protein which regulates gene expression by promoting mRNA decay,46 the consequences of ZFP36L2 dysfunction in CRC remain largely unknown. Our study showed that mutated ZFP36L2 (mostly frameshift) resulted in loss of its protein expression, conferring enhanced metastatic potential of CRC cells. Together, these data implicate ZFP36L2 mutation as a potential key driver event in CRC metastasis, but its causal role for CRC progression should be clarified in future functional investigations.
Several limitations should be noted. First, our metastatic samples were not all treatment naïve. Except those from patient C01, most samples taken from secondary operations or later were obtained after adjuvant therapy. However, these samples did not demonstrate an increased number of somatic mutations compared with those obtained before treatment (online supplemental table S2). Thus, adjuvant therapy does not appear to affect phylogenic reconstruction, which is consistent with a previous study.11 Second, as our samples were surgically resected specimens, our sample size was limited (eg, 1 case for diaspora model), and we were unable to analyse metastases that occurred later with no surgical indications. Thus, we cannot determine the origin of these inoperable metastases, especially in cases with polyphyletic seeding. Due to the diversity of metastatic models and the difference in timing of diagnosis, the metastatic models might follow a sequential appearance rather than mutual exclusiveness. Moreover, the inference of migration pattern and clone origin may be greatly influenced by the lack of sampling of primary tumour. For instance, model inference would be problematic for patient C06 if clone 5 (with FLT3 mutation) was missed in the single primary tumour, indicating the importance of enough sampling. Further studies with a larger sampling design are needed to explore the existence of new or mixed models.
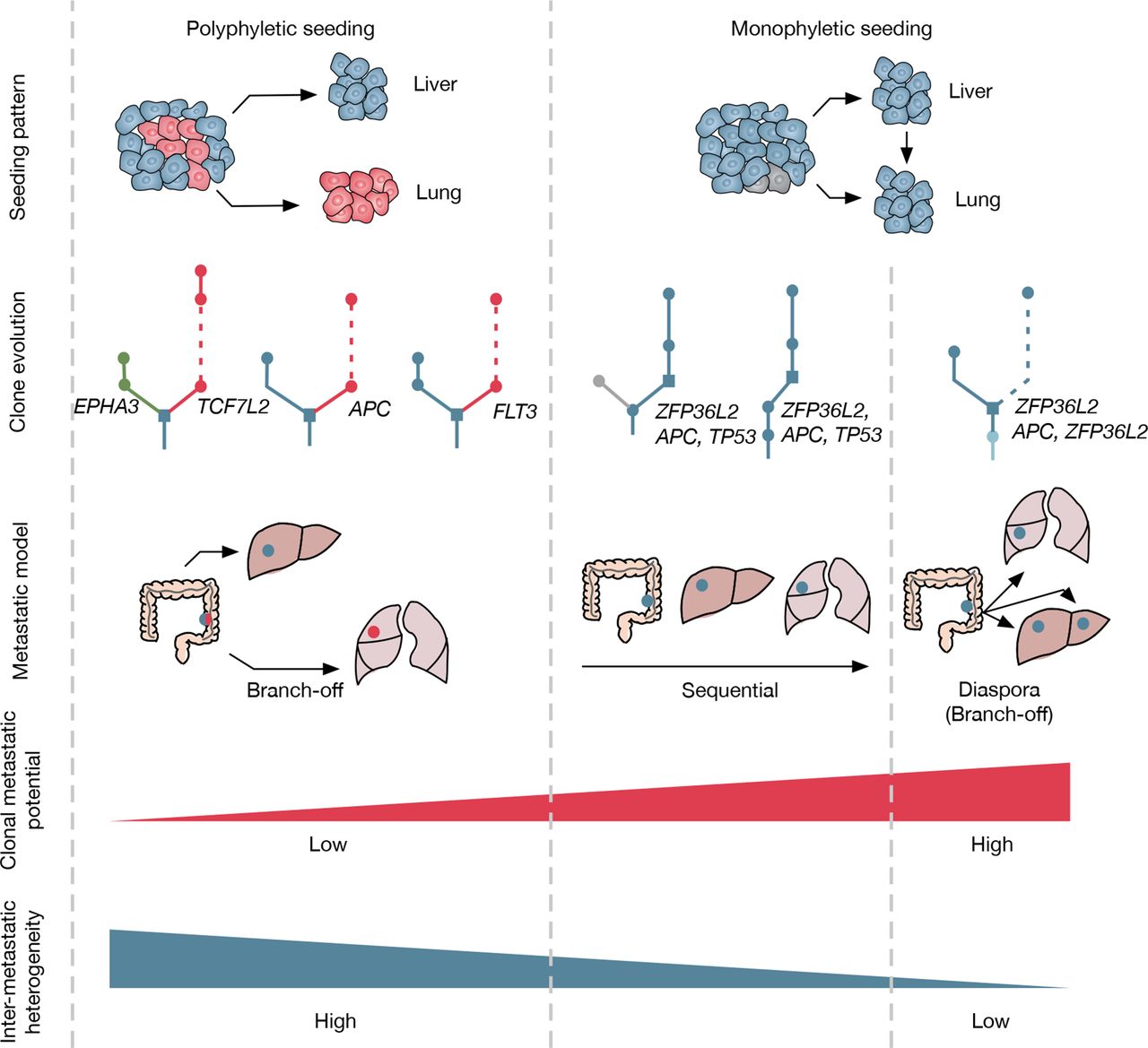
Since therapeutic strategies and treatment outcomes of CRC patients mostly depend on the monitoring of distant metastasis, it is crucial to understand the systemic nature of metastasis and the consequent patterns. Although organ-specific patterns of metastasis are proposed for several cancer types based on anatomy, the defined organ tropisms are not rigid and limited to experimental settings and clinical observations. Here, we described the genomic evolution of CRC in the scenario of systemic metastasis. Through the use of phylogenic analysis, our study provides genomic evidence for the classical sequential model, as well as two novel models (branch-off and diaspora) of metastasis in CRC (figure 7). Moreover, ZFP36L2 is identified as a potential driver gene involved in metastatic progression of CRC. Given the diversity of metastatic models and intermetastatic driver gene heterogeneity, biopsy and sequencing of metastases may be considered for therapeutic decision making in patients with CRC, especially for those with systemic or late postoperative metastasis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of metastatic models in this study seeding patterns for polyphyletic and monophyletic cases were illustrated. Red cells refer to tumour clones acquired additional driver mutations during the growth of the primary tumour. For clone evolution, different colours were labelled for liver or lung branch if metastases origin from advanced tumour clones with additional driver mutations. Three metastatic models were concluded eventually, including the branch-off model (lung metastasis originated from primary tumour, polyphyletic seeding), the sequential model (lung metastasis originated from liver metastasis, monophyletic seeding), and the diaspora model (lung metastasis originated from primary tumour, monophyletic seeding). Clonal metastatic potential was inferred based on the time elapsed from primary surgery to metastasis diagnosis. Intermetastatic heterogeneity refers to the probability of driver gene heterogeneity among liver and lung metastasis.
Data availability statement
Data are available on reasonable request. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, HX (xuheng81916@scu.edu.cn). Most data supporting the findings of this study are available within the article and onlin supplemental files. The raw sequencing data from this study have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, under accession numbers HRA000046 (BioProject: PRJCA001380) that can be accessed at http://bigd.big.ac.cn/gsa-human. All other data are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Ethics Committee of West China Hospital, Sichuan University (2018(280); 2019(338)).
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
H-NC, YS and FL contributed equally.
Contributors HX, Z-GZ and H-NC designed the study, HYZ collected and prepared clinical samples, H-NC, W-HZ, YQ, ZW, HL, YZ, LieY and J-KH gathered the clinical data, H-NC, YS, SYZ, MYC and XYX analysed the data, FL, XL, MCL, QLL, ZNX and QQH proceeded the experiments, XHF, BL, HBH, CHH, YP, WC, LZD, LiY, WZ, BD and YL vouch for the data and the analysis, YQW, HX and Z-GZ supervised the project. H-NC, HX, YS, SYZ and FL wrote the paper, all authors approved and decided to publish the paper.
Funding This work was supported by (1) National Natural Science Foundation of China (82073246, 81673452, 81702378, 81821002 and 81902437); (2) National key research development programme of China (2016YFC0905000 and 2016YFC0906000); (3) China Postdoctoral Science Foundation (2019T120845, 2018M643496, 2019M653428 and 2019M653418); (4) Sichuan Science and Technology Programme (2019YFS0263); (5) Post-Doctor Research Project, West China Hospital, Sichuan University (19XJ0075, 2018HXBH007 and 18HXBH024); (6) 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYYC20003, 2016105, ZYJC18004).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.